
ABSTRACT
Epidemiological and clinical studies have shown that exposure to particulate matter (PM) is associated with an increased incidence of lung cancer and metastasis. However, the underlying mechanism remains unclear. Here, we demonstrated the central role of PM-induced neutrophil recruitment in promoting lung cancer metastasis. We found that reactive oxygen species (ROS)-mediated alveolar epithelial macroautophagy/autophagy was essential for initiating neutrophil chemotaxis and pre-metastatic niche formation in the lungs in response to PM exposure. During PM-induced autophagy, the E3 ubiquitin ligase TRIM37 was degraded and protected TRAF6 from proteasomal degradation in lung epithelial cells, which promoted the NFKB-dependent production of chemokines to recruit neutrophils. Importantly, ROS blockade, autophagy inhibition or TRAF6 knockdown abolished PM-induced neutrophil recruitment and lung metastasis enhancement. Our study indicates that host lung epithelial cells and neutrophils coordinate to promote cancer metastasis to the lungs in response to PM exposure and provides ideal therapeutic targets for metastatic progression.
Abbreviations: ACTA2/α-SMA: actin alpha 2, smooth muscle, aorta; ATII: alveolar type II; Cho-Traf6 siRNA: 5'-cholesterol-Traf6 siRNA; EMT: epithelial-mesenchymal transition; HBE: human bronchial epithelial; HCQ: hydroxychloroquine; MAPK: mitogen-activated protein kinase; NAC: N-acetyl-L-cysteine; NFKB: nuclear factor of kappa light polypeptide gene enhancer in B cells; NS: normal saline; PM: particulate matter; ROS: reactive oxygen species; TRAF6: TNF receptor-associated factor 6; TRIM37: tripartite motif-containing 37.
Introduction
The impact of atmospheric pollution on health is widely recognized as an increasingly serious public health issue [Citation1,Citation2]. Extensive epidemiological data show that exposure to atmospheric pollution, especially atmospheric particulate matter (PM), not only exacerbates airway inflammatory diseases such as asthma and chronic obstructive pulmonary disease (COPD) but is also associated with significant increases in the morbidity and mortality of lung cancer, especially among nonsmokers [Citation3–6]. Long-term exposure to high concentrations of PM accelerates lung function decline and is estimated to be responsible for approximately 500,000 lung cancer deaths worldwide [Citation1,Citation7]. Previous experimental studies demonstrated that inhaled PM can be deposited in different compartments of the respiratory tract and interact with epithelial cells and resident immune cells [Citation8,Citation9]. Inflammatory responses, airway remodeling and epithelial-mesenchymal transition (EMT) are thought to play important roles in the PM-induced development of pulmonary disease [Citation10–13]. In addition, several reports have indicated that PM can promote tumor cell invasion in vitro via alterations in global microRNA expression [Citation14,Citation15]. However, the effects of PM exposure on the development of lung metastasis and the specific molecular mechanisms underlying these effects remain poorly defined.
Multiple studies have demonstrated that exposure to PM increases oxidative stress and autophagy (an evolutionarily conserved process of self-cellular component recycling) in lung epithelial cells [Citation12,Citation13,Citation16–18]. Autophagy plays dual roles in tumor development via complex mechanisms that suppress the tumorigenic process during the early stages of malignancy but also promote the progression of advanced cancer [Citation19–21]. Autophagy can promote both the spread of malignant cancer cells through the circulation [Citation22] and the colonization of target organs by metastatic cells [Citation23]. Therefore, we aimed to explore whether PM-induced autophagy plays a central role in the mechanism through which PM exposure affects lung metastasis.
As a member of the TRAF (TNF receptor-associated factor) family, TRAF6 was first identified as a key mediator of inflammatory signaling [Citation24,Citation25]. Recently, the roles of TRAF6 in autophagy and cancer have attracted great interest. On the one hand, autophagy induced by the activation of TLR4 or TLR3 increases the K63-linked ubiquitination of TRAF6, thus activating the MAPK and NFKB pathways and facilitating lung cancer cell invasion [Citation26]. On the other hand, TRAF6 catalyzes the K63-linked polyubiquitination of LC3B and inhibits colorectal cancer metastasis through the regulation of selective autophagic CTNNB1/β-catenin degradation [Citation27]. In addition, various signaling pathways affected by TRAF6 have been implicated in cancer metastatic processes. For example, the TRAF6-ATM-H2AX signaling axis promotes tumorigenesis and metastasis through the activation of HIF1A/HIF1α signaling [Citation28]. Considering the functional roles of TRAF6 in mediating autophagy and inflammation-associated metastasis, we sought to determine the effects of TRAF6 on lung metastasis during PM exposure.
Here, we demonstrate that PM exposure promotes lung metastasis by recruiting neutrophils to the lungs. We further demonstrate that reactive oxygen species (ROS)-induced autophagy is essential for PM-induced neutrophil infiltration and metastatic progression. The PM-induced autophagic degradation of TRIM37 led to the accumulation of TRAF6 in airway epithelial cells, thus inducing NFKB pathway activation and inflammatory chemokine production. The inhibition of ROS activity or autophagy, as well as silencing TRAF6, attenuated the PM-induced enhancement of lung metastasis. Therefore, our study provides new insight into the PM exposure associated enhancement of metastasis and may contribute to the development of novel therapeutic strategies for cancer.
Results
PM exposure promotes lung pre-metastatic niche formation and lung metastasis
To determine the effect of PM exposure on lung metastasis, PM was administered via intranasal instillation, while B16-F10 melanoma cells or Lewis lung carcinoma (LLC) cells were injected via the tail vein into C57BL/6 mice on the indicated days (). We found that PM markedly increased the lung metastasis of B16-F10 and LLC cells ( and S1A), as evidenced by an increase in lung metastatic foci in mice exposed to PM ( and S1B). In addition, PM exposure promoted the seeding of mCherry+ B16-F10 cells in the lungs (Figure S1C,D). We also evaluated spontaneous lung metastasis of subcutaneously implanted 4T1 breast tumors in BALB/c mice (). After surgical removal of the primary tumor, PM treatment resulted in more severe lung metastasis of 4T1 tumor (). These data indicate that exposure to PM increases the risk of tumor metastasis to the lungs.
Figure 1. PM exposure promotes lung pre-metastatic niche formation and lung metastasis. (A-C) Schematic illustration of the PM exposure (100 µg) time points and the establishment of lung metastatic tumors (A). Representative images (left) and quantitative analysis (right) of the lung metastasis of B16-F10 tumor cells in normal saline (NS)- or PM (100 µg)-treated mice, as detected by luciferase-based bioluminescence imaging (B). Representative lung appearance (left) and statistical analysis (right) of lung nodules in NS- or PM (100 µg)-treated mice bearing B16-F10 tumor cell lung metastases (C). (D-E) Schematic illustration of the establishment of 4T1 spontaneous lung metastatic models and the PM exposure (100 µg) timepoints (D). H&E-stained lung sections (left) and quantification (right) of the lung metastatic burden in NS- or PM (100 µg)-treated mice bearing 4T1 tumor lung metastases. Scale bar: 50 µm (E). (F-G) Quantitative PCR analysis of pro-metastatic genes (F), representative images of ACTA2/α-SMA-stained lung sections (left) and quantitative analysis of ACTA2/α-SMA expression levels (right) (G) in the lungs of mice after 5 exposures to PM (100 µg) over 10 days. Scale bars: 100 μm. The error bars indicate the mean ± SD values (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001 (unpaired Student’s t test). The data are one representative experiment of three independent experiments. Each independent experiment had three samples (n = 3) per group, and each dot represents one sample.
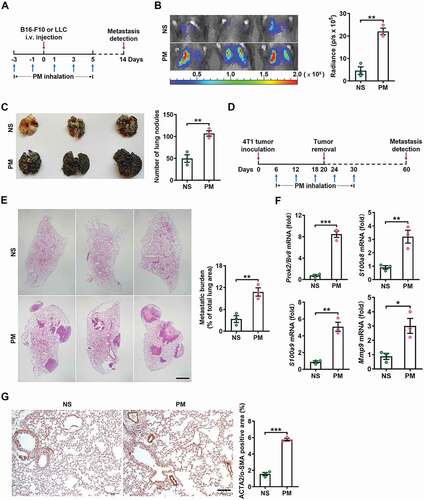
The “seed and soil” hypothesis proposes that an environment conducive to metastasis, referred to as the pre-metastatic niche, is required for the dissemination and colonization of tumor cells in distant target organs [Citation29]. To investigate whether PM can promote pre-metastatic niche formation in the lungs, we measured the expression of characteristic niche genes, including Prok2/Bv8, S100a8, S100a9 and Mmp9, which have been reported to promote tumor cell migration, invasion and colonization at metastatic sites [Citation30]. The levels of these genes were significantly increased in the lungs of PM-treated mice (), and the gene expression levels of S100a8 and S100a9 were mainly upregulated in PTPRC/CD45+ ITGAM/CD11b+ GSR/Gr1+ neutrophils (Figure S1E). Furthermore, the protein expression levels of ACTA2/α-SMA (actin alpha 2, smooth muscle, aorta), a marker of cancer-associated fibroblasts (CAFs) and EMT, were upregulated in the lungs of PM-treated mice () [Citation31,Citation32]. However, among EMT-related genes, PM exposure only affected the transcription of Acta2/Asma, as we detected no upregulation of other EMT-related markers at the transcript level (Figure S1F). In vitro, PM exposure did not affect the expression of any EMT-related genes except for Acta1/Asma or affect cell morphology (Figure S1G,H). Thus, PM exposure promotes pre-metastatic niche formation in the lungs.
PM promotes lung metastasis via a neutrophil-dependent mechanism
Immune cells induce pre-metastatic niche formation by remodeling the local tumor microenvironment [Citation33,Citation34]. Therefore, we analyzed the differences in the lung immune cell landscape between normal saline (NS)-treated and PM-treated mice. PM exposure significantly increased the proportion of ITGAM/CD11b+ myeloid cells (Figure S2A). Further analysis showed that the proportion of ITGAM/CD11b+ GSR/Gr1+ neutrophils but not ITGAM/CD11b+ ADGRE1/F4/80+ macrophages was greatly increased in response to PM (). In addition, immunofluorescence staining revealed a significant increase in LY6G+ neutrophil accumulation in the lungs of PM-treated mice (). PM exposure did not affect the percentages of CD19+ B cells, KLRB1C/NK1.1+ NK cells ITGAX/CD11c+ MHC-II+ dendritic cells, CD4+ T cells or CD8+ T cells, (Figure S2B). These results indicate that PM exposure promotes the recruitment of neutrophils to the lungs.
Figure 2. PM promotes lung metastasis via a neutrophil-dependent mechanism. (A) The frequency of neutrophils and macrophages in the lungs of C57BL/6 mice was examined by flow cytometry after NS or PM (100 µg) exposure for 24 h. (B) Immunofluorescence staining (left) and quantitative analysis (right) of LY6G+ neutrophils in the lungs of NS- or PM (100 µg)-treated mice. Scale bar: 100 μm. (C) The gene expression levels of Cxcl1, Cxcl2, Cxcl5, Cxcl8, Ccl3 and Cxcl12 in the lung tissues of mice exposed to PM (100 µg) for 12 h were analyzed by quantitative PCR. (D) The protein expression levels of CXCL2 and IL8 in the BALF of mice exposed to PM (100 µg) were analyzed by ELISA. (E) The gene expression levels of Cxcl1, Cxcl2 and Cxcl5 and the frequency of neutrophils in lung tissues after PM (100 µg) exposure for the indicated durations were analyzed by quantitative PCR and flow cytometry. (F) The protein expression levels of VEGF in lung tissues were measured by ELISA. (G) Immunofluorescence analysis of the presence of extravasated dextran in lung tissues after NS or PM (100 µg) exposure. Scale bars: 100 μm. (H) The frequency of neutrophils in the lungs of mice after NS or PM (100 µg) exposure and anti-KDR/VEGFR-2 treatment was determined by flow cytometry. (I) Bioluminescent images (left) and quantitative analysis (right) of B16-F10 cell lung metastasis in mice after NS or PM (100 µg) exposure and neutrophil depletion with anti-GSR/Gr1. The error bars indicate the mean ± SD values (n = 3). A, B, C, D and F *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant (unpaired Student’s t test). E, H and I *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant (one-way ANOVA test). The data are one representative experiment of three independent experiments. Each independent experiment had three samples (n = 3) per group, and each dot represents one sample.
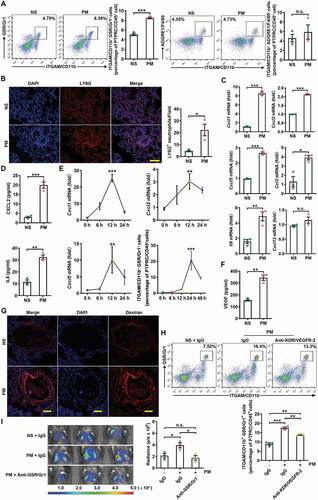
We next assessed the expression levels of Cxcl1, Cxcl2, Cxcl5, Cxcl12, Ccl3 and Il8, which are the chemokines responsible for neutrophil chemotaxis. We found that the gene expression levels of Cxcl1, Cxcl2, Cxcl5, Ccl3 and Il8 but not Cxcl12 were significantly upregulated in the lungs of PM-treated mice (), and elevated protein expression levels of CXCL2 and IL8 were also observed in bronchoalveolar lavage fluid (BALF) (). While we observed no increases in the transcript levels of Cxcr1, Cxcr2 or Cxcr4, the transcript level of Ccr2 was significantly increased in neutrophils in the lungs of PM-treated mice (Figure S2C). In addition, the expression levels of the pro-inflammatory cytokines Il6 and Il1b were upregulated in lung tissues after PM exposure (Figure S2D). The kinetics of these changes in chemokine and neutrophil levels were analyzed and indicated that the upregulation of chemokine expression occurred earlier than the increase in neutrophil accumulation, which is consistent with the causal relationship between these events (). Thus, PM exposure promotes neutrophil recruitment by upregulating the production of chemokines.
Cytokines, including VEGF, PGF/PLGF, CSF1/M-CSF, CSF2/GM-CSF, and CSF3/G-CSF, have been implicated in the mobilization of myeloid cells [Citation35]. We found that PM greatly enhanced the expression level of Vegfa but failed to increase the expression of Pgf/Plgf, Csf1/M-csf, Csf2/Gm-csf, or Csf3/G-csf in the lungs (Figure S2E). Elevated VEGF protein expression was also observed in the lungs ().
VEGF is also involved in tumor-induced neoangiogenesis [Citation36,Citation37]. We detected more PECAM1/CD31+ endothelial cells in the lungs of PM-treated mice than in those of NS-treated mice (Figure S2F). We next analyzed vascular leakage, which is an early event in pre-metastatic niche formation in the lungs. PM exposure markedly enhanced lung vascular permeability, as evidenced by the presence of extravasated dextran (). To examine the role of VEGF in PM-induced neutrophil recruitment, VEGF was blocked by anti-KDR/VEGFR-2. Anti-KDR/VEGFR-2 significantly reduced the PM-induced recruitment of neutrophils into the lungs (). Therefore, VEGF is involved in PM-induced neutrophil recruitment into the lungs.
Neutrophils support pre-metastatic niche formation and metastatic progression [Citation38–41]. Thus, we determined the effects of neutrophils on PM-induced lung metastasis. After anti-GSR/Gr1-mediated neutrophil depletion, PM no longer promoted the lung metastasis of B16-F10 cells (). In NOD-Prkdcscid il2rg−/- mice, which have extensive immune cell defects but an abundance of neutrophils, PM exposure enhanced the lung metastasis of B16-F10 cells, which was accompanied by neutrophil accumulation in the lungs (Figure S2G,H). Consistent with its effect on neutrophil recruitment, anti-KDR/VEGFR-2 treatment also impaired the PM-induced lung metastasis of B16-F10 cells (Figure S2I). Taken together, these results show that PM induces lung metastasis via neutrophil recruitment.
PM-induced lung metastasis is dependent on autophagy
Airborne PM induces autophagy in human lung A549 cells and human bronchial epithelial (HBE) cells [Citation12,Citation16], and autophagy is related to IL8 secretion [Citation16]. Therefore, we investigated whether autophagy participated in PM-induced lung metastasis. Consistent with previous publications, PM dose-dependently induced the conversion of LC3B-I to LC3B-II in HBE cells (Figure S3A). In HBE cells with mRFP-GFP-LC3 overexpression, we found increased numbers of autophagosomes (yellow puncta) and autolysosomes (red puncta) after PM exposure, excluding the possibility that the increase in autophagosomes resulted from dysfunctional autophagic flux (Figure S3B). Moreover, in mice expressing the autophagy reporter GFP-LC3, PM significantly increased the number of GFP-LC3 puncta in the lungs (). Thus, PM exposure induces autophagy in lung epithelial cells.
Figure 3. PM-induced lung metastasis is dependent on autophagy. (A) Expression of GFP-LC3 and EPCAM and the numbers of GFP puncta in the lungs of mice exposed to NS or PM (100 µg) for 24 h. Scale bars: 100 μm. (B) The frequency of neutrophils in the lungs of lc3b-/- mice and their wild-type (WT) littermates after PM (100 µg) exposure was examined by flow cytometry. (C) Bioluminescent images (left) and quantitative analysis (right) of B16-F10 cell lung metastasis in lc3b-/- mice and their WT littermates after NS or PM (100 µg) exposure. (D) Quantitative PCR analysis of pro-metastatic genes in the lungs of NS- or PM (100 µg)-treated lc3b-/- mice or their WT littermates. (E) The protein expression levels of CXCL2 and IL8 in BALF of Sftpc-CreER × Atg5fl/fl mice and Sftpc-CreER mice after exposure to PM (100 µg) were analyzed by ELISA. (F) The frequency of neutrophils in the lungs of Sftpc-CreER × Atg5fl/fl mice and Sftpc-CreER mice after PM (100 µg) exposure was examined by flow cytometry. (G) Bioluminescent images (top) and quantitative analysis (bottom) of B16-F10 cell lung metastasis in Sftpc-CreER × Atg5fl/fl mice and Sftpc-CreER mice after NS or PM (100 µg) exposure. (H) Bioluminescent images (left) and quantitative analysis (right) of B16-F10 cell lung metastasis in NS- or PM (100 µg)-exposed mice with or without HCQ (60 mg kg−1) treatment. The error bars indicate the mean ± SD values (n = 3). A **P < 0.01 (unpaired Student’s t test). B to H *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant (two-way ANOVA test). The data are one representative experiment of three independent experiments. Each independent experiment had three samples (n = 3) per group, and each dot represents one sample.
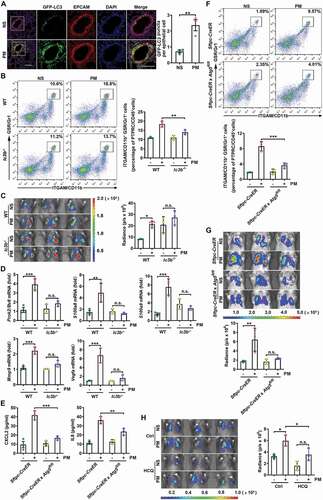
To confirm the role of autophagy in PM-induced neutrophil recruitment, we determined the number of neutrophils in the lungs of lc3b-/- and Becn1+/- mice, which have autophagy deficiency in the lungs and other organs. While PM exposure elicited an increased accumulation of neutrophils in the lungs of autophagy competent mice, it exhibited less significant induction of neutrophil infiltration into the lungs of lc3b-/- and Becn1+/- mice ( and S3C). It should be noted that lc3b-/- and Becn1+/- mice had higher levels of metastasis than their littermates in the absence of PM exposure. Correspondingly, PM did not promote the lung metastasis of B16-F10 cells in lc3b-/- or Becn1+/- mice ( and S3D). In addition, the secretion of CXCL2 and IL8 was significantly reduced in the lungs of PM-treated lc3b-/- and Becn1+/- mice (Figure S3E,F). The PM-induced increases in Prok2/Bv8, S100a8, S100a9, Mmp9 and Vegfa transcripts were also abolished in the lungs of lc3b-/- and Becn1+/- mice ( and S3G). These results demonstrate that autophagy is required for pre-metastatic niche formation by recruiting neutrophils.
Next, we evaluated how autophagy in epithelial cells was involved in neutrophil recruitment. We confirmed that PM induced more GFP-LC3 puncta in EPCAM+ epithelial cells than in EPCAM− cells (). Compared with those of EPCAM− cells in the lungs, higher chemokine gene expression levels were induced in EPCAM+ epithelial cells by PM (Figure S3H). By using Sftpc-CreER(Esr1) × Atg5fl/fl mice, in which autophagy is specifically deficient in alveolar type II (ATII) cells, we found that the PM-mediated induction of CXCL2 and IL8 protein expression was largely impaired, accompanied by a decrease in neutrophil accumulation in the lungs compared with those of Sftpc-CreER mice (). In addition, the PM-induced lung metastasis of B16-F10 cells was greatly attenuated in Sftpc-CreER × Atg5fl/fl mice (). Collectively, these data indicate that increased autophagy in airway epithelial cells is important for PM-induced neutrophilia and subsequent lung metastasis.
To clarify whether autophagy inhibition is a therapeutic strategy for PM-related lung metastasis in the clinic, we further explored the impact of hydroxychloroquine (HCQ), a pharmacological autophagy inhibitor that is effective in the treatment of malaria and lupus erythematosus in clinic [Citation42,Citation43], on PM-induced lung metastasis. We found that HCQ showed significant inhibitory effects on PM-induced lung metastasis and pro-metastatic gene expression ( and S3I). Thus, HCQ is a promising drug for treating PM-related lung metastasis.
PM-induced ROS are responsible for enhanced autophagy and lung metastasis
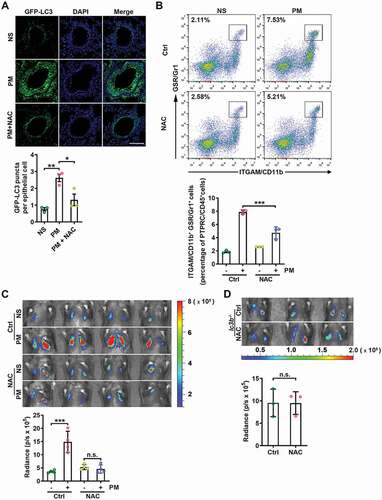
PM has been reported to induce both ROS and autophagy in lung epithelial cells [Citation16,Citation44], and ROS can promote autophagy [Citation45,Citation46]. Therefore, we hypothesized that PM induces autophagy in airway epithelial cells via ROS. First, we confirmed that ROS were increased in PM-treated HBE cells (Figure S4A). The blockade of intracellular ROS with the antioxidant N-acetyl-L-cysteine (NAC) abolished the PM-induced conversion of LC3B-I to LC3B-II, autophagic flux, and increases in the protein expression levels of CXCL2 and IL8 in HBE cells (Figure S4B–D). Moreover, NAC treatment also decreased the number of GFP-LC3 puncta in the lungs of GFP-LC3 mice exposed to PM (). NAC treatment significantly reduced PM-induced neutrophil recruitment to the lungs and abrogated the enhancement of B16-F10 cell lung metastasis in response to PM exposure (). In addition, NAC had no significant effect on the PM-induced lung metastasis of B16-F10 cells in lc3b−/- mice (). These data demonstrate that PM-induced lung metastasis is mediated by ROS-induced autophagy.
Figure 4. PM-induced ROS are responsible for enhanced autophagy and lung metastasis. (A) GFP-LC3 expression and the numbers of GFP puncta in the airway epithelial cells of mice exposed to NS or PM (100 µg) with or without NAC (275 mg kg−1) treatment for 24 h. Scale bar: 100 μm. (B) The frequency of neutrophils in the lungs of NS- or PM (100 µg)-exposed mice with or without NAC (275 mg kg−1) treatment was examined by flow cytometry. (C) Bioluminescent images (top) and quantitative analysis (bottom) of B16-F10 cell lung metastasis in NS- or PM (100 µg)-exposed mice with or without NAC (275 mg kg−1) treatment. (D) Bioluminescent images (top) and quantitative analysis (bottom) of B16-F10 cell lung metastasis in PM (100 µg)-exposed lc3b−/- mice with or without NAC (275 mg kg−1) treatment. The error bars indicate the mean ± SD values (n = 3 or 4). A *P < 0.05; **P < 0.01 (one-way ANOVA test). B and C ***P < 0.001; n.s., not significant (two-way ANOVA test). D n.s., not significant (unpaired Student’s t test). The data are one representative experiment of three independent experiments. Each independent experiment had three or four samples (n = 3 or 4) per group, and each dot represents one sample.
PM-induced TRAF6 accumulation contributes to lung metastasis by activating the NFKB pathway
The MAPK and NFKB signaling pathways contribute to the induction of inflammatory chemokines [Citation47,Citation48]. We next examined whether these pathways affected PM-induced increases in the levels of related inflammatory chemokines in airway epithelial cells. Both the MAPK (JNK, ERK, p38) and NFKB (RELA/p65) signaling pathways were obviously activated in PM-treated HBE cells (). However, only the NFKB pathway specific inhibitor BAY11-7082 completely abolished the PM-mediated upregulation of CXCL2 and IL8 expression in HBE cells (), indicating the decisive role of NFKB pathway in this process.
Figure 5. PM-induced TRAF6 accumulation contributes to lung metastasis by activating the NFKB pathway. (A) Western blot analysis of the indicated proteins in HBE cells exposed to NS or PM (100 μg/ml) for the indicated times. (B) The protein expression levels of CXCL2 and IL8 in the culture supernatants of HBE cells exposed to PM (100 μg/ml) and treated with the indicated inhibitor (MAPK/JNK inhibitor, SP600125, 10 μM; MAPK/ERK inhibitor, U0126, 10 μM; MAPK/p38 inhibitor, SB203580, 10 μM or NFKB (RELA/p65) inhibitor, BAY11-7082, 10 μM) were analyzed by ELISA. (C) Western blot analysis of TRAF6 and TRAF2 in HBE cells exposed to NS or PM (100 μg/ml) for the indicated times. (D) Representative immunofluorescence images of TRAF6 expression in HBE cells treated with NS or PM (100 μg/ml) for 24 h. (E) The protein expression levels of CXCL2 and IL8 in the culture supernatants of HBE cells exposed to PM (100 μg/ml) for 24 h after negative control (NC) siRNA or Traf6 siRNA transfection were analyzed by ELISA. (F) Images and quantitative analysis of TRAF6 expression in the lungs of mice exposed to NS or PM (100 μg) for 24 h. Scale bar: 100 μm. (G) Bioluminescent images (left) and quantitative analysis (right) of B16-F10 cell lung metastasis in Cho-Traf6 siRNA-treated mice with NS or PM (100 μg) exposure. The error bars indicate the mean ± SD values (n = 3). B andF **P < 0.01; ***P < 0.001; n.s., not significant (unpaired Student’s t test). E andG *P < 0.05; **P < 0.01 (two-way ANOVA test). The data are one representative experiment of three independent experiments. Each independent experiment had three samples (n = 3) per group, and each dot represents one sample.
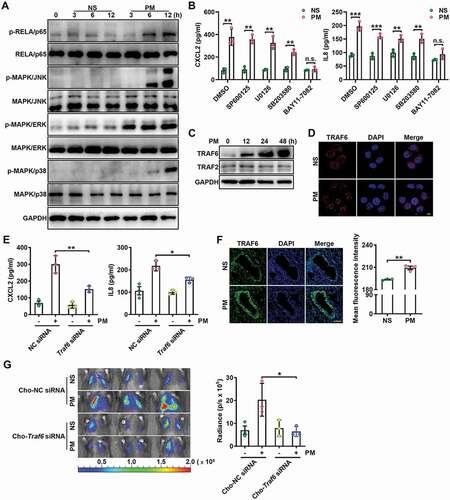
Both TRAF2 and TRAF6 are adaptor molecules that assemble active NFKB signaling scaffolds. We found that PM exposure induced significant accumulation of the TRAF6 protein but not the TRAF2 protein in HBE cells (). In addition, the nuclear localization of TRAF6 increased in HBE cells in response to PM exposure (). Consistently, knockdown of TRAF6 but not TRAF2 blocked NFKB pathway activation in PM-treated HBE cells (Figure S5A,B). More directly, knockdown of TRAF6 attenuated the PM-induced enhancement of CXCL2 and IL8 secretion in HBE cells (). Furthermore, ST2825, a specific MYD88 dimerization inhibitor, could not reverse PM-mediated enhancement of CXCL2 and IL8 production (Figure S5C), suggesting that MYD88 is not upstream of TRAF6 in this process. In addition, PM exposure significantly enhanced TRAF6 protein expression levels in the lungs ().
To further investigate the role of TRAF6 in PM-induced lung metastasis, we administered cholesterol-conjugated Traf6 small interfering RNA (Cho-Traf6 siRNA) to mice before PM exposure and confirmed that Cho-Traf6 siRNA effectively knocked down TRAF6 expression in the lungs (Figure S5D). Cho-Traf6 siRNA treatment inhibited the PM-induced lung metastasis of B16-F10 cells (). Accordingly, in mice transfected with Cho-Traf6 siRNA, PM-induced increases in CXCL2 and IL8 protein expression levels and neutrophil numbers in the lungs were reduced (Figure S5E,F). These results demonstrate that PM promotes lung metastasis via TRAF6-mediated NFKB pathway activation.
PM-induced autophagy prevents the proteasome-dependent degradation of TRAF6
Next, we sought to explore the mechanism by which PM induces TRAF6 accumulation in airway epithelial cells. First, we confirmed that PM did not affect Traf6 gene expression levels in HBE cells (Figure S6A), suggesting that TRAF6 was post transcriptionally regulated. Treatment with the proteasome inhibitor MG132 increased TRAF6 levels and abrogated the PM-mediated accumulation of TRAF6 in HBE cells (). K63-linked ubiquitination of TRAF6 is related to its activation, whereas K48-linked ubiquitination is associated with TRAF6 degradation by the proteasome [Citation49–51]. We found that PM exposure caused a distinct decrease in both the total and K48-linked ubiquitination of TRAF6, although K63-linked ubiquitination was unchanged (Figure S6B), further supporting the occurrence of proteasome-dependent TRAF6 degradation.
Figure 6. PM-induced autophagy prevents the proteasome-dependent degradation of TRAF6. (A) Western blot analysis of TRAF6 protein expression levels in HBE cells treated with PM (100 μg/ml) for 24 h with or without the proteasome inhibitor MG132 (10 μM) (top), and statistical analysis of the relative protein expression levels (bottom). (B) Western blot analysis of TRAF6 protein expression levels in HBE cells treated with PM (100 μg/ml) for 24 h with or without the lysosome inhibitor Baf A1 (10 nM) (top), and statistical analysis of the relative protein expression levels (bottom). (C) Western blot analysis of LC3B-I and LC3B-II levels in HBE cells treated with PM (100 μg/ml) for 24 h with or without MG132 (10 μM) (top), and statistical analysis of the relative protein expression levels (bottom). (D) Representative fluorescence images (top) and quantitative analysis of GFP+ and RFP+ puncta (bottom) in HBE cells transfected with mRFP-GFP-LC3-expressing vectors for 24 h and then treated with NS or PM (100 μg/ml) in the presence of MG132 (10 μM). Scale bar: 5 μm. The error bars indicate the mean ± SD values (n = 3). A to C *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant (two-way ANOVA test). D *P < 0.05; **P < 0.01 (unpaired Student’s t test). The data are one representative experiment of three independent experiments. Each independent experiment had three samples (n = 3) per group, and each dot represents one sample.
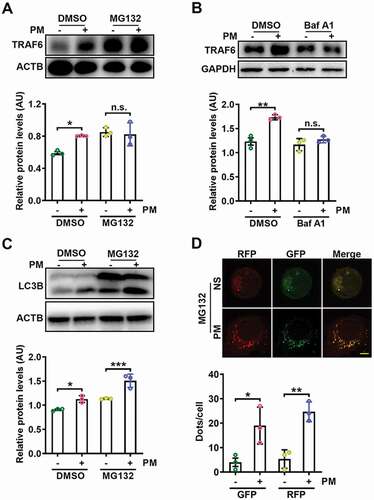
Then, we examined the relationship between PM-induced autophagy and TRAF6 degradation. We found that bafilomycin A1 (Baf A1), an autophagosome inhibitor, abrogated PM-mediated TRAF6 accumulation in HBE cells (). This effect was also observed in HBE cells with ATG3 knockdown (Figure S6C). Moreover, Baf A1 treatment abolished the inhibitory effect of PM on the K48-linked ubiquitination of TRAF6 and reversed PM-induced enhancement of NFKB activation (Figure S6D,E). Since both the proteasome and autophagy mediate TRAF6 degradation, we further investigated which of these contributors was the upstream factor. Even in the presence of MG132, PM increased both the conversion of LC3B-I to LC3B-II and autophagic flux () in HBE cells, suggesting that autophagy induction precedes proteasome-mediated TRAF6 degradation. Thus, these data indicate that PM-induced autophagy reduces the proteasomal degradation of TRAF6.
Autophagy-mediated TRIM37 degradation inhibits TRAF6 degradation via the ubiquitin-proteasome pathway
Since the PM-induced accumulation of TRAF6 was found to be due to a decrease in the autophagy-dependent degradation of TRAF6 by proteasomes, we surmised that an E3 ubiquitin ligase controls the proteasomal degradation of ubiquitylated TRAF6 and that TRAF6 degradation is strongly controlled by PM-induced autophagy. Protein profiling and STRING protein interaction database analysis revealed that interactions may occur among TRAF6, SQSTM1/p62, and the E3 ubiquitin ligase TRIM37 (Figure S7A). Indeed, we confirmed that exogenous TRIM37 and TRAF6 interacted with each other regardless of PM treatment (). Moreover, the interaction between endogenous TRAF6 and TRIM37 was mainly observed in the nucleus and was independent of PM exposure (). We next evaluated whether TRIM37 mediates TRAF6 ubiquitination. TRIM37 overexpression obviously induced the K48-linked but not K63-linked ubiquitination of TRAF6 (). Correspondingly, TRIM37 overexpression led to reduced TRAF6 levels, and this reduction was abolished by MG132 (). These results suggest that TRIM37 acts as an E3 ligase that mediates the degradation of TRAF6 via the ubiquitin-proteasome pathway.
Figure 7. Autophagy-mediated TRIM37 degradation inhibits TRAF6 degradation via the ubiquitin-proteasome pathway. (A) Coimmunoprecipitation and western blot analysis of HEK293T cells co-transfected with Flag-TRAF6 and HIS-TRIM37, stimulated with increasing concentrations of PM (25, 50, 100 and 200 μg/ml) for 12 h, and treated with MG132 (10 μM) for 6 h before harvest. (B) Representative fluorescence images of the colocalization of TRAF6 and TRIM37 in HBE cells treated with NS or PM (100 μg/ml) for 12 h in the presence of MG132 (10 μM). Scale bar: 5 μm. (C) Western blot analysis of the ubiquitination of TRAF6 in HEK293T cells co-transfected with Flag-TRAF6 and HA-K48-Ub or HA-K63-Ub with or without HIS-TRIM37 and treated with MG132 (10 μM) for 6 h before harvest. (D) Western blot analysis of TRAF6 proteins in HBE cells transfected with HIS-TRIM37 or the empty vector and treated with DMSO or MG132 (10 μM) for 6 h before harvest. (E-F) Western blot analysis of TRAF6 protein levels in HBE cells transfected with HIS-TRIM37 or the empty vector (E) or with NC siRNA or Trim37 siRNA (F) and then exposed to NS or PM (100 μg/ml) for 24 h. (G) Representative immunofluorescence images (left) and quantification of the colocalization (right) of LC3B and TRIM37 in HBE cells treated with NS or PM (100 μg/ml) for 12 h with or without Sqstm1 siRNA transfection in the presence of Baf A1 (10 nM). Scale bar: 5 μm. (H) Western blot analysis of TRIM37 and TRAF6 protein levels in HBE cells transfected with NC siRNA or Sqstm1 siRNA, and then exposed to NS or PM (100 μg/ml) for 24 h. (I) Western blot analysis of TRIM37 and TRAF6 protein levels in HBE cells transfected with NC siRNA or Atg3 siRNA and then exposed to NS or PM (100 μg/ml) for 24 h. The error bars indicate the mean ± SD values (n = 3). **P < 0.01; n.s., not significant (one-way ANOVA test). The data are one representative experiment of three independent experiments. Each independent experiment had three samples (n = 3) per group, and each dot represents one sample.
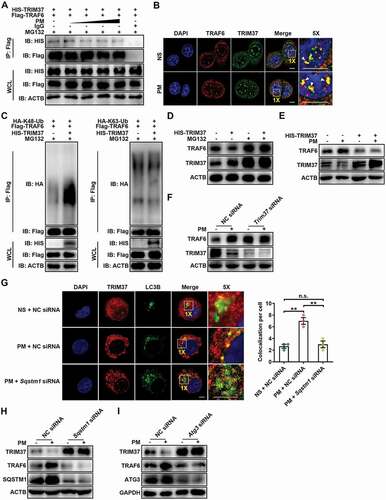
Next, we examined the role of TRIM37 in PM-induced TRAF6 degradation. PM hardly increased TRAF6 in HBE cells with TRIM37 overexpression or with TRIM37 knockdown (). Furthermore, PM failed to reduce the K48-linked ubiquitination of TRAF6 after TRIM37 knockdown (Figure S7B). Consistently, CXCL2 and IL8 levels were not increased by PM in HBE cells with TRIM37 knockdown (Figure S7C). PM-induced NFKB activation was abolished in TRIM37-overexpressing HBE cells (Figure S7D). These results demonstrate that the PM-induced accumulation of TRAF6 is TRIM37 dependent.
Finally, we investigated whether autophagy is involved in TRIM37 degradation. Similar to PM, the autophagy inducer rapamycin sharply reduced TRIM37 levels in HBE cells (Figure S7E), indicating that TRIM37 is degraded via autophagy. Ubiquitinated proteins are sorted into autophagosomes via SQSTM1 [Citation52]. Accordingly, increased TRIM37 ubiquitination and increased interactions between SQSTM1 and TRIM37 were observed after PM exposure (Figure S7F,G). The localization of TRIM37 with autophagosomes was also elevated in PM-treated HBE cells but was impaired by SQSTM1 knockdown (). In addition, SQSTM1 knockdown notably increased TRIM37 protein expression levels and attenuated TRAF6 accumulation and NFKB activation in PM-treated HBE cells ( and S7H). Additionally, we found that ATG3 knockdown or Baf A1 treatment markedly increased TRIM37 levels in PM-treated HBE cells ( and S7I). Accordingly, the increases in CXCL2 and IL8 levels observed in PM-treated HBE cells were reduced by ATG3 knockdown (Figure S7J). Finally, Kaplan-Meier analysis showed that TRIM37 correlated with a good prognosis in non‐small‐cell lung cancer (NSCLC) patients, indicating that TRIM37 exhibits antitumor effects during the progression of NSCLC (Figure S7K). Collectively, these results indicate that PM induces autophagy, enhancing TRIM37 degradation, and preventing the degradation of TRAF6 via the ubiquitin-proteasome pathway.
Discussion
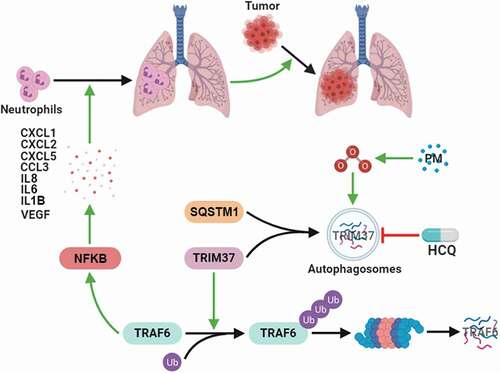
Although numerous epidemiological studies have shown that exposure to pollutant particles increases tumor incidence and mortality, few experimental studies have been conducted to investigate the mechanisms by which PM exposure promotes metastatic progression. In this study, we revealed a novel mechanism by which PM mediates lung metastasis by promoting TRAF6 protein accumulation through ROS-induced autophagy-dependent TRIM37 degradation in alveolar epithelial cells, which results in enhanced NFKB pathway activation and chemokine production, thus promoting pre-metastatic niche formation in the lungs via neutrophil recruitment (). Our work provides new knowledge regarding PM-induced lung metastasis and potential strategies for preventing cancer progression in patients who are frequently exposed to PM.
Figure 8. Working model of the mechanism by which PM controls lung metastasis. PM exposure triggers autophagy in the airway epithelium through increased ROS levels. The E3 ligase TRIM37 is sorted into autophagosomes via SQSTM1 in response to PM-induced autophagy. Autophagy-dependent TRIM37 degradation results in TRAF6 protein accumulation by inhibiting the K48-linked ubiquitination of TRAF6, thereby promoting NFKB pathway activation and inflammatory chemokine production. Finally, neutrophils are recruited to the lungs, thus fostering pre-metastatic niche formation in the lungs and lung metastasis.
Chronic inflammation can modify the local tissue microenvironment to increase the risk of cancer progression [Citation53,Citation54]. Previous publications have demonstrated that inflammatory neutrophils promote tumor cell colonization and metastasis in lung tissues [Citation38–41,Citation55]. Interestingly, in this study, PM-associated lung neutrophilia promoted tumor metastasis to the lungs, and neutrophil depletion abolished this effect. However, the elimination of neutrophils may not be clinically feasible because it could cause systemic neutropenia. We further demonstrated that autophagy deficiency and anti-KDR/VEGFR-2 therapy could reduce neutrophil infiltration and metastatic progression in PM-exposed mice. Specifically, we found that anti-KDR/VEGFR-2 therapy partially decreased PM-induced neutrophil recruitment and metastatic behavior. However, HCQ, an autophagy inhibitor, almost completely eliminated the PM-mediated enhancement of neutrophil migration and metastatic colonization. In addition to inhibiting autophagy during RNA virus infection or autoimmune diseases, HCQ can exert inhibitory effect on inflammation via inhibiting TLR7/9 signaling [Citation56,Citation57]. Specifically, HCQ directly binds to nucleic acids from infecting RNA viruses or endogenous nucleic acids released from apoptotic or necrotic cells within the endosome, thereby preventing nucleic acids from binding to TLRs. In our study, PM did not stimulate TLR7/9 signaling similar to pathogen-derived and synthetic nucleic acids, and TLR7/9 signaling was not involved in PM-induced inflammatory chemokine production. Thus, we believe that HCQ reduces PM-induced metastasis by inhibiting autophagy rather than TLR7/9-related inflammation. Therefore, the application of HCQ or anti- KDR/VEGFR-2 therapy may reduce the PM-associated risk of metastasis and have potential clinical value.
The roles of autophagy within the tumor microenvironment are very complicated [Citation58]. It has been reported that ultrafine PM can be endocytosed into airway epithelial cells and trigger autophagy [Citation16,Citation59]. In the present study, lung metastasis was significantly enhanced in lc3b-/- and Becn1+/- mice, which have systemic autophagic defects, compared with their WT littermates. We suspect that this effect is the result of systemic autophagy deficiency; thus, the detailed mechanism is complicated. Autophagy itself is an important conservative cellular response to maintain homeostasis. Systemic autophagy deficiency destroys the body’s homeostasis, which might enable tumor cells to escape immune surveillance and allow metastasis. To evaluate whether autophagy is involved in regulating PM-induced neutrophil recruitment and lung metastasis, without altering system-level autophagy or disrupting systemic homeostasis, we generated Sftpc-CreER × Atg5fl/fl mice, since PM-induced autophagy mainly occurred in ATII cells. The basal level of metastasis in Sftpc-CreER × Atg5fl/fl mice was similar to that in control mice without PM treatment. More importantly, autophagy defects – whether specifically in ATII cells or throughout the body – completely abolished the PM-induced enhancement of neutrophil recruitment and lung metastasis. Overall, these results indicated that in PM-induced lung metastasis, autophagy in airway epithelial cells was deleterious. Moreover, our findings provide evidence that autophagy inhibition in ATII cells may have therapeutic advantages for PM-induced lung metastasis.
In addition to endocytosis, PM-induced cellular responses may occur via direct interactions with the lipid layer of cellular membranes, cell surface receptor activation, or indirectly through ROS formation [Citation60,Citation61]. Previous publications have demonstrated that PM enhances ROS activity in lung epithelial cells [Citation44], and accumulating evidence has implicated ROS in the induction of autophagy [Citation45,Citation46]. The role of ROS in cancer progression is currently considered either deleterious or protective [Citation62–66]. Hence, we studied the roles of ROS in mediating PM-induced autophagy and metastatic behavior. Administration of the antioxidant NAC reduced autophagy and lung metastasis in PM-exposed mice. This anti-metastatic effect of NAC was abolished in lc3b-/- mice. These data suggest that ROS play a protumor role in PM-associated metastasis by inducing autophagy and that antioxidants may reduce PM-related metastatic risk. In this study, the mechanism of PM-induced ROS production was not explored in detail. ROS might be produced by triggering cellular redox machinery that could be activated by organic chemicals or transition metals carried by PM [Citation60].
The ubiquitin/proteasomal and autophagy/lysosomal protein degradation systems are involved in the progression of inflammation and cancer via the dynamic regulation of cellular organelle and protein levels [Citation19,Citation67–69]. The newly identified E3 ubiquitin ligase TRIM37 is involved in various biological processes [Citation70–72]. We detected an interaction between TRIM37 and TRAF6 and found that PM treatment led to the autophagic degradation of TRIM37 in HBE cells, which reduced the proteasomal degradation of TRAF6; this process led to the accumulation of TRAF6, thus promoting NFKB pathway activation and chemokine production. TRAF6 silencing greatly alleviated PM-induced NFKB pathway activation and abrogated the PM-mediated enhancement of experimental metastasis. Therefore, our data revealed a mechanism by which PM induces TRAF6 accumulation via autophagic degradation of TRIM37, consequently initiating NFKB pathway activation and pro-metastatic inflammatory responses.
Collectively, our findings enhance our understanding of the mechanism by which environmental air pollutants, such as PM, contribute to pre-metastatic niche formation in the lungs and may shed light on potential therapeutic interventions.
Materials and methods
Mice and cell lines
Male C57BL/6 J and BALB/c (6- to 8-week-old) mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd (SCXK 2017–0005). NOD-Prkdcscid il2rg-/- mice were purchased from Biocytogen (110,586). lc3b-/- and Becn1+/- mice were kindly provided by Prof. Zhihua Chen (Zhejiang University, Hangzhou, China). GFP-LC3 mice were kindly provided by Dr. Lionel Apetoh (Université de Bourgogne Franche Comté, France). Atg5fl/fl mice were purchased from RIKEN BRC Experimental Animal Division (RBRC02975). Sftpc-CreER mice were kindly provided by Prof. Kuijun Chen (Third Military Medical University, Chongqing, China). Sftpc-CreER × Atg5fl/fl mice were generated by crossing Atg5fl/fl mice with Sftpc-CreER transgenic mice. Successful Cre-mediated deletion was confirmed by PCR and western blot after mice were intraperitoneally injected with tamoxifen (1 mg/100 μl; Sigma, T5648-1 G) in olive oil for three consecutive days. Mice were maintained under specific pathogen-free conditions, and experimental protocols were reviewed and approved by the Ethics Committee for Animal Studies at Zhejiang University. The PCR primers used for mouse genotyping are listed in Table S1.
HBE, human embryonic kidney HEK293T and murine mammary adenocarcinoma 4T1 cells were purchased from the American Type Culture Collection (CRL-2078, CRL-1573.3, CRL-2539). Luciferase-labeled murine LLC-luci and murine melanoma B16-F10-luci cells were obtained from PerkinElmer (BW124734, BW119267). HBE, 4T1 and LLC-luci cells were cultured in RPMI-1640 medium (Gibco, 11,875,119) supplemented with 10% (v:v) fetal bovine serum (FBS; Gibco, 10,099,141). HEK293T and B16-F10-luci cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 11,965,092) supplemented with 10% FBS. All cells were maintained in a humidified incubator at 37°C with 5% (vol:vol) CO2. All cell lines were routinely tested and confirmed to be negative for Mycoplasma contamination with a Mycoplasma Detection Kit (Lonza, LT07-318).
In vitro PM exposure
Vacuum-packed PM was produced by burning 2-monochlorophenol (2-MCP), a common by-product of waste combustion, with fly ash containing Cu(II)O at 230°C (defined as MCP230), as previously described [Citation44]. PM used in this study was confirmed to be negative for lipooligosaccharide contamination, and a particle size less than 0.2 μm was verified by transmission electron microscopy and flow cytometry [Citation44]. PM was suspended and sonicated in sterile saline at a final concentration of 2 mg/ml. When HBE and HEK293T cells reached 70–80% confluence, they were pretreated with or without the specific inhibitors described below for 30 min and were then exposed to 100 μg/ml PM. The following inhibitors were used: the proteasome inhibitor MG132 (10 μM; Selleck, S2619), lysosome inhibitor Baf A1 (10 nM, Selleck, S1413), MTOR inhibitor rapamycin (10 nM or 100 nM, Selleck, S1039), ROS inhibitor NAC (1 mM, Sigma, A7250), MAPK/ERK inhibitor U0126 (10 μM, Selleck, S1102), MAPK/JNK-specific inhibitor SP600125 (10 μM, Selleck, S1460), MAPK/p38-specific inhibitor SB203580 (10 μM, Selleck, S1076), NFKB-specific inhibitor BAY11-7082 (10 μM, Selleck, S2913) and MYD88 inhibitor ST2825 (10 μM, MCE, HY-50,937).
Plasmid and siRNA transfection
HBE and HEK293T cells were transfected with plasmids using JetPEI® Transfection Reagent (Polyplus Transfection, 101–10 N) according to the manufacturer’s protocol. Transient transfection of siRNA (Table S2) into HBE and HEK293T cells was performed with INTERFERin® Transfection Reagent (Polyplus Transfection, 409–10) according to the manufacturer’s instructions.
Experimental lung metastasis models and in vivo PM treatment
To investigate the effect of PM exposure on experimental lung metastasis, PM was prepared as described for the in vitro experiments, and mice were treated with 100 μg PM (in 50 μl of saline) by intranasal instillation every other day for a total of 5 times. Control mice were administered the same volume of saline. After the second exposure to PM, C57BL/6 J, Becn1+/- mice and lc3b-/- mice were injected with 1 × 106 luciferase-labeled LLC or B16-F10 cells (in 200 μl of phosphate-buffered saline (PBS; Gibco, 10,010,023) via the tail vein. Cells were resuspended in calcium- and magnesium-free PBS and filtered through a 40-μm mesh filter immediately prior to injection. Experimental lung metastasis was monitored for 2 weeks after tail vein injection. Mice were anesthetized with 1% pentobarbital sodium and intraperitoneally injected with luciferin (Promega, P1043) at a dose of 100 μg/kg of body weight. Images were acquired with the IVIS Spectrum In vivo Imaging System (PerkinElmer) 10 min after luciferin injection, to ensure consistent photon flux. Total photon flux in the lung area was analyzed with Living Image software (PerkinElmer).
To establish the spontaneous lung metastasis model, 5 × 105 4T1 cells were subcutaneously injected into the shaved right flanks of mice 6 days before the initiation of PM exposure. Cells were resuspended in calcium- and magnesium-free PBS and filtered through a 40-μm mesh immediately prior to injection. The mice were then treated with PM by nasal drip every 6 days for a total of 5 times, and primary tumors were surgically removed 20 days after cell injection. The metastatic tumor burden was measured by standard hematoxylin and eosin (H&E) staining of paraffin-embedded lung sections 60 days after cell injection. Images were acquired with an Olympus BX53 inverted microscope (Olympus, Melville, NY).
Lung tissue dissociation and flow cytometry
Lung tissues were dissected from mice, washed extensively in PBS, minced and incubated with 2 mg/ml collagenase type I (Worthington Biochemical Corp., LS004214), 2 mg/ml collagenase type IV (Worthington Biochemical Corp., LS004210), and 0.1 mg/ml DNase (Sigma, 04536282001) in RPMI-1640 medium for 60 min at 37°C with shaking (150 r.p.m.). The cell suspension was filtered through a 70-μm cell strainer, and red blood cells were lysed using red blood cell lysis buffer (Sigma, 11,814,389,001). For flow cytometry, single-cell suspensions were stained with a combination of different fluorophore-conjugated monoclonal antibodies () for 30 min at 4°C. Compensation was performed using fluorescence-minus-one (FMO) controls. Dead cells were excluded from analysis with Fixable Viability Dye eFluorTM (Invitrogen, 65–0863-14 or 65–0867-14). Samples were evaluated on a CytoFlex flow cytometer (Beckman Coulter, Brea, CA, USA), and the data were analyzed with FlowJo software (Tree Star, Ashland, OR). For cell sorting, we used a MoFlo Astrios EQ instrument (Beckman Coulter) to achieve purities > 95%. The following cell types were identified using the following combinations of cell markers: CD4+ or CD8+ T cells (PTPRC/CD45+ CD4+ or CD8+), B cells (PTPRC/CD45+ CD19+), NK cells (PTPRC/CD45+ KLRB1C/NK1.1+), macrophages (PTPRC/CD45+ ITGAM/CD11b+ ADGRE1/F4/80+), dendritic cells (PTPRC/CD45+ ITGAX/CD11c+ MHC-II+), neutrophils (PTPRC/CD45+ ITGAM/CD11b+ GSR/Gr1+), and epithelial cells (PTPRC/CD45− EPCAM+).
Table 1. Antibodies for flow cytometry, immunoblotting and immunofluorescence
Lung permeability experiments and immunofluorescence staining
PM was suspended and sonicated in sterile saline at a final concentration of 2 mg/ml. C57BL/6 J mice were treated with 100 μg of PM (in 50 μl of saline) by nasal drip each day for 4 consecutive days. Control mice were treated with the same volume of NS. Twenty-four hours after PM treatment, the mice were injected with 2 mg of Texas Red lysine-fixable dextran 70,000 MW (Invitrogen, D1830) via the tail vein. One hour after dextran injection, the mice were euthanized and perfused with 40 ml of PBS. The lungs were dissected and fixed overnight with a mixture of 2% paraformaldehyde and 20% sucrose. For immunofluorescence staining, the lungs were cut into 8-µm-thick sections and applied to glass slides. After being fixed and stained with 4'6-diamidino-2-phenylindole (DAPI; Sigma, D9542), the sections were washed with PBS and examined with an Olympus IX83-FV3000 confocal microscope (Olympus Corp, Tokyo, Japan).
Quantitative real-time PCR
Total RNA was extracted from cells or lung homogenates using TRIzol reagent (Thermo Fisher Scientific, 10,296,028) and reverse transcribed into complementary DNA (cDNA) with a cDNA synthesis kit (Takara, 639,506) according to the manufacturer’s instructions. Real-time PCR was conducted using SYBR Green (Vazyme, Q221-01) and specific primers on a Roche LightCycler® 480II platform (Roche Diagnostics, USA). All primers used are listed in Table S1. The following thermal cycling conditions were used for PCR: 1 cycle at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 60°C for 34 s. The data were analyzed by the 2−ΔΔCt method.
Immunoblot analysis
For western blot, proteins in cell lysates were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) on 8–12% gels and transferred to polyvinylidene difluoride membranes (Roche, 03010040001). Each membrane was blocked with 5% (wt:vol) bovine serum albumin (BSA; Sigma, B2064) in TBST buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween-20 [Solarbio, T8220]) and then incubated with appropriate primary antibodies overnight at 4°C. After being incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies for 1 h, each membrane was scanned using a Tanon 4500 imaging system (Shanghai, China).
For immunoprecipitation, cell lysates were prepared in low-salt buffer containing phosphatase and protease inhibitors (Sangon Biotech, A500850; Sangon Biotech, A600869; Thermo Scientific™, 36,978) and were incubated with the indicated primary antibodies and Protein A/G beads (Santa Cruz Biotechnology, sc-2003) at 4°C overnight with gentle shaking. Flag-, His-, or HA-labeled proteins were immunoprecipitated with the corresponding beads and boiled with 2× SDS loading buffer (MKBio, MP2210) before being analyzed according to the immunoblotting protocol. Uncropped scanned images of membranes are presented in the affiliated Source Data file. The antibodies were diluted with NCM Universal Antibody Diluent (New Cell & Molecular Biotech Co. Ltd., WB500D).
For the ubiquitination assay, cells were lysed with cold SDS lysis buffer (50 mM Tris-HCl, pH 6.8, 150 mM NaCl, 10% glycerol, 1% SDS) containing protease inhibitors. Before being diluted 10-fold with dilution buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 [MKBio, MS4311]), the samples were boiled with SDS buffer for 5 min. After being incubated at 4°C for 30 min, the diluted lysates were centrifuged at 12,000 g for 15 min at 4 C, and the supernatants were subjected to immunoprecipitation with the indicated antibodies. All antibodies and corresponding dilutions used are listed in .
In vivo antibody-mediated neutralization and drug treatment
The neutralization of VEGF-KDR/VEGFR signaling and neutrophils with specific antibodies was performed based on previously published methods [Citation38,Citation73]. During the establishment of experimental lung metastasis models, the antibody-mediated neutralization of VEGF-KDR/VEGFR signaling or depletion of GSR/Gr1+ cells was performed by intraperitoneal injection of 800 μg of anti-mouse KDR/VEGFR-2 (DC101; Bio X Cell, BE0060) or 100 μg of anti-GSR/Gr1 (clone RB6-8C5; Bio X Cell, BE0075) antibodies, respectively, per mouse every 3 days before the first PM inhalation. Rat IgG1(HRPN, Bio X Cell, BE0088) and rat IgG2b (LTF-2, Bio X Cell, BE0090) was used as the isotype control. For in vivo autophagy inhibition or ROS blockade, HCQ (60 mg kg−1 in 200 μl of PBS) or NAC (275 mg kg−1 in 200 μl of PBS) was intraperitoneally injected into mice every other day concurrent with PM exposure. To analyze PM-induced neutrophil recruitment, an anti-mouse KDR/VEGFR-2 antibody or NAC was injected 24 h before PM exposure.
Knockdown of TRAF6 expression in vivo
For TRAF6 knockdown in vivo, Cho-Traf6 siRNA and NC siRNA were synthesized (GenePharma, Shanghai, China) for in vivo RNA delivery. During the establishment of experimental lung metastasis models, mice were administered 20 μg of cholesterol-conjugated siRNA by nasal drip on days −5, −4, −2, +2, and +6 after intravenous tumor cell injection. The successful knockdown of TRAF6 expression in vivo was confirmed by western blot (Figure S5D). To analyze PM-induced neutrophil recruitment, mice were administered 20 μg of cholesterol-conjugated siRNA by nasal drip for two consecutive days. Twenty-four hours after the last injection, mice were exposed to PM by nasal drip for 24 h.
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of CXCL2 and IL8 in human cell culture supernatants and the concentrations of CXCL2, IL8 and VEGF in mouse BALF supernatants were measured with ELISA kits according to the manufacturers’ protocols. An ELISA kit for human IL8 (431,501) was purchased from BioLegend. ELISA kits for human CXCL2 (900-M120), murine CXCL2 (900-M152), and murine VEGF (900-M99) were purchased from PeproTech. An ELISA kit for murine IL8 (MM200) was purchased from Jiangsu Meimian Industrial Co. Absorbance values were measured with a SynergyMx M5 microplate reader (Molecular Devices).
Immunohistochemical (IHC) and immunofluorescence staining
Paraformaldehyde-fixed paraffin-embedded murine lung tissue samples were sectioned (4 µm) and subjected to IHC and immunofluorescence staining. The expression of PECAM1/CD31 and α-SMA was assessed by IHC staining with different primary antibodies according to the manufacturer’s instructions (). The expression of LY6G and EPCAM in paraffin-embedded mouse lung sections was assessed by immunofluorescence staining with different primary antibodies. Images of the sections were acquired, and the positive areas were analyzed.
To examine GFP-LC3 in cells, mice were euthanized and lung tissues were fixed with 4% paraformaldehyde, embedded in OCT (Solarbio, 4583) compound and cut into 6-μm sections on a cryotome. The slides were washed with PBS and stained with DAPI. The GFP signals in the sections were examined with an Olympus IX83-FV3000 confocal microscope (Olympus Corp, Tokyo, Japan), and the relative fluorescence intensity values were determined with ImageJ software.
HBE cells were seeded on microscope cover slips (Invitrogen, H18200), fixed with precooled methanol for 10 min and permeabilized with 0.1% Triton X-100 for 10 min. After being blocked with 5% BSA for 1 h at room temperature, the cells were incubated at 4°C overnight with different combinations of primary antibodies (). The primary antibodies were then detected using DyLight 488- and DyLight 549-labeled secondary antibodies (Abcam, ab96931 and ab96921). Nuclei were stained with DAPI. The fluorescence signals of stained HBE cells were sequentially acquired in single optical sections with an Olympus IX83-FV3000 confocal microscope (Olympus Corp, Tokyo, Japan). The relative fluorescence intensity values were determined with ImageJ software.
In vitro autophagic flux assay
To evaluate autophagic flux, HBE cells were transfected with the mRFP-GFP-LC3 plasmid for 24 h and were then treated with PM with or without 10 μM MG132 or 1 mM NAC for 12 h. The fluorescence signals of transfected HBE cells were sequentially acquired in single optical sections with an Olympus IX83-FV3000 confocal microscope (Olympus Corp, Tokyo, Japan). The relative fluorescence intensity values were determined with ImageJ software.
Detection of ROS levels
To determine ROS levels, HBE cells were treated with PM for 12 h, collected and incubated with 2',7'-dichlorofluorescin diacetate (DCFH-DA; 10 mM; Beyotime, S0033S) for 20 min. The levels of fluorescence were then measured by flow cytometry.
Statistical analysis
All data are expressed as the mean ± SD. Statistical analyses were performed with GraphPad Prism 7.0 software. The differences between two groups of data were analyzed by unpaired Student’s t test, and differences among three or more groups were analyzed by one-way analysis of variance (ANOVA) or two-way ANOVA followed by a post hoc Tukey test. Overall survival and post-progression survival were assessed by Kaplan-Meier analysis. A value of P < 0.05 was considered statistically significant.
Supplemental Material
Download Zip (18.8 MB)Acknowledgments
We thank the Core Facilities of Zhejiang University School of Medicine for technical support. We are grateful to Mrs. SS. Liu for her help with confocal laser scanning microscopy, and Xinghui Song for her help with fluorescence-activated cell sorting (FACS).
Disclosure statement
No potential conflict of interest was reported by the author(s). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Cohen AJ, Brauer M, Burnett R, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389(10082):1907–1918.
- Liu C, Chen R, Sera F, et al. Ambient particulate air pollution and daily mortality in 652 cities. N Engl J Med. 2019;381:705–715.
- Eckel SP, Cockburn M, Shu YH, et al. Air pollution affects lung cancer survival. Thorax. 2016;71:891–898.
- Lippi G, Mattiuzzi C. Particulate matter pollution and lung cancer: a worldwide perspective. Clin Respir J. 2020;14:179–180.
- Tseng CH, Tsuang BJ, Chiang CJ, et al. The relationship between air pollution and lung cancer in nonsmokers in Taiwan. J Thorac Oncol. 2019;14:784–792.
- Zhang ZY, Zhu DW, Cui B, et al. Association between particulate matter air pollution and lung cancer. Thorax. 2020;75:85–87.
- Lee KK, Bing R, Kiang J, et al. Adverse health effects associated with household air pollution: a systematic review, meta-analysis, and burden estimation study. Lancet Glob Health. 2020;8:e1427–e34.
- Asgharian B, Price OT, Oldham M, et al. Computational modeling of nanoscale and microscale particle deposition, retention and dosimetry in the mouse respiratory tract. Inhal Toxicol. 2014;26:829–842.
- Zhao QJ, Chen H, Yang T, et al. Direct effects of airborne PM2.5 exposure on macrophage polarizations. BBA-Gen Subj. 2016;1860:2835–2843.
- Thevenot PT, Saravia J, Jin N, et al. Radical-containing ultrafine particulate matter initiates epithelial-to-mesenchymal transitions in airway epithelial cells. Am J Respir Cell Mol Biol. 2013;48(2):188–197.
- Wang PL, Thevenot P, Saravia J, et al. Radical-containing particles activate dendritic cells and enhance Th17 inflammation in a mouse model of asthma. Am J Resp Cell Mol. 2011;45(5):977–983.
- Deng X, Zhang F, Rui W, et al. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol In Vitro. 2013;27(6):1762–1770.
- Xu X, Wang H, Liu S, et al. Aodengqimuge, et al. TP53-dependent autophagy links the ATR-CHEK1 axis activation to proinflammatory VEGFA production in human bronchial epithelial cells exposed to fine particulate matter (PM2.5). Autophagy. 2016;12:1832–1848.
- Li XB, Lv Y, Gao N, et al. microRNA-802/Rnd3 pathway imposes on carcinogenesis and metastasis of fine particulate matter exposure. Oncotarget. 2016;7:35026–35043.
- Lu YY, Lin Y, Ding DX, et al. MiR-26a functions as a tumor suppressor in ambient particulate matter-bound metal-triggered lung cancer cell metastasis by targeting LIN28B-IL6-STAT3 axis. Arch Toxicol. 2018;92:1023–1035.
- Chen ZH, Wu YF, Wang PL, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12:297–311.
- Deng X, Zhang F, Wang L, et al. Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis. 2014;19:1099–1112.
- Deng XB, Feng NN, Zheng M, et al. PM2.5 exposure-induced autophagy is mediated by lncRNA loc146880 which also promotes the migration and invasion of lung cancer cells. BBA-Gen Subj. 2017;1861:112–125.
- Onorati AV, Dyczynski M, Ojha R, et al. Targeting autophagy in cancer. Cancer. 2018;124:3307–3318.
- Rao S, Tortola L, Perlot T, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056.
- White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42–46.
- Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–622.
- Peng YF, Shi YH, Shen YH, et al. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS One. 2013;8:e74407.
- Ishida T, Mizushima S, Azuma S, et al. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996;271:28745–28748.
- Cao Z, Xiong J, Takeuchi M, et al. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446.
- Zhan Z, Xie X, Cao H, et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257–268.
- Wu H, Lu XX, Wang JR, et al. TRAF6 inhibits colorectal cancer metastasis through regulating selective autophagic CTNNB1/β-catenin degradation and is targeted for GSK3B/GSK3β-mediated phosphorylation and degradation. Autophagy. 2019;15:1506–1522.
- Rezaeian AH, Li CF, Wu CY, et al. A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1α activation, tumorigenesis and metastasis. Nat Cell Biol. 2017;19:38–51.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101.
- Wu CF, Andzinski L, Kasnitz N, et al. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int J Cancer. 2015;137:837–847.
- Scherz-Shouval R, Santagata S, Mendillo ML, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578.
- Yamamura Y, Asai N, Enomoto A, et al. Akt-Girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer Res. 2015;75:813–823.
- Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. 2016;30:668–681.
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437.
- Marigo I, Dolcetti L, Serafini P, et al. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179.
- Cella CA, Minucci S, Spada F, et al. Dual inhibition of mTOR pathway and VEGF signalling in neuroendocrine neoplasms: from bench to bedside. Cancer Treat Rev. 2015;41:754–760.
- Ruan J, Hajjar K, Rafii S, et al. Angiogenesis and antiangiogenic therapy in non-Hodgkin’s lymphoma. Ann Oncol. 2009;20:413–424.
- Coffelt SB, Kersten K, Doornebal CW, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348.
- Liu Y, Gu Y, Han Y, et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell. 2016;30:243–256.
- Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227.
- Quail DF, Olson OC, Bhardwaj P, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol. 2017;19:974–987.
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, et al. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis. 2010;69:20–28.
- Olsen NJ, Schleich MA, Karp DR. Multifaceted effects of hydroxychloroquine in human disease. Semin Arthritis Rheum. 2013;43:264–272.
- Balakrishna S, Lomnicki S, McAvey KM, et al. Environmentally persistent free radicals amplify ultrafine particle mediated cellular oxidative stress and cytotoxicity. Part Fibre Toxicol. 2009;6:11.
- Lin WJ, Kuang HY. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy. 2014;10:1692–1701.
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38.
- Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692.
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362.
- Hu HB, Sun SC. Ubiquitin signaling in immune responses. Cell Res. 2016;26:457–483.
- Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011;21:22–39.
- Zhao W, Wang L, Zhang M, et al. E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J Immunol. 2012;188:2567–2574.
- Donohue E, Balgi AD, Komatsu M, et al. Induction of covalently crosslinked p62 oligomers with reduced binding to polyubiquitinated proteins by the autophagy inhibitor verteporfin. PLoS One. 2014;9:e114964.
- McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16:717–727.
- Peinado H, Zhang H, Matei IR, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17:302–317.
- Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–417.
- Kuznik A, Bencina M, Svajger U, et al. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. 2011;186:4794–4804.
- Lamphier M, Zheng W, Latz E, et al. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol. 2014;85:429–440.
- Rybstein MD, Bravo-San Pedro JM, Kroemer G, et al. The autophagic network and cancer. Nat Cell Biol. 2018;20:243–251.
- Wu YF, Li ZY, Dong LL, et al. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy. 2020;16:435–450.
- Ovrevik J, Refsnes M, Lag M, et al. Triggering mechanisms and inflammatory effects of combustion exhaust particles with implication for carcinogenesis. Basic Clin Pharmacol Toxicol. 2017;121(Suppl 3):55–62.
- Ovrevik J, Refsnes M, Lag M, et al. Activation of proinflammatory responses in cells of the airway mucosa by particulate matter: oxidant- and non-oxidant-mediated triggering mechanisms. Biomolecules. 2015;5:1399–1440.
- Ubellacker JM, Tasdogan A, Ramesh V, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585:113–118.
- Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–496.
- Piskounova E, Agathocleous M, Murphy MM, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191.
- Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238.
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947.
- Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737.
- Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer. 2018;18:69–88.
- Wu G, Song L, Zhu J, et al. An ATM/TRIM37/NEMO axis counteracts genotoxicity by activating nuclear-to-cytoplasmic NF-kappaB signaling. Cancer Res. 2018;78:6399–6412.
- Meitinger F, Ohta M, Lee KY, et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature. 2020;585:440–446.
- Yeow ZY, Lambrus BG, Marlow R, et al. Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature. 2020;585:447–452.
- Ding X, Lucas T, Gp M, et al. Distinct functions of epidermal and myeloid-derived VEGF-A in skin tumorigenesis mediated by HPV8. Cancer Res. 2015;75:330–343.