
ABSTRACT
The sole proteases of the macroautophagy/autophagy machinery, the ATG4s, contribute to autophagosome formation by cleaving Atg8-family protein members (LC3/GABARAPs) which enables Atg8-family protein lipidation and de-lipidation. Our recent work reveals that ATG4s can also promote phagophore growth independently of their protease activity and of Atg8-family proteins. ATG4s and their proximity partners including ARFIP2 and LRBA function to promote trafficking of ATG9A to mitochondria during PINK1-PRKN mitophagy. Through the development of a 3D electron microscopy framework utilizing FIB-SEM and artificial intelligence (termed AIVE: Artificial Intelligence-directed Voxel Extraction), we show that ATG4s promote ER-phagophore contacts during the lipid-transfer phase of autophagosome biogenesis, which requires ATG2B and ATG9A to support phagophore growth. We also discovered that ATG4s are not essential for removal of Atg8-family proteins from autolysosomes, but they can function as deubiquitinase-like enzymes to counteract the conjugation of Atg8-family proteins to other proteins, a process that we have termed ATG8ylation (also known as LC3ylation). These discoveries demonstrate the duality of the ATG4 family in driving autophagosome formation by functioning as both autophagy proteases and trafficking factors, while simultaneously raising questions about the putative roles of ATG8ylation in cell biology.
Since the discovery of ATG4s, their contribution to autophagy has been solely attributed to proteolytic processing of the ubiquitin-like Atg8-family proteins. The de-lipidation function of human ATG4s in living cells is poorly understood while the overall interplay of ATG4s and Atg8-family proteins in human cells has largely remained unclear. We set out to tackle these knowledge gaps and in doing so uncovered unexpected roles for ATG4s, including a proteolytic and Atg8-family protein independent role in autophagosome formation, and a role in counteracting the attachment of Atg8-family proteins onto other proteins (termed ATG8ylation, or LC3ylation).
Using multigene knockout cell lines, our initial analyses set out to address the in vivo activity of each ATG4 family member toward each Atg8-family protein member, as well as the specificity of ATG4s during PINK1-PRKN mitophagy and the role of de-lipidation [Citation1]. The first unexpected result came about during the de-lipidation analyses. Using GABARAPL1-G, a GABARAP mutant that can be lipidated in the absence of ATG4 cleavage, we observed robust removal of GABARAPL1-G from autolysosomes in the absence of ATG4s both during PINK1-PRKN mitophagy and starvation-induced autophagy. This finding indicates that the protease activity of ATG4s is not essential for Atg8-family protein de-lipidation/removal from autolysosomes. Therefore, it is likely that additional mechanisms involved in the removal/recycling of lipidated Atg8-family proteins from autolysosomes remain to be discovered (). We speculate that alternative proteases are involved, or that lysosome biogenesis pathways such as autophagic lysosome reformation may contribute to Atg8-family protein removal/recycling.
Figure 1. The multifaceted functions of human ATG4s. The canonical function of ATG4s is to process Atg8-family proteins to prime these proteins for lipidation onto phagophore membranes (1). However, during PINK1-PRKN mitophagy, ATG4s also regulate ATG9A trafficking to mitochondria via their proximal partners, ARFIP2 and LRBA, to promote ER-phagophore contacts which are established by ATG2-ATG9A. This function of ATG4s is independent of their protease activity and of Atg8-family proteins (2). ATG4s can function as deubiquitin-like enzymes to regulate levels of ATG8ylation through a process named de-ATG8ylation (3) but are not essential for the removal of lipidated Atg8-family proteins from autolysosomes (4)
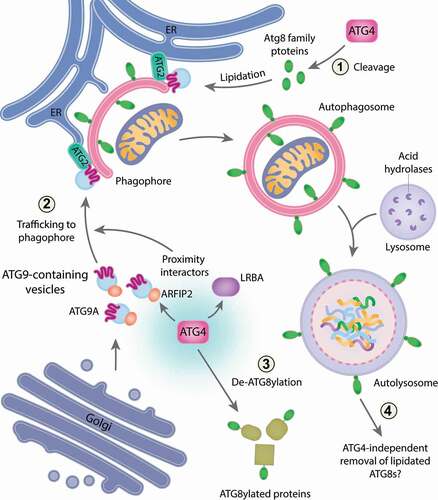
Along with Robin Ketteler’s laboratory, we discovered that the ubiquitin-like Atg8-family proteins can be covalently linked to other cellular proteins in a post-translational modification we have termed ATG8ylation (). In the absence of ATG4 protease activity, ATG8ylation levels are increased, and this can be reversed by the re-introduction of ATG4s. Therefore, ATG4s can function as deubiquitinase-like enzymes to remove Atg8-family proteins from ATG8ylated protein targets (de-ATG8ylation) (). Our analyses also identified ATG16L1 as a substrate of ATG8ylation. Given that ATG16L1 plays roles in interferon signaling and inflammation, it would be interesting to examine the potential role of ATG8ylation in modulating ATG16L1’s activity during these processes. Additionally, systematically determining the substrate profile of ATG8ylation for each Atg8-family protein member can provide new clues about potential cellular roles of ATG8ylation and which pathways it can contribute to. This would be an important step to clarify whether ATG8ylation is a spurious event, or a specifically regulated process involved in cell biology.
The second unexpected result from the study came about during analyses of autophagosome formation. Based on our previous work, cells lacking Atg8-family proteins generate small autophagosomes which fail to undergo fusion with lysosomes during mitophagy. If ATG4s function exclusively via Atg8-family proteins, then it would be expected that cells lacking ATG4s should have a similar phenotype. However, ATG4 family knockout cells fail to form autophagosomes. Instead, cells lacking ATG4s can only generate phagophores that rarely grow into fully formed autophagosomes. This result pointed toward an additional role for ATG4s during autophagosome biogenesis that does not require Atg8-family proteins. To test this possibility, we knocked out genes encoding the ATG4 family in Atg8-family protein gene hexa KO cells to create an ATG4 Atg8-family protein gene deca KO (hereafter deca KO). Knockout of all ATG4s in Atg8-family protein gene hexa KO cells recapitulates the phenotype of ATG4 tetra KO cells, supporting the idea that ATG4s can function to promote phagophore growth independently of Atg8-family proteins. Notably, the function of ATG4s in driving phagophore growth is also independent of their protease activity.
We next sought to understand how ATG4s promote phagophore growth by mapping the ATG4 proximity network using APEX2 biotinylation labeling. ATG4 proximity partners include factors that regulate protein transport, and membrane and lipid trafficking including ATG9 vesicles. Indeed, we discovered that ATG9A recruitment to mitochondria during PINK1-PRKN mitophagy is defective in deca KO cells compared to controls. The ATG4 proximity partners, ARFIP2 and LRBA, are also involved in the regulation of ATG9A trafficking and are required for efficient PINK1-PRKN mitophagy. ARFIP2 has previously been shown to play a role in ATG9A trafficking during starvation-induced autophagy. However, the analyses revealed a hitherto unknown function for LRBA, a protein associated with immune deficiency and autoimmunity, in ATG9A trafficking. Whether LRBA’s role in mitophagy contributes to disease pathogenesis remains to be determined.
Given that the ATG9-ATG2 axis has previously been shown to mediate lipid transfer from the ER to phagophores, we hypothesized that ATG4s promote ER-phagophore contacts to drive phagophore growth. To address this hypothesis, Focused Ion Beam-Correlative Light Electron Microscopy (FIB-CLEM) was used to visualize phagophore structures in 3D. To enable an accurate, unbiased, and rapid (compared to traditional approaches) quantitative assessment of phagophore structures, we developed an artificial intelligence driven framework to automate reconstruction of the volumetric electron microscopy data (termed AIVE (Artificial Intelligence-directed Voxel Extraction)). GFP-ATG2B labeled phagophores in cells lacking ATG4s cells are smaller and have less ER contacts relative to their size. This supports a model in which ATG4s, through ATG9A trafficking, promote ER-phagophore contacts required for phagophore expansion. However, the work raises additional questions around how ATG4s might function during ATG9A trafficking. One possibility is that ATG4s act as scaffolds which bring trafficking factors together with ATG9A vesicles.
Collectively, our study has shown additional roles for the ATG4 family during autophagosome formation which sit outside of the Atg8-family protein lipidation system. By doing so we demonstrate that ATG4s are protease-dependent and -independent autophagy factors. The discoveries open the door for further investigations into understanding how ATG9A is trafficked and identifying further mechanisms of Atg8-family protein removal from autolysosomes. The ATG4 proximity networks also indicate that ATG4s may play additional cellular roles that lie outside the sphere of autophagosome formation, while the potential cellular functions of ATG8ylation await to be discovered.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- Nguyen TN, Padman BS, Zellner S, et al. ATG4 family proteins drive phagophore growth independently of the LC3/GABARAP lipidation system. Mol Cell. 2021;81(9):2013–2030 e9. .