
ABSTRACT
Nonalcoholic fatty liver disease (NAFLD) affects a quarter of the global population. However, its pathogenesis is not completely understood. In our recent study, we have demonstrated that in a high-fat diet-induced liver steatosis model, the activation of SREBF1/SREBP-1c (sterol regulatory element binding transcription factor 1) directly upregulates Mir216a transcription, which inhibits CTH/CSE (cystathionase (cystathionine gamma-lyase)) expression and its function in hydrogen sulfide (H2S) production. Reduced H2S production suppresses the sulfhydration of ULK1 (unc-51 like autophagy activating kinase 1), consequently inhibiting autophagic flux and lipid droplet turnover. A single substitution mutation (C951S) in ULK1 or the silencing of CTH impairs ULK1 sulfhydration-mediated lipophagy, thereby promoting hepatic steatosis in mice. Interestingly, the sulfhydration of ULK1 increases its intrinsic kinase activity to modulate autophagy at both initiation and progression stages of autophagic catabolic flux. This study reveals that SREBF1/SREBP-1c contributes to hepatic lipid accumulation through its combined effect of increased lipid synthesis coupled with decreased lipid degradation mediated by autophagic dysregulation.
Nonalcoholic fatty liver disease (NAFLD) is one of the most crucial metabolic disorders worldwide and is closely associated with obesity and diabetes. However, current strategies for the treatment of NAFLD are limited, owing to the incomplete knowledge regarding the pathogenesis of NAFLD. The most characteristic pathological feature of hepatic steatosis is the presence of excessive lipid droplets (LDs), which is primarily caused by prolonged disturbances in liver lipid homeostasis due to an imbalance in uptake, synthesis, degradation, or secretion. Understanding the factors that trigger this imbalance may, therefore, be crucial in developing more efficient therapeutic strategies for NAFLD.
SREBFs (sterol regulatory element binding transcription factors) are master transcriptional factors that regulate the expression of genes required for maintaining lipid homeostasis. SREBFs consist of two major isoforms, SREBF1/SREBP-1a/SREBP-1c, and SREBF2/SREBP-2. SREBF1, the predominant hepatic isoform of SREBFs, preferentially activates the synthesis of fatty acids and triglycerides (TGs), which is stimulated by a hyperinsulinemic state caused as a result of insulin resistance, a hallmark of metabolic diseases, including NAFLD. During hyperinsulinemia, autophagy-mediated lysosomal LD degradation, referred to as lipophagy, is suppressed in the liver, leading to hepatic steatosis with LD accumulation. This observation indicates an intimate link between SREBF1 activity and lipophagy in lipid homeostasis. In our current study, we reveal that lipid synthesis and degradation in the liver are reciprocally controlled by SREBF1 through two different mechanisms [Citation1].
Activation of SREBF1 and lipogenic genes is observed in the livers of mice subjected to a high-fat diet (HFD), whereas autophagic flux is suppressed, as evidenced by increased LC3-II and SQSTM1/p62. However, the HFD-induced dysregulation of autophagy is completely abolished in srebf1/srebp-1c knockout mice, thus protecting these mice against hepatic steatosis. SREBF1 directly promotes the expression of Mir216a, which suppresses the expression of CTH/CSE (cystathionase (cystathionine gamma-lyase)), one of the two main metabolic enzymes responsible for synthesis of hydrogen sulfide (H2S) in the liver. H2S is a gasotransmitter that acts by modifying the cysteine residues of the target proteins, a phenomenon referred to as sulfhydration. Using a modified biotin switch assay, we examined the sulfhydration of ULK1, an autophagy regulatory kinase. Furthermore, we observed that a ULK1 mutant, harboring a C951S substitution in the C-terminal domain, suppresses sulfhydration in response to an H2S donor or starvation ().
Figure 1. Negative regulation of autophagy by SREBF1. Activation of CTH-H2S signaling in the liver causes the sulfhydration of ULK1 at C951. This modification stabilizes the ULK1-ATG13-RB1CC1/FIP200 complex and increases its kinase activity in the autophagy initiation stage. However, the activation of SREBF1-Mir216a in a hepatic steatosis model inhibits CTH-H2S signaling-mediated ULK1 sulfhydration, increasing UVRAG-RUBCN association, which blocks the fusion of autophagosomes with lysosomes. In contrast, SREBF1 deficiency leads to ULK1 sulfhydration-mediated autophagic flux, promoting lipid degradation and preventing hepatic steatosis
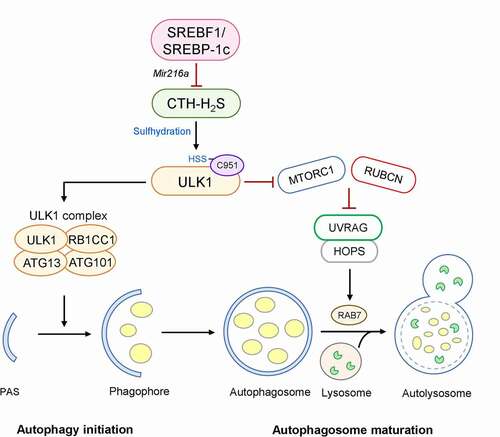
ULK1 plays a critical role in multiple stages of autophagy, such as initiation, nucleation, and fusion. Sulfhydration of ULK1 is required for the formation of both autophagosomes and autolysosomes. In the early stage of autophagy, sulfhydration of ULK1 stabilizes its interaction with its binding partners ATG13 and RB1CC1/FIP200, allowing the complete exertion of its kinase activity. In the late stage of autophagy, this modification decreases MTORC1 signaling, consequently blocking the interaction of UVRAG (UV radiation resistance associated gene) and RUBCN/Rubicon, which negatively regulates the maturation and fusion of autophagosomes. The free form of UVRAG can interact via homotypic fusion and protein sorting, promoting the fusion of an autophagosome with a lysosome. In contrast, the ULK1C951S mutant increases UVRAG and RUBCN interaction, thereby causing impaired hepatic autophagic flux, both in vitro and in vivo (). Interestingly, SREBF1 deficiency enhances the fusion of autophagosomes with lysosomes and LDs, leading to an accelerated LD breakdown; however, when CTH is silenced in HFD-fed srebf1/srebp-1c knockout mice, ULK1 sulhfydration and autophagic flux are impaired, and hepatic lipid accumulation is increased.
Summarily, in hepatic steatosis, SREBF1 increases lipid synthesis via the direct activation of lipogenic genes at the transcriptional level and inhibits lipid catabolism through the activation of Mir216a, which decreases CTH-H2S signaling and ULK1-stimulated autophagy. This finding provides new insight into the regulation of autophagy by the SREBF1-Mir216a axis and H2S signaling (protein sulfhydration) in the pathogenesis of NAFLD.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
Reference
- Nguyen TTP, Kim DY, Lee YG, et al. SREBP-1c impairs ULK1 sulfhydration-mediated autophagic flux to promote hepatic steatosis in high-fat-diet-fed mice. Mol Cell. 2021 Jun 30. DOI:https://doi.org/10.1016/j.molcel.2021.06.003.