
ABSTRACT
Macroautophagy/autophagy is primarily considered as a degradative pathway via the lysosome, yet the secretory functions of autophagy proteins have recently been unveiled. Autophagy proteins have been implicated in metabolic organ development, homeostasis and function, and deficiency in autophagy is associated with metabolic disorders. However, the molecular mechanisms by which autophagy proteins regulate energy metabolism and insulin sensitivity were unclear. We previously showed that systemic activation of autophagy by a hyperactive BECN1F121A mutant reduces insulin storage in islets but improves insulin sensitivity systemically. In our recent study, we found that BECN1 functions in adipose tissue to systemically regulate energy metabolism. Adipose-specific expression of BECN1F121A is sufficient to improve systemic insulin sensitivity without negatively affecting pancreatic insulin storage. We demonstrated that BECN1 interacts with exocyst subunit proteins and facilitates the secretion of an adipokine, ADIPOQ (adiponectin, C1Q and collagen domain containing), in adipose tissue. Thus, our findings suggest that BECN1 regulates insulin sensitivity in a non-degradative and non-cell autonomous manner by facilitating ADIPOQ secretion. Our study also highlighted the distinct functions of autophagy proteins in different metabolic tissues.
Maintaining systemic energy homeostasis is critical to prevent metabolic disorders, such as type 2 diabetes (T2D). T2D is a chronic metabolic disease characterized by hyperglycemia, pancreatic β cell dysfunction, and insulin resistance in metabolic organs. The role and mechanism of autophagy in insulin sensitivity and energy metabolism remain poorly understood. In the past 15 years, the role of autophagy in metabolism was primarily studied using autophagy-deficient mice. To better elucidate the function of the autophagy machinery in metabolic regulation, we generated an autophagy-hyperactive knock-in mouse model, carrying a single amino acid substitution, F121A, in the BH3 domain of the key autophagy protein BECN1 (BECN1F121A). The advantage of the BECN1F121A model is that unlike other autophagy-activating (gain-of-function) mice or compounds that induce autophagy supraphysiologically, the BECN1F121A mutant disrupts a nutrient-dependent BECN1-BCL2 interaction and activates autophagy to a physiological level. Using this mouse model, we previously found that autophagy plays distinct roles in insulin-producing β cells and insulin-responsive metabolic organs such as liver and skeletal muscle. In response to high-fat diet feeding, BECN1F121A mice are more glucose intolerant than wild-type (WT) mice, due to decreased glucose-stimulated insulin secretion caused by excess autophagic degradation of insulin granules (vesicophagy) in pancreatic β cells. However, these BECN1F121A mutant mice are not insulin resistant. Instead, their systemic insulin sensitivity is improved compared to WT mice, which cannot be explained by insulin storage defects. Our current study [Citation1] addressed this question, and resolved why global autophagy hyperactivation impairs, while adipose-specific autophagy activation improves, glucose tolerance.
Besides being more insulin sensitive, BECN1F121A mice also show higher activation of AMPK signaling in metabolic organs (liver and muscle) than WT mice. Importantly, the serum from BECN1F121A mice is sufficient to activate AMPK in cultured WT cell lines, but primary cells isolated from BECN1F121A mice, once cultured in vitro, do not show activated AMPK, indicating the presence of a circulating factor that activates AMPK in BECN1F121A mice. By profiling of metabolic hormones, we identified the circulating factor as ADIPOQ, an adipose-derived hormone (adipokine). ADIPOQ can activate the AMPK and PPARA (peroxisome proliferator activated receptor alpha) signaling pathways via binding ADIPOQ receptors, and exert versatile anti-diabetic effects in muscle, liver, and vascular systems. We found that adipose-specific BECN1F121A expression is sufficient to increase serum ADIPOQ levels, non-adipose AMPK activity, and systemic insulin sensitivity, which is similar to whole-body BECN1F121A expression. Notably, different from global BECN1F121A mice, because BECN1F121A is not expressed and thus vesicophagy is not induced in β cells of adipose-specific BECN1F121A mice, glucose tolerance is also improved in these mice.
We explored multiple possible mechanisms of BECN1-mediated ADIPOQ secretion. Basal autophagy removes unnecessary organelles in preadipocytes and is required for the development of white adipose tissue. However, we found that hyperactive autophagy does not accelerate the process. Both body weight and adipose tissue mass of BECN1F121A mice are similar to that of WT mice. In addition, neither confocal microscopy nor biochemical autophagosome isolation experiments indicate the presence of ADIPOQ inside autophagosomes, suggesting that ADIPOQ is not secreted by an autophagosome-based unconventional secretory pathway as is IL1B. Instead, we found that the subcomplex I proteins of the exocyst, a multi-subunit complex involved in vesicle trafficking, EXOC2/SEC5, EXOC3/SEC6 and EXOC4/SEC8, but not the subcomplex II proteins such as EXOC8/EXO84 and EXOC7/EXO70, are novel binding partners of BECN1, through its coiled-coil domain. The subcomplex I subunit proteins also show stronger binding affinity with BECN1F121A than with WT BECN1 by co-immunoprecipitation. Because the BECN1F121A mutant cannot bind and be inhibited by BCL2, we propose that increased BECN1-exocyst binding affinity is a result of increased protein trafficking of BECN1 from the mitochondria and ER pool to ADIPOQ vesicles, and not due to structural or conformational changes induced by the F121A mutation per se ().
Figure 1. Proposed model of BECN1-regulated adiponectin secretion in adipose tissue. When released from BCL2 inhibition, BECN1 is translocated from the ER-Golgi-mitochondrial pools to ADIPOQ vesicles. There it interacts with the exocyst EXOC3/SEC6-EXOC4/SEC8 subcomplex and promotes adiponectin secretion, leading to improvement of systemic insulin sensitivity by activating the AMPK signaling pathway in metabolic organs
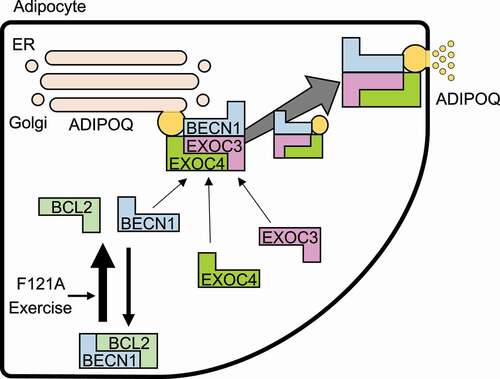
The role of exocyst proteins in adipose tissue has been understudied. We found that exocyst complex subunits do not function in the same manner during secretion of different adipokines. For example, shRNA knockdown of Exoc3/Sec6 almost completely blocks ADIPOQ secretion from cultured adipocytes, but only marginally reduces the secretion of LEP (leptin), another adipokine regulating appetite and energy balance. Knockdown of Exoc4/Sec8 also inhibits ADIPOQ secretion, which is likely mediated via EXOC3/SEC6, because Exoc4/Sec8 knockdown also decreases EXOC3/SEC6 protein levels. Based on recent publications, EXOC4/SEC8 and EXOC3/SEC6 form a tight subcomplex in the exocyst through the coiled-coil region of each protein; thus, depleting one subunit may well affect the stability of another. In contrast, although interacting with BECN1, EXOC2/SEC5 is not required for secretion of ADIPOQ in adipocytes. This is reminiscent of published findings in Drosophila showing that Sec5 deficiency does not affect the release of neurotransmitters. We speculate that EXOC2/SEC5 may be partially functionally redundant with BECN1 in certain secretory cells, and therefore its function can be compensated by the BECN1-EXOC3/SEC6 interaction. To further understand how BECN1-exocyst proteins regulate ADIPOQ secretion, spatiotemporal analyses by live-cell imaging, as well as structural studies among the SEC proteins, will be necessary.
Of note, the BECN1-exocyst pathway shows specificity in adipokine regulation. We found that different from ADIPOQ, serum levels of LEP and RETN (resistin; another adipokine) are indistinguishable between adipose-specific BECN1F121A mice and control WT mice. Blocking the BECN1-EXOC3/SEC6 interaction also does not affect LEP secretion from cultured adipocytes. This is in line with two of our observations: first, the localizing region of ADIPOQ vesicles is distinct from that of LEP vesicles in primary adipocytes, with the former in the cell center and the latter in the peripheral area; and second, there is a lower colocalization of BECN1 with LEP vesicles than with ADIPOQ vesicles by confocal imaging. Furthermore, because Exoc3/Sec6 knockdown blocks ADIPOQ secretion but only reduces LEP secretion to a limited extent, we speculate that different adipokine vesicles may be associated with different sets of exocyst proteins. This may be an important mechanism to ensure that adipokine secretion is timely and specifically regulated, because otherwise, all adipokines would be secreted simultaneously once the exocyst pathway is activated, which is not what is observed in cultured adipocytes or in vivo. In terms of BECN1, it is known to localize at the trans-Golgi network (TGN) where secretory proteins are sorted. BECN1 may be specifically involved in the sorting of ADIPOQ at the TGN and in the segregation of ADIPOQ secretory vesicles by interacting with the exocyst subcomplex I proteins. Besides ADIPOQ, LEP and RETN, adipose tissue secretes a variety of other hormones. Further studies are needed to test the above hypotheses, and demonstrate how BECN1, and its binding partners in the class III phosphatidylinositol 3-kinase autophagy complex, function in the secretory pathway in adipocytes and various other endocrine cells.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Kuramoto K, Kim YJ, Hong JH, et al. The autophagy protein Becn1 improves insulin sensitivity by promoting adiponectin secretion via exocyst binding. Cell Rep. 2021;35(8):109184. Epub 2021/ 05/27. PubMed PMID: 34038729; PMCID: PMC8177967. doi: https://doi.org/10.1016/j.celrep.2021.109184.