
ABSTRACT
Non-canonical autophagy pathways decorate single-membrane vesicles with Atg8-family proteins such as MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3). Phagosomes containing the bacterial pathogen Listeria monocytogenes (L.m.) can be targeted by a non-canonical autophagy pathway called LC3-associated phagocytosis (LAP), which substantially contributes to the anti-listerial activity of macrophages and immunity. We here characterized a second non-canonical autophagy pathway targeting L.m.-containing phagosomes, which is induced by damage caused to the phagosomal membrane by the pore-forming toxin of L.m., listeriolysin O. This pore-forming toxin-induced non-canonical autophagy pathway (PINCA) was the only autophagic pathway evoked in tissue macrophages deficient for the NADPH oxidase CYBB/NOX2 that produces the reactive oxygen species (ROS) that are required for LAP induction. Similarly, also bone marrow-derived macrophages (BMDM) exclusively targeted L.m. by PINCA as they completely failed to induce LAP because of insufficient production of ROS through CYBB, in part, due to low expression of some CYBB complex subunits. Priming of BMDM with proinflammatory cytokines such as TNF and IFNG/IFNγ increased ROS production by CYBB and endowed them with the ability to target L.m. by LAP. Targeting of L.m. by LAP remained relatively rare, though, preventing LAP from substantially contributing to the anti-listerial activity of BMDM. Similar to LAP, the targeting of L.m.-containing phagosomes by PINCA promoted their fusion with lysosomes. Surprisingly, however, this did not substantially contribute to anti-listerial activity of BMDM. Thus, in contrast to LAP, PINCA does not have clear anti-listerial function suggesting that the two different non-canonical autophagy pathways targeting L.m. may have discrete functions.
Abbreviations: actA/ActA: actin assembly-inducing protein A; ATG: autophagy-related; BMDM: Bone marrow-derived macrophages; CALCOCO2/NDP52: calcium-binding and coiled-coil domain-containing protein 2; CYBA/p22phox: cytochrome b-245 light chain; CYBB/NOX2: cytochrome b(558) subunit beta; E. coli: Escherichia coli; IFNG/IFNγ: interferon gamma; L.m.: Listeria monocytogenes; LAP: LC3-associated phagocytosis; LGALS: galectin; LLO: listeriolysin O; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; NCF1/p47phox: neutrophil cytosol factor 1; NCF2/p67phox: neutrophil cytosol factor 2; NCF4/p67phox: neutrophil cytosol factor 4; Peritoneal macrophages: PM; PINCA: pore-forming toxin-induced non-canonical autophagy; plc/PLC: 1-phosphatidylinositol phosphodiesterase; PMA: phorbol 12-myristate 13-acetate; RB1CC1/FIP200: RB1-inducible coiled-coil protein 1; ROS: reactive oxygen species; S. aureus: Staphylococcus aureus; S. flexneri: Shigella flexneri; SQSTM1/p62: sequestosome 1; S. typhimurium: Salmonella typhimurium; T3SS: type III secretion system; TNF: tumor necrosis factor; ULK: unc-51 like autophagy activating kinase; PM: peritoneal macrophages; WT: wild type.
Introduction
Macroautophagy/autophagy, a cellular pathway targeting cytoplasmic components for degradation [Citation1], is a key event in host-pathogen interaction [Citation2]. Both canonical and non-canonical autophagy pathways targeting invading microbes have been identified. The canonical autophagy pathway targeting invading microbes is referred to as xenophagy. A cup-shaped structure called the phagophore expands until it completely surrounds the microbe and encloses it in a characteristic double-membrane vesicle called an autophagosome. A key event in autophagosome generation and maturation is its decoration with Atg8-family proteins such as MAP1LC3/LC3 (microtubule-associated proteins 1 light chain 3) by the autophagic machinery. Non-canonical autophagy pathways targeting invading microbes utilize some, but not all, components of the autophagic machinery to decorate single-membrane vesicles with LC3 [Citation3]. LC3-associated phagocytosis (LAP) is such a non-canonical autophagy pathway [Citation4]. LAP is induced by activation of specific surface receptors of phagocytes such as macrophages and results in formation of so-called LAPosomes. LAPosomes, in many aspects, resemble conventional phagosomes but are decorated with LC3, a process that strictly depends on reactive oxygen species (ROS) produced by the NADPH oxidase CYBB/NOX2. As compared to conventional phagosomes, LAPosomes usually [Citation5] show enhanced fusion with lysosomes and hence acquisition of microbicidal molecules. In consequence, LAP significantly contributes to elimination of a number of different pathogens [Citation4].
Listeria monocytogenes (L.m.) is a Gram-positive bacterial pathogen with specialized escape mechanisms to avoid killing and degradation in conventional phagosomes [Citation6]. L.m. use their pore-forming toxin listeriolysin O (LLO) and the phospholipases C plcA/PLCA and plcB/PLCB to destroy the phagosomal membrane and escape into the cytosol before the phagosome fuses with lysosomes and thereby acquires the microbicidal molecules that are crucial for the killing and degradation of phagocytosed microbes. In the cytosol, L.m. use their virulence factors plcA, plcB [Citation7] and actA/ActA [Citation8,Citation9] to protect themselves from being targeted, killed and degraded by xenophagy.
We recently have demonstrated that LAP substantially contributes to the anti-listerial activity of tissue macrophages and thereby to anti-listerial immunity of mice [Citation10,Citation11]. LAP of L.m. is induced through a signaling pathway emanating from the receptor ITGB2/ß2 integrin MAC-1 (integrin subunit beta 2)-ITGAM/CR3/integrin αmβ2 (integrin subunit alpha M) that induces SMPD1 (spingomyelin phosphodiesterase 1)-mediated changes in membrane lipid composition that facilitate assembly and activation of CYBB. CYBB-derived ROS then induce LC3 recruitment to L.m.-containing phagosomes. By promoting fusion of L.m.-containing phagosomes with lysosomes, LAP increases exposure of L.m. to bactericidal molecules, thereby enhancing the anti-listerial activity of macrophages and immunity of mice.
It is important to note that only matured tissue macrophages are able to target L.m. by LAP [Citation10]. In bone marrow-derived macrophages (BMDM), by contrast, LC3 recruitment to L.m. instead occurs through a non-canonical form of autophagy that is induced by LLO-inflicted damage to the phagosomal membrane [Citation10,Citation12,Citation13]. We will refer to this pore-forming toxin-induced form of non-canonical autophagy as “PINCA” throughout this manuscript. Whereas LAP clearly exerts antimicrobial function [Citation4], the significance of PINCA for host cell defense remained unresolved.
In this study, we have addressed the potential role of PINCA in anti-listerial defense of macrophages. Furthermore, we define the defect of BMDM that prevents them from targeting L.m. by LAP.
Results
BMDM exclusively target L.m. by PINCA
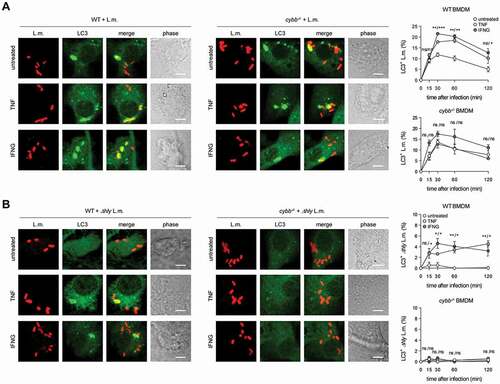
In wild-type (WT) BMDM, about 10–15% of phagocytosed L.m. resided in LC3-positive vesicles in the first hour after infection (). Afterward, colocalization of L.m. with LC3 dropped markedly, which may reflect the previously reported escape of L.m. from LC3-positive vesicles early after infection [Citation13]. To investigate whether LC3 recruitment to L.m. was by canonical or non-canonical autophagy, we used BMDM deficient for the ULK complex components ULK1 and ULK2 (ulk1−/- ulk2−/-) or RB1CC1/FIP200 (rb1cc1−/-). The ULK complex is required for initiation of canonical forms of autophagy such as xenophagy but not for that of non-canonical forms of autophagy [Citation1]. LC3 recruitment to L.m. was completely independent of ULK1 and ULK2 () and of RB1CC1 (), indicating exclusive targeting of L.m. by non-canonical autophagy. For LAP, ROS production by CYBB is essentially required [Citation10,Citation14,Citation15]. Colocalization of L.m. with LC3 was completely unaltered in cybb−/- BMDM (), showing that LC3 recruitment to L.m. was not by LAP. L.m. damage the phagosomal membrane through their pore-forming toxin LLO, which perforates and eventually destroys the phagosomal membrane [Citation16]. Of note, L.m. deficient for LLO (Δhly L.m.; LLO is encoded by the hly gene) did not colocalize with LC3 at all (), indicating that LLO-inflicted membrane damage is a prerequisite for LC3 recruitment to L.m. in BMDM. Other L.m. virulence factors such as plcA, plcB and actA did not influence LC3 recruitment to L.m. (Fig. S1). Together, these data show that LC3 recruitment to L.m. in BMDM occurs exclusively through a non-canonical form of autophagy that is induced by LLO-inflicted damage to the phagosomal membrane, i.e. by PINCA.
Figure 1. (Continued)
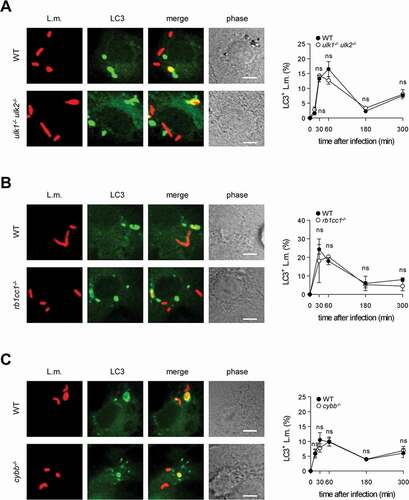
Figure 1. BMDM exclusively target L.m. by PINCA. (A-D) GFP-LC3 transgenic BMDM of the indicated genotypes were infected with wt (A-C) or Δhly L.m. (D) for the indicated periods of time. LC3+ L.m. were quantified by immunofluorescence microscopy. Data are shown as mean ± SEM of six (A), two (B), seven (C) or three (D) independent experiments. Representative micrographs from 1 h after infection are shown. Scale bar: 4 µm. ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < .0001.
Similar to L.m., Staphylococcus aureus (S. aureus) can damage the phagosomal membrane [Citation17]. To this end, S. aureus express a number of different pore-forming toxins. Similar to L.m., S. aureus elicited LC3 conversion in BMDM in a CYBB-, ULK1-/ULK2- and RB1CC1-independent manner (Fig. S2A-C). This was abrogated in S. aureus deficient for Agr, the global regulator of S. aureus virulence that also controls expression of the pore-forming toxins (ΔAgr S. aureus) [Citation18]. These data suggest that, similar to L.m., S. aureus in BMDM does not trigger canonical autophagy or LAP but PINCA. By contrast, Salmonella typhimurium (S. typhimurium) and Shigella flexneri (S. flexneri) do not form pores in the phagosomal membrane but insert the needlelike structures of their type III secretion systems (T3SS). In epithelial cells, this leads to T3SS-induced rupture of the vacuole, which induces tagging of the ruptured membrane by galectins, recruitment of receptor proteins such as CALCOCO2/NDP52 or SQSTM1/p62 and targeting by selective autophagy [Citation19,Citation20]. In BMDM, S. typhimurium and S. flexneri did not elicit LC3 conversion (Fig. S2A), indicating that, in line with current literature [Citation21–25] this pathway is not induced in macrophages.
Targeting by PINCA impedes the ability of L.m. to damage the phagosomal membrane
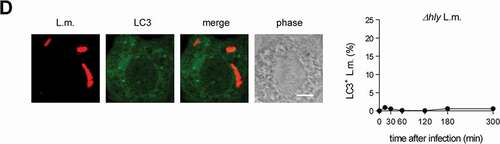
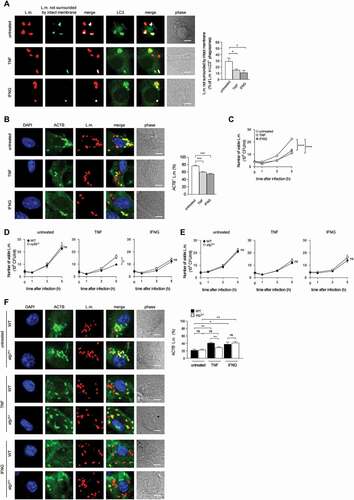
Having established PINCA as the exclusive mechanism for recruitment of LC3 to L.m. in BMDM, we next aimed to characterize this pathway in more detail. First, we investigated whether PINCA serves to reduce or repair LLO-inflicted damage to the phagosomal membrane. To visualize L.m. that managed to damage the membrane of their phagosome, we used a differential staining method based on selective permeabilization of the plasma membrane followed by antibody staining of L.m. (see [Citation26] and Materials and Methods for details). This procedure resulted in specific staining of L.m. residing in perforated phagosomes or that had managed to destroy the phagosomal membrane and escape into the cytosol. L.m. in intact phagosomes, by contrast, remained unstained by this procedure, allowing us to discriminate L.m. residing in intact phagosomes from those that had managed to damage the membrane of their phagosome. Using this method, we found that L.m. colocalizing with LC3 were stained less often for having damaged the membrane of their phagosome than LC3-negative L.m. indicating that LC3-positive phagosomes containing L.m. were damaged less often as respective LC3-negative phagosomes (). Also in tissue macrophages such as peritoneal macrophages (PM), LC3-positive phagosomes containing L.m. were damaged less often than respective LC3-negative phagosomes (). In PM, L.m. are targeted by LAP [Citation10]. However, potentially not exclusively as a subpopulation of LC3-positive phagosomes containing L.m. was clearly damaged suggesting that either even LAP cannot completely prevent L.m. from damaging the phagosomal membrane or that PM target L.m. not only by LAP but to some degree also by PINCA.
Figure 2. (Continued)
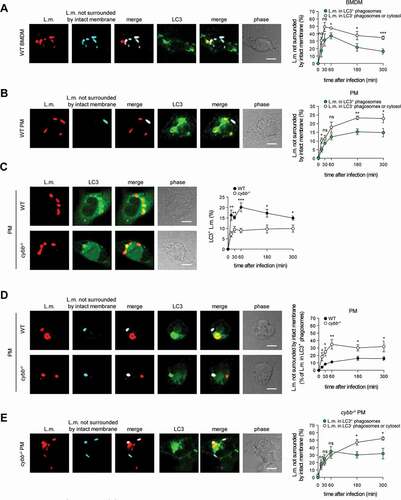
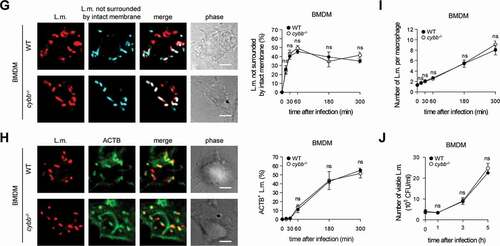
Figure 2. Targeting by PINCA impedes the ability of L.m. to damage the phagosomal membrane. BMDM or PM were infected with L.m. for the indicated periods of time. (A and B) L.m. in GFP-LC3 transgenic BMDM (A) or PM (B) that managed to damage the membrane of their phagosome were identified by differential staining. The percentages of L.m. that had managed to damage the membrane of their phagosome were quantified by immunofluorescence microscopy separately for L.m. colocalizing with LC3 and LC3-negative L.m. (for technical reasons the latter includes L.m. residing in perforated LC3−phagosomes as well as L.m. that had managed to destroy the phagosomal membrane and escape into the cytosol). (C) LC3+ L.m. in PM from GFP-LC3 transgenic WT and cybb−/- mice were quantified by immunofluorescence microscopy. (D and F) L.m. in PM (D) or BMDM (F) from GFP-LC3 transgenic WT or cybb−/- mice that had managed to damage the membrane of their LC3+ phagosome were identified by differential staining. The respective percentages of damaged phagosomes among LC3+ phagosomes were quantified by immunofluorescence microscopy. (E and G) L.m. in PM (E) or BMDM (G) from GFP-LC3 transgenic cybb−/- mice that had managed to damage the membrane of their phagosome were identified by differential staining. The percentages of L.m. that had managed to damage the membrane of their phagosome were quantified by immunofluorescence microscopy separately for L.m. colocalizing with LC3 and LC3-negative L.m. Data are shown as mean ± SEM of five to eight independent experiments. Representative micrographs from 3 h (A, B, E, G) or 1 h (C, D, F) after infection are shown. Scale bar: 4 µm. ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
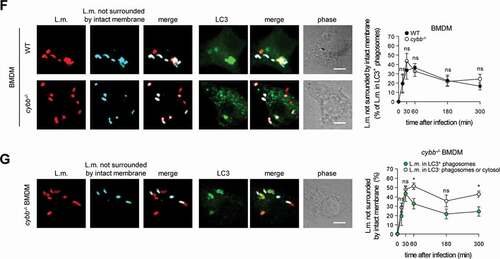
Of note, LC3 is recruited to L.m. to some extent also in PM deficient for CYBB and therefore also for LAP ( and [Citation10]). Yet, while LC3 recruitment to L.m. by LAP in WT PM is independent of LLO, LC3 recruitment to L.m. in CYBB-deficient PM strictly depends on LLO expression [Citation10]. Thus, LC3 recruitment to L.m. in CYBB-deficient PM is exclusively induced by PINCA, just like it is the case in BMDM. Accordingly, focusing solely on LC3-positive phagosomes revealed that the LC3-positive phagosomes formed by PINCA in cybb−/- PM were damaged much more often than the LC3-positive phagosomes formed in WT PM (). By contrast, analysis of damaged LC3-positive phagosomes in BMDM, which are formed exclusively by PINCA, revealed no difference in damage frequency between WT and cybb−/- (). Of note, when considering all damaged phagosomes in WT or cybb−/- PM or BMDM, respectively, LC3-positive phagosomes were always damaged less often than LC3-negative phagosomes (). These data indicate that either targeting of L.m.-containing phagosomes by PINCA results in membrane damage repair or that not only targeting by LAP but also that by PINCA impedes the ability of L.m. to damage the phagosomal membrane.
Targeting of L.m.-containing phagosomes by PINCA enhances their fusion with lysosomes
Indeed, LC3-positive phagosomes containing L.m. fused more often with lysosomes not only in WT PM, which are able to use the particularly microbicidal pathway of LAP against L.m., but also in cybb−/- PM () and in WT and cybb−/- BMDM (), which exclusively target L.m. by PINCA. Thus, similar to LAP, the targeting of L.m.-containing phagosomes by PINCA promotes their fusion with lysosomes.
Figure 3. Targeting of L.m.-containing phagosomes by PINCA enhances their fusion with lysosomes. BMDM or PM were infected with L.m. for the indicated periods of time. (A and B) Colocalization of L.m. with LC3, lysosomal fluid phase markers and ACTB was analyzed by immunofluorescence microscopy in PM (A) or BMDM (B) from GFP-LC3 transgenic WT or cybb−/- mice. Where indicated, ACTB+ L.m. were excluded from analysis to enable precise comparison of the acquisition of lysosomal fluid phase markers by LC3+ vs. LC3− L.m.-containing phagosomes. (C) ACTB+ L.m. in WT and atg7−/- BMDM at 5 h after infection were quantified by immunofluorescence microscopy. (D) Bacterial burden of WT and atg7−/- BMDM was determined by plating on blood agar plates. Data are shown as mean ± SEM of three (A-C) or six (D) independent experiments. Representative micrographs from 5 h after infection are shown. Scale bar: 4 µm. ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
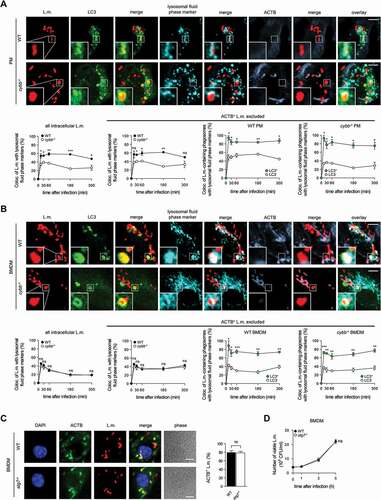
Because LAP enhances anti-listerial activity of macrophages by promoting phagolysosomal fusion, we next investigated whether PINCA contributes to anti-listerial activity of BMDM. In atg7−/- BMDM, which are deficient for the membrane association of Atg8-family proteins that is required for all forms of autophagy [Citation3] and therefore also for recruitment of LC3 to L.m [Citation10]., the number of L.m. that managed to escape from the phagosome into the cytosol and recruit ACTB () as well as the overall bacterial burden () were unaltered. Combined with our data that PINCA is the only autophagic pathway targeting L.m. in BMDM ()), these data indicate that PINCA does not substantially contribute to the anti-listerial activity of BMDM.
BMDM fail to induce LAP of L.m. due to insufficient ROS production by CYBB
In contrast to PINCA, LAP clearly promotes the anti-listerial activity of macrophages [Citation10]. Therefore, we contemplated whether the limitation of BMDM to PINCA could be overcome, i.e. whether BMDM could be endowed with the ability to target L.m. by LAP instead of PINCA. To be able to address this question, we first defined the defect that prevents BMDM from targeting L.m. by LAP.
For induction of LAP, ROS production by CYBB is essential [Citation10,Citation14,Citation15]. BMDM produced substantially less ROS through CYBB than PM (). This was not only the case for ROS production in response to L.m. infection but also for that in response to pharmacological stimulation with phorbol 12-myristate 13-acetate (PMA) indicating a generally reduced capacity of BMDM to produce ROS by CYBB. Indeed, expression of CYBB itself and also that of other subunits of the CYBB complex such as CYBA/p22phox and NCF1/p47phox was substantially lower in BMDM than in PM (). Therefore, we reasoned that, unlike PM, BMDM may not produce enough ROS by CYBB to induce LAP and thereby restrict escape of L.m. from the phagosome. In this respect, deficiency in producing ROS by CYBB impaired the ability of PM ()) but not that of BMDM ()) to prevent L.m. from damaging the phagosomal membrane, escaping into the cytosol, recruiting ACTB and proliferating.
Figure 4. (Continued)
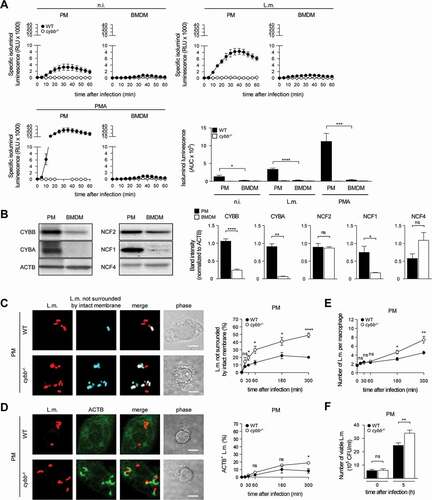
Figure 4. BMDM fail to induce LAP in response to L.m. infection due to insufficient ROS production by CYBB. (A) ROS production by WT and cybb−/- PM or BMDM after L.m. infection or pharmacological stimulation with PMA was measured using isoluminol. The kinetics of ROS production and the AUC as a measure for the total amount of ROS produced are shown. (B) Expression levels of CYBB, CYBA, NCF2/p67phox, NCF1 and NCF4 in PM and BMDM were analyzed by western blot and quantified by densitometry. (C-J) WT and cybb−/- PM (C-F) or BMDM (G-J) were infected with L.m. for the indicated periods of time. (C and G) L.m. that had managed to damage the membrane of their phagosome were identified by differential staining and quantified by immunofluorescence microscopy. (D and H) ACTB+ L.m. were quantified by immunofluorescence microscopy. (E and I) The number of L.m. per macrophage was quantified by immunofluorescence microscopy. (F and J) Bacterial burden was determined by plating on blood agar plates. Data are shown as mean ± SEM of seven (A), three (B, D, H) or six to seven (C, E, F, G, I, J) independent experiments. Representative micrographs from 5 h after infection and immunoblots are shown. Scale bar: 4 µm. n.i., not infected; ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
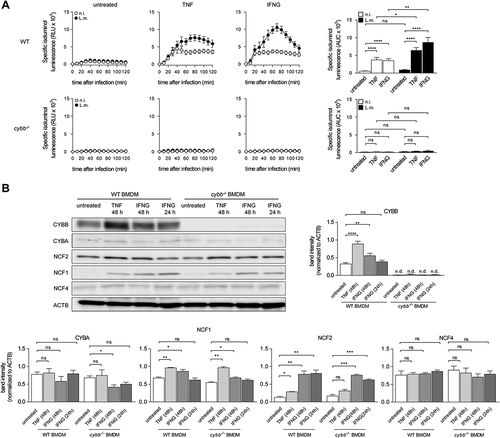
To test our hypothesis that BMDM fail to induce LAP because they do not produce sufficient amounts of ROS by CYBB, we next sought to explore if increasing ROS production by CYBB would endow BMDM with the ability to induce LAP and thereby restrict escape of L.m. from the phagosome. The capability to produce ROS by CYBB is markedly increased after activation of macrophages by proinflammatory stimuli [Citation27]. We decided to activate the BMDM by priming with proinflammatory cytokines such as TNF and IFNG/IFNγ, which are not only potent activators of macrophages but also key factors in anti-listerial immunity [Citation28]. Priming of BMDM with TNF or IFNG indeed markedly increased ROS production by CYBB (). In part, this was because priming increased expression of CYBB and other subunits of the CYBB complex such as NCF1 and NCF2 (). Yet, priming with IFNG increased ROS production by CYBB to a higher degree than TNF, whereas expression of CYBB was increased to a much lower degree by IFNG than by TNF. Thus, also other mechanisms than upregulation of CYBB expression contributed to the enhancement of ROS production by CYBB by priming with TNF or IFNG.
Figure 5. Priming of BMDM with pro-inflammatory cytokines enhances ROS production through CYBB. WT and cybb−/- BMDM were either left untreated or stimulated for 48 h with TNF or for 24 h or 48 h with IFNG and then infected with L.m. where indicated. (A) ROS production after infection was measured using isoluminol. The kinetics of ROS production and the AUC as a measure for the total amount of ROS produced are shown. (B) Expression levels of CYBB, CYBA, NCF2, NCF1 and NCF4 in naïve and primed BMDM were analyzed by western blot and quantified by densitometry. Data are shown as mean ± SEM of six (A) and three to thirteen (B) independent experiments. Representative immunoblots are shown. n.i., not infected; ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Of note, not only ROS production by CYBB but also recruitment of LC3 to L.m. was markedly increased by priming of BMDM with TNF or IFNG (). This occurred in a CYBB-dependent manner indicating that the increase in LC3 recruitment to L.m. was by LAP. Indeed, priming with TNF or IFNG endowed BMDM with the ability to recruit LC3 also to phagosomes containing LLO-deficient L.m. (). cybb−/- BMDM remained incapable of recruiting LC3 to phagosomes containing LLO-deficient L.m. indicating that this ability was a result of the increased ROS production through CYBB by primed BMDM. Thus, the enhancement of ROS production by CYBB resulting from the priming with TNF or IFNG endowed BMDM with the ability to recruit LC3 to L.m. through a non-canonical autophagy pathway independent of LLO-inflicted damage to the phagosomal membrane, i.e. with the ability to target L.m. by LAP. As compared to PM, primed BMDM managed to target only a relatively small percentage of L.m. by LAP (compare with Fig. S3 and [Citation10]), though.
Figure 6. Enhancing ROS production through CYBB by priming with pro-inflammatory cytokines endows BMDM with the ability to target L.m. by LAP. WT and cybb−/- BMDM were either left untreated or stimulated for 48 h with TNF or for 24 h with IFNG and then infected with wt (A) or Δhly (B) L.m. for the indicated periods of time. LC3+ L.m. were quantified by immunofluorescence microscopy. Data are shown as mean ± SEM of at least three independent experiments. Representative micrographs from 1 h after infection are shown. Scale bar: 4 µm. ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
The limited ability to target L.m. by LAP only makes a minor contribution to the anti-listerial activity of primed BMDM
LC3-positive phagosomes containing L.m. were damaged less often in BMDM primed with TNF or IFNG than in naïve BMDM () suggesting that the newly acquired ability to recruit LC3 to L.m.-containing phagosomes by LAP may promote the ability of BMDM to restrict L.m. escape. Indeed, priming of BMDM with TNF or IFNG significantly reduced the percentage of L.m. that managed to escape into the cytosol and recruit ACTB () and enhanced their anti-listerial activity (). Importantly, priming with TNF or IFNG can increase several effector mechanisms in macrophages [Citation29–31].
Figure 7. The limited ability to target L.m. by LAP only makes a minor contribution to the anti-listerial activity of primed BMDM. BMDM of the indicated genotypes were either left untreated or stimulated for 48 h with TNF or for 24 h with IFNG and then infected with L.m. for the indicated periods of time. (A) L.m. that had managed to damage the membrane of their LC3+ phagosome at 1 h after infection were identified by differential staining. The respective percentages of damaged phagosomes among LC3+ phagosomes were quantified by immunofluorescence microscopy. (B) ACTB+ L.m. at 5 h after infection were quantified by immunofluorescence microscopy. (C-E) Bacterial burden of WT, cybb−/- and atg7−/- BMDM was determined by plating on blood agar plates. (F) ACTB− L.m. at 5 h after infection were quantified by immunofluorescence microscopy. Data are shown as mean ± SEM of five (A and D), four (B and F), ten (C) or six (E) independent experiments. Representative micrographs from 1 h (A) or 5 h (B and F) after infection are shown. Scale bar: 4 µm. ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
To address the question whether the enhanced anti-listerial activity of BMDM primed with TNF or IFNG was due to the newly acquired ability to target L.m. by LAP, increased production of ROS by CYBB or a combination of both, we analyzed the effect of priming on the anti-listerial activity of atg7−/- and cybb−/- BMDM. Priming with TNF enhanced the anti-listerial activity of cybb−/- BMDM, but not to a similar degree as in WT BMDM (), indicating that TNF priming enhances the anti-listerial activity of BMDM in part through the increase in production of ROS by CYBB (). Priming with IFNG enhanced the anti-listerial activity of cybb−/- BMDM to a similar degree as in WT BMDM indicating that the increase in production of ROS by CYBB in BMDM primed with IFNG is not required for their enhanced anti-listerial activity. Possibly, the lack of ROS production by CYBB in cybb−/- BMDM primed with IFNG can be compensated by other antimicrobial mechanisms that are also enhanced under these circumstances [Citation30,Citation31]. Priming of atg7−/- BMDM with TNF or IFNG enhanced their anti-listerial activity to a similar degree as in WT BMDM ( and S4). Thus, the newly acquired ability of primed BMDM to target L.m. by LAP does not substantially contribute to their anti-listerial activity. This suggests that either the deficiency for LAP can be compensated by other antimicrobial mechanisms that are also enhanced by priming with TNF or IFNG [Citation29–31] or that the relatively small number of L.m. that are targeted by LAP in primed BMDM ()) is not sufficient to have LAP make an impact on overall anti-listerial activity of BMDM. In support of the latter, not only naïve but also TNF-primed and IFNG-primed atg7−/- PM showed impaired restriction of listerial proliferation (Fig. S5), indicating that LAP also contributes to anti-listerial activity of primed PM. Moreover, although still the majority of L.m. managed to escape from the phagosome into the cytosol and recruit ACTB, TNF priming markedly increased the percentage of L.m. that did not manage to escape from the phagosome and recruit ACTB in WT but not atg7−/- BMDM (). Thus, the ability to target L.m. by LAP enhanced restriction of L.m. escape to some degree also in BMDM.
Taken together, these data show that enhancing the production of ROS by CYBB endows BMDM with the ability to target L.m. by LAP. However, the still relatively rare targeting of L.m. by LAP in BMDM primed with TNF or IFNG is not sufficient to have LAP make a strong impact on their overall anti-listerial activity. Thus, even primed BMDM largely remain restricted to PINCA, which does not contribute to anti-listerial activity, though.
Discussion
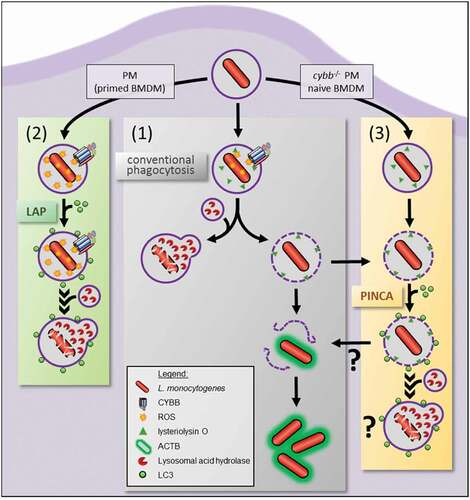
L.m. are targeted by multiple autophagic pathways. While L.m. effectively avoid targeting by xenophagy, they can be targeted by at least two non-canonical autophagy pathways, LAP and PINCA (). In tissue macrophages such as PM and in vivo, L.m. are targeted by LAP [Citation10]. Our data now show that (naïve) BMDM, the most commonly used primary macrophage model, fail to induce LAP and instead exclusively target L.m. by PINCA. Thus, L.m. are targeted by a completely different non-canonical autophagy pathway in BMDM than in vivo.
Figure 8. Intracellular paths of L.m. phagocytosed by macrophages. L.m.-containing phagosomes in macrophages can follow three different intracellular paths. (1) L.m. either manage to damage the phagosomal membrane resulting in escape into the cytosol, recruitment of ACTB and proliferation or are killed and degraded after fusion of their phagosome with lysosomes. (2) CYBB ROS-dependent recruitment of LC3 to the phagosome by LAP promotes its fusion with lysosomes and thereby enhances L.m. killing and degradation. (3) Damaged phagosomes can be targeted by PINCA, which promotes their fusion with lysosomes. Surprisingly, however, this does not result in enhanced killing and degradation of L.m. leaving the precise function of PINCA an open question. Naïve BMDM and cybb−/- PM only can target L.m. by PINCA because ROS production through CYBB is insufficient for LAP induction. BMDM primed with proinflammatory cytokines such as TNF and IFNG and WT PM, by contrast, produce sufficient amounts of ROS to target L.m. by LAP.
Of note, our data indicate that tissue macrophages such as PM may not exclusively target L.m. by LAP because a subpopulation of LC3-positive phagosomes containing L.m. was clearly damaged. This suggests that either even LAP cannot completely prevent L.m. from damaging the phagosomal membrane or that L.m. are targeted by PINCA to some extent also in PM. In support of the latter, LC3 is recruited to L.m. also in CYBB-deficient PM, which cannot induce LAP, indicating targeting by PINCA at least under these conditions. To decipher the precise relative contributions of LAP and PINCA to LC3 recruitment to L.m., specific factors for LAP vs. PINCA will have to be identified first, though.
LAP substantially enhances the anti-listerial activity of macrophages by promoting phago-lysosomal fusion [Citation10]. Of note, our data indicate that also PINCA promotes fusion of L.m.-containing phagosomes with lysosomes. Surprisingly, however, this did not seem to substantially contribute to anti-listerial activity. At least in BMDM, which exclusively target L.m. by PINCA, the number of L.m. that managed to escape from the phagosome into the cytosol, recruit ACTB and proliferate was not increased in ATG7-deficient BMDM. However, a very large proportion of L.m. in BMDM manage to escape into the cytosol, which may mask any positive effect targeting of some L.m. by PINCA may have. This may be different in PM, in which only few L.m. manage to escape from the phagosome. In PM, L.m. are targeted by LAP and as discussed above, very likely, also PINCA. Because both LAP and PINCA depend on ATG7, the question whether only LAP or also PINCA contributes to anti-listerial activity of PM can unfortunately not be answered using atg7−/- PMs. Thus, this question can only be answered upon identification of factors specific for LAP vs. PINCA and generation of respective knockout lines. For now, LAP remains the only autophagic pathway that clearly contributes to the elimination of L.m. by macrophages.
The precise function of PINCA remains an open question. Importantly, pathogens such as L.m. and S. aureus, which damage the phagosomal membrane through pore-forming toxins, elicited PINCA whereas pathogens such as S. typhimurium and S. flexneri, which damage the phagosome by insertion of the needlelike structures T3SS into the phagosomal membrane, did not. Several studies have shown that, in epithelial cells, T3SS-induced rupture of the vacuole by S. typhimurium initiates selective autophagy targeting of S. typhimurium [Citation19,Citation32,Citation33]. This targeting by canonical autophagy is strictly dependent on ULK complex recruitment to the damaged membranes tagged by LGALS8/galectin-8 and receptor proteins such as CALCOCO2 [Citation33,Citation34]. However, BMDM did not show induction of selective autophagy (or any other form of autophagy) after infection with S. typhimurium, at least not until 180 min after infection (Fig. S2A). This is in line with current literature that S. typhimurium represses autophagy induction in macrophages [Citation22–24]. It is quite possible that the presence of ubiquitin at a subpopulation of LC3-positive L.m.-containing phagosomes that we observed indicates tagging of damaged phagosomes by galectins leading to recruitment of receptor proteins such as CALCOCO2 and SQSTM1 and then LC3 as it is the case during targeting of damaged endomembranes by selective autophagy [Citation34–36]. However, in sharp contrast to selective autophagy [Citation10], LC3 recruitment to damaged L.m.-containing phagosomes occurred normally in macrophages deficient for ULK1/ULK2 or RB1CC1 indicating that PINCA involves mechanisms of LC3 recruitment that are different from those leading to the targeting of S. typhimurium in ruptured vacuoles in epithelial cells. In line with this, data from Weng et al. indicate that LGALS3/galectin-3 and LGALS8 are not required for autophagic targeting of L.m. in macrophages [Citation37]. Whether LC3 recruitment by PINCA depends on ubiquitination and subsequent recruitment of receptor proteins such as CALCOCO2 and SQSTM1 is an important open question for future studies. Concerning the function of PINCA, membrane damage caused to vacuoles containing S. typhimurium by the SPI-1 T3SS has been shown to be repaired by canonical autophagy [Citation36]. Targeting of Listeria-containing phagosomes by PINCA may represent a similar attempt of macrophages to repair damage to the phagosomal membrane caused by L.m. that, however, largely fails because LLO, in conjunction with PlcA and PlcB, is too efficient in destroying the phagosomal membrane. Nevertheless, our data show that LC3-positive L.m.-containing phagosomes are damaged less often than conventional phagosomes. Studies using L.m. that express only low amounts of LLO [Citation38] or using agents that cause more mild damage to the phagosomal membrane will be needed to answer the question of whether the dedicated function of PINCA is to repair damaged phagosomal membranes. It is tempting to speculate, though, that LC3-positive membranes may be recruited to damaged phagosomes to patch them. Alternatively, LC3 may recruit one or multiple of the growing number of proteins containing a LC3-interacting region [Citation39] to the damaged phagosome. Such LC3-interacting region-containing proteins may tag the damaged phagosome as requiring special attention, directly promote fusion with lysosomes or exert another, yet to be identified, function of PINCA. Of note, non-canonical autophagy pathways that decorate endosomes suffering ionic imbalances [Citation40] or containing aggregated proteins such as Aβ amyloids [Citation41] with LC3 have been identified. Thus, it seems conceivable that decoration of vesicles with LC3 by non-canonical autophagy generally serves to tag the vesicle as requiring special attention.
Our data show that the reason why BMDM fail to induce LAP in response to L.m. infection is their insufficient production of ROS by CYBB due to low expression of CYBB and some of the other subunits of the CYBB complex. Of note, BMDM have been shown to induce LAP in response to fungal stimuli such as Aspergillus fumigatus, zymosan particles and to polystyrene beads coated with Toll-like receptor agonists [Citation15,Citation42]. In our hands, however, BMDM did not show any induction of LAP in response to apathogenic bacteria such as Escherichia coli K12 (E. coli.), zymosan particles or polystyrene beads coated with TLR (toll-like receptor) agonists (Fig. S6 and own unpublished data). This may be the consequence of subtle differences in the protocol for generation of BMDM. It is tempting to speculate that such differences may influence the amount of ROS that can be produced by CYBB and hence the ability of BMDM to induce LAP. In support of this idea, Lam et al. have observed some degree of CYBB-dependent targeting of L.m. by non-canonical autophagy in BMDM [Citation43], indicating that ROS production through CYBB by BMDM in this particular case was sufficient for induction of LAP. Other groups, including ourselves, however, have shown that autophagic targeting of L.m. in BMDM strictly depends on LLO (i.e. is exclusively by PINCA) [Citation10,Citation12,Citation13]. Only after ROS production through CYBB was increased by priming of BMDM with proinflammatory cytokines such as TNF and IFNG, BMDM were able to target L.m. by LAP.
The percentage of L.m. targeted by LAP in BMDM primed with TNF or IFNG remained relatively small. Nevertheless, the percentage of L.m. that did not manage to escape into the cytoplasm and remained entrapped in phagosomes was increased in a LAP-dependent manner in TNF-primed BMDM. However, still more than half of the L.m. were cytosolic at 5 h after infection (as opposed to about 10% in naïve PM ( and [Citation10]), indicating that even in primed BMDM a large proportion of L.m. manage to escape from the phagosome. In consequence, the newly acquired (limited) ability of primed BMDM to target L.m. by LAP was not sufficient to prevent L.m. from proliferating in these cells. Of note, the overall anti-listerial activity of BMDM was markedly increased after priming with TNF or IFNG even in CYBB- or ATG7-deficient BMDM indicating that other antimicrobial mechanisms than ROS production and autophagic targeting dominated [Citation29–31]. Studies comparing the effects of cytokines on BMDM in vitro to the response of PM to PAMPs in vivo revealed that the majority of regulated genes is different, and classical and alternative macrophage activation in vitro do not match in vivo M1/M2 polarization [Citation44]. Therefore, optimized priming conditions that increase targeting of L.m. by LAP to a higher degree than TNF or IFNG may enable BMDM to more effectively use LAP against L.m. but these remain to be identified. Alternatively, the generally reduced anti-listerial activity of BMDM as compared to PM may also affect the otherwise highly efficient killing of L.m. in LAPosomes.
BMDM are a commonly used model to investigate the antimicrobial mechanisms of macrophages. However, our data show that BMDM are inappropriate for analysis of LAP, at least in the context of L.m. infection. PM are much more suitable as they reflect the in vivo situation [Citation10]. Moreover, PM express higher levels of LC3 and respond to L.m. infection with a more robust induction of LC3 conversion and recruitment than BMDM (Fig. S7). For analysis of PINCA and elucidation of its precise function, by contrast, BMDM may be optimal as PINCA is the only autophagic pathway triggered by L.m. in these cells.
Materials and methods
Mice
GFP-LC3 transgenic mice, GFP-LC3 transgenic cybb−/- mice, Atg7fl/fl mice expressing Cre recombinase under the Lyz2/LyzM promotor (atg7[MYEL-KO]) and GFP-LC3 transgenic Ulk1fl/fl, Ulk2fl/fl or Rb1cc1fl/fl mice expressing Cre recombinase under the Lyz2 promotor (ulk1[MYEL-KO] ulk2[MYEL-KO] or rb1cc1[MYEL-KO]) have been described previously [Citation10].
All mice were backcrossed at least ten times to the C57BL/6 background. Animals were kept under specific pathogen-free conditions at the animal facilities of the University of Cologne, Cologne, Germany. Experiments were conducted in accordance with the Animal Protection Law of Germany and in compliance with the Ethics Committee at the University of Cologne. For experiments, 8–20 weeks old mice were used.
Isolation of PM
Mice were sacrificed by cervical dislocation and peritoneal cells were collected by peritoneal lavage using 8 mL ice-cold PBS (Sigma-Aldrich, D8537-500ML). Red blood cells were lysed in 5 mL 0.2% NaCl (Roth GmbH, 9265.2) in H2O for 30 s. Then, isotonic conditions were reconstituted by addition of 5 mL 1.6% NaCl in H2O. After red blood cell lysis, macrophages were enriched from peritoneal cells by magnetic-activated cell sorting using ITGAM/CD11b MicroBeads (Miltenyi Biotec, 130–049-601) according to the instructions of the manufacturer. Viable cells were determined using Trypan blue (Sigma-Aldrich, T8154-100ML) exclusion in a Neubauer chamber (LO Laboroptik, 1,100,000). Where indicated, PM were stimulated with 10 ng/mL TNF (R&D Systems, 410-MT-010/CF) or 10 ng/mL IFNG (R&D Systems, 485-MI-100/CF) for the indicated period of time.
Generation of BMDM
Mice were sacrificed by cervical dislocation. Bone marrow cells were isolated from tibias and femurs and cultured for 6–7 d in VLE-RPMI 1640 medium (Biochrom GmbH, FG1415) supplemented with 10% FCS (Biowest, S1810), penicillin-streptomycin (100 U/mL and 100 μg/mL, respectively; Sigma-Aldrich, P0781-100ML), 10 mM HEPES (Biochrom GmbH, L1613; pH 7.3), 1 mM sodium pyruvate (Biochrom GmbH, L0473), 20 ng/mL recombinant CSF1/M-CSF (Peprotech, 315–02). Antibiotics were removed 24 h prior to the experiment [Citation45]. After differentiation, BMDM were harvested using 2 mM EDTA (Roth, 8043.2) in PBS and viable cells were determined using Trypan blue exclusion in a Neubauer chamber. Where indicated, BMDM were stimulated with 10 ng/mL TNF or 10 ng/mL IFNG for the indicated period of time.
Bacteria
In vivo-passaged Listeria monocytogenes (L.m.), strain EGD-e (serotype 1/2a), and its isogenic deletion mutants Δhly, ΔActA, ΔPlcA and ΔPlcB were cultured at 37°C in brain-heart-infusion (BHI) medium (Beckton Dickinson GmbH, 256,120) until mid-log phase as described previously [Citation10]. Salmonella typhimurium SL1344 (S. typhimurium) and the deletion mutant ΔssaV (both kindly provided by Julia Fischer, University Hospital Cologne, Cologne, Germany) were cultured in BHI medium until mid-log phase at 37°C. Escherichia coli K12 DH5-alpha (E.c), Staphylococcus aureus JE2 (S. aureus) and the MW2 deletion mutant ΔAgr (both kindly provided by Oleg Krut, Paul-Ehrlich-Institut, Langen, Germany) and Shigella flexneri M90T (S. flexneri) (kindly provided by Thomas Kufer, University of Hohenheim, Hohenheim, Germany) were cultured at 37°C in Luria broth (LB) medium (10 g/L Tryptone [OXOID Deutschland GmbH, LP0042, 10 g/L NaCl [Merck KGaA, 13,423], 5 g/L yeast extract [OXOID Deutschland GmbH, LB0021]) until mid-log phase. After adjustment to the desired density, serial dilutions of the inocula were plated on blood agar plates (OXOID Deutschland GmbH, PB5306A) to verify the colony-forming units (CFU).
Pulse-chase labeling of lysosomes with fluid phase markers
PM or BMDM were incubated for 1 h with 1.25 × 1011 red-fluorescent latex beads with a diameter of 20 nm (Thermo Fisher Scientific, F8786) in DMEM (Biochrom GmbH, FG0435) supplemented with 10% FCS or VLE-RPMI 1640 medium supplemented with 10% FCS, 10 mM HEPES, 1 mM sodium pyruvate and 20 ng/ml recombinant M-CSF1, respectively, (pulse). Non-endocytosed 20 nm latex beads were removed by washing three times with PBS. Afterward, cells were incubated for 2 h at 37°C in DMEM supplemented with 10% FCS or VLE-RPMI 1640 medium supplemented with 10% FCS, 10 mM HEPES, 1 mM sodium pyruvate and 20 ng/ml recombinant CSF1, respectively, (chase) leading to accumulation of the 20 nm latex beads in lysosomes [Citation46].
Immunofluorescence microscopy
PM or BMDM were infected with wt, Δhly, ΔActA, ΔPlcA or ΔPlcB L.m. at a multiplicity of infection (MOI) of 0.5 or co-incubated with E.c. or zymosan particles (Thermo Fisher Scientific, Z2849; labeled with Alexa Fluor 594 succinimidyl ester as described previously [Citation46]) at a MOI of 1. Infection was synchronized by centrifugation at 850 x g, 4°C for 5 min. At the indicated time points after infection, cells were fixed with 3% paraformaldehyde (Merck KGaA, 70,129,447) in PBS for 20 min at room temperature (RT). After blocking with 3% BSA (Sigma-Aldrich, A9418-500 G) in PBS for 15 min at RT, extracellular L.m. or E.c. were stained with polyclonal antibodies against L.m. (US Biologicals, L2650-01A; 1:250) or polyclonal antibodies against LPS (Acris Antibodies, BP2235; 1:100), respectively, in 3% BSA in PBS for 30 min or 1 h respectively, at RT and a fluorophore-conjugated secondary antibody. Next, cells were permeabilized with 0.1% saponin (Sigma-Aldrich, S7900) in PBS for 15 min at RT and blocked for 15 min at RT with 3% BSA in 0.1% saponin in PBS. L.m. or E.c were stained in 3% BSA and 0.1% saponin in PBS as described above but with a different fluorophore-conjugated secondary antibody. Through this differential staining, intracellular L.m. or E.c. are stained by one fluorophore only whereas extracellular L.m. or E.c. are stained by two fluorophores. Where indicated, cells were stained for 1 h with monoclonal antibody against mono- and polyubiquitinylated conjugates (Enzo, BML-PW8810; 1:200) and a fluorophore-conjugated secondary antibody or filamentous ACTB with fluorescently labeled Phalloidin (Thermo Fisher Scientific, A12379; 1:1,000) for 30 min and nuclei were visualized with DAPI (Thermo Fisher Scientific, 62,248; 1:10,000) or NucGreen Dead 488 (Thermo Fisher Scientific R37109;1:20). Samples were mounted on glass microscope slides (Engelbrecht Medizin & Labortechnik GmbH, 11,102) using ProLong Gold antifade reagent (Thermo Fisher Scientific, P36934) and examined using a FluoView1000 confocal laser scanning microscope (Olympus). At least 100 infected cells were analyzed per sample. Extracellular L.m. and E.c. were excluded from analysis, except for ACTB colocalization experiments.
Identification of damaged L.m.-containing phagosomes by differential staining
Differential permeabilization of cellular membranes was performed as described before [Citation26] with modifications. At the indicated time points after infection, cells were washed three times with ice-cold KHM buffer (110 mM potassium acetate, 20 mM HEPES, 2 mM MgCl2; pH 7.3). For selective permeabilization of the plasma membrane, cells were incubated for precisely 1 min with 50 µg/mL digitonin (Sigma-Aldrich, D5628-1 G) in KHM buffer. After three wash steps with ice-cold KHM buffer, cells were incubated with polyclonal antibodies against L.m. (US Biologicals, L2650-01A; 1:250) in 3% BSA in KHM buffer for 15 min on ice. After washing three times with ice-cold KHM buffer, cells were incubated with a fluorophore-conjugated secondary antibody in 3% BSA in KHM buffer for 15 min on ice. Then, cells were washed three times with PBS and fixed using 3% paraformaldehyde in PBS for 20 min at RT. This procedure results in selective staining of L.m. that are accessible for the antibody after selective permeabilization of the plasma membrane, i.e. extracellular L.m., L.m. that managed to escape into the cytosol and L.m. that reside in vesicles with perforated membranes. To stain all L.m., i.e. also those enclosed in unperforated vesicles, cells were permeabilized with 0.1% saponin in PBS and L.m. were stained as described above albeit with a fluorophore of a different color and in 0.1% saponin in PBS. Through this differential staining, L.m. in intact phagosomes are stained only by one fluorophore whereas extracellular L.m., cytosolic L.m. and L.m. in perforated vesicles are stained by two fluorophores. Samples were mounted on glass microscope slides using ProLong Gold antifade reagent and examined using a FluoView1000 confocal laser scanning microscope. At least 100 infected cells were analyzed per sample. Extracellular L.m. were excluded from analysis.
Analysis of bacterial burden
PM and BMDM were seeded in triplicates at a density of 150,000 cells/well and infected with L.m. at a MOI of 1 (or 0.5, 0.25 or 0.1, where indicated). Infection was synchronized by centrifugation at 850 x g, 4°C for 5 min and cells were incubated in Hanks’ balanced salt solution (HBSS) with Ca2+ and Mg2+ (Gibco, 14,025–050) supplemented with 5% normal mouse serum (Innovative Research, IMSCD1-COMPL) at 37°C. At the indicated time points after infection, cells were lysed in 0.1% Triton X-100 (Sigma-Aldrich, 8787100ML) in PBS and serial dilutions of lysates were plated on blood agar plates using a spiral plater (IUL instruments). CFU/mL were determined with an automatic colony counter (IUL instruments).
Quantification of extracellular ROS
PM and BMDM were seeded in triplicates at a density of 50,000 cells/well in a white 96 F non-treated microwell plate (Thermo Fisher Scientific, 236,105). After infection with L.m. at a MOI of 1 or stimulation with 1 ng/mL PMA (Sigma-Aldrich, P8139-1 mg), cells were incubated at 37°C in 50 µM isoluminol (Sigma-Aldrich, A8264) and 3.2 U/mL HRP (Merck Millipore, 516,531–5KU) in HBSS. Isoluminol luminescence was measured at 60 s intervals using a Tristar2 multimode plate reader LB 942 (Berthold Technologies).
Western blotting
PM and BMDM were infected with L.m., S. aureus or S. typhimurium, their respective deletion mutants Δhly, ΔAgr and ΔssaV, S. flexneri or were co-incubated with E.c. at a MOI of 5 or zymosan particles at a MOI of 3, lysed in RIPA lysis buffer (50 mM Tris HCl [Roth GmbH, 9090.3], 150 mM NaCl, 0.1% Nonidet P-40 [Applichem GmbH, A1694,0250], 0.5% sodium deoxycholate [Sigma-Aldrich, D6750-25 G], 1% SDS [Roth GmbH, CN30.3]; pH 7.5) supplemented with 0.5% benzonase endonuclease (Merck Millipore, 70,746–3) and protease and phosphatase inhibitor cocktails (Roche, 11,697,498,001 and 04906837001, respectively) for 10 min at RT and then incubated at 95°C for 10 min. Protein concentration was determined using Pierce BCA Protein Assay Kit according to the manufacturer´s instructions (Thermo Fisher Scientific, 23,225). Equal amounts of protein in Laemmli buffer (12 mM Tris-HCl, 5% glycerol [Sigma-Aldrich, 49,781], 2% β-mercaptoethanol [Roth GmbH, 4227.1], 0.4% SDS, 0.04% bromophenol blue [Roth, A512.1]) were denatured at 95°C for 10 min and subjected to standard SDS-PAGE and western blotting. Monoclonal antibodies against CYBB (Santa Cruz Biotechnology, sc-130,543; 1:1,000), CYBA (Santa Cruz Biotechnology, sc-130,550; 1:1,000), NCF4 (Santa Cruz Biotechnology, sc-48,376; 1:1,000), NCF1 (Santa Cruz Biotechnology, sc-17,845; 1:1,000) and NCF2 (Santa Cruz Biotechnology, sc-374,510; 1:1,000) and ACTB (Sigma-Aldrich, A2228; 1:10,000) were diluted in 5% BSA in TBS-T (15 mM NaCl, 1 mM Tris [Roth GmbH, 4855.3], 0.1% Tween20 [Roth GmbH, 9127.2]-). Polyclonal antibodies against LC3B (Sigma-Aldrich, L7543; 1:500) and horseradish peroxidase-conjugated goat anti-mouse (Sigma-Aldrich, A0168; 1:5,000) or rabbit (Sigma-Aldrich, A0545; 1:5,000) IgG secondary antibodies were diluted in 5% nonfat dry milk (Roth GmbH, T145.3) in TBS-T. Specific band intensity was quantified by densitometry using ImageJ software (National Institutes of Health) and normalized to ACTB.
Statistical analysis
For statistical analysis, data were subjected to unpaired two-tailed Student’s t test. P values of less than 0.05 were considered significant.
Supplemental Material
Download Zip (970.7 KB)Acknowledgments
We thank the teams of the animal facility of the Center of Molecular Medicine Cologne and the Dezentrales Tierhaltungsnetzwerk of the University of Cologne for support in animal caretaking.
Disclosure statement
The authors declare that they have no competing interests.
Supplementary materials
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–1836.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013 Oct;13(10):722–737.
- Dupont N, Nascimbeni AC, Morel E, et al. Molecular mechanisms of noncanonical autophagy. Int Rev Cell Mol Biol. 2017;328:1–23.
- Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Semin Cell Dev Biol. 2020 May;101:68–76.
- Cemma M, Grinstein S, Brumell JH. Autophagy proteins are not universally required for phagosome maturation. Autophagy. 2016 Sep;12(9):1440–1446.
- Ray K, Marteyn B, Sansonetti PJ, et al. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol. 2009 May;7(5):333–340.
- Tattoli I, Sorbara MT, Yang C, et al. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013 Nov 27;32(23):3066–3078.
- Yoshikawa Y, Ogawa M, Hain T, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009 Oct;11(10):1233–1240.
- Mitchell G, Ge L, Huang Q, et al. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun. 2015 May;83(5):2175–2184.
- Gluschko A, Herb M, Wiegmann K, et al. The beta2 Integrin Mac-1 induces protective LC3-Associated phagocytosis of Listeria monocytogenes. Cell Host Microbe. 2018 Mar 14;23(3):324–337 e5.
- Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis initiated by integrin ITGAM-ITGB2/Mac-1 enhances immunity to Listeria monocytogenes. Autophagy. 2018;14(8):1462–1464.
- Meyer-Morse N, Robbins JR, Rae CS, et al. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One. 2010 Jan 6;5(1):e8610.
- Mitchell G, Cheng MI, Chen C, et al. Listeria monocytogenes triggers noncanonical autophagy upon phagocytosis, but avoids subsequent growth-restricting xenophagy. Proc Natl Acad Sci U S A. 2018 Jan 9;115(2):E210–E217.
- Huang J, Canadien V, Lam GY, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009 Apr 14;106(15):6226–6231.
- Martinez J, Malireddi RK, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015 Jul;17(7):893–906.
- Osborne SE, Brumell JH, Listeriolysin O. from bazooka to Swiss army knife. Philos Trans R Soc London Ser B. 2017 Aug 5;372:1726.
- Flannagan RS, Heit B, Heinrichs DE. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens. 2015 Nov 27;4(4):826–868.
- Lee AS, de Lencastre H, Garau J, et al. Methicillin-resistant Staphylococcus aureus. Nat Rev Dis Primers. 2018 May 31;4(1):18033.
- Birmingham CL, Smith AC, Bakowski MA, et al. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006 Apr 21;281(16):11374–11383.
- Dupont N, Lacas-Gervais S, Bertout J, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009 [ 2009 Aug 20];6(2):137–149.
- Gogoi M, Shreenivas MM, Chakravortty D. Hoodwinking the Big-Eater to Prosper: the Salmonella-macrophage paradigm. J Innate Immun. 2019;11(3):289–299.
- Owen KA, Meyer CB, Bouton AH, et al. Activation of focal adhesion kinase by Salmonella suppresses autophagy via an Akt/mTOR signaling pathway and promotes bacterial survival in macrophages. PLoS Pathog. 2014;10(6):e1004159.
- Ganesan R, Hos NJ, Gutierrez S, et al. Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS Pathog. 2017;13(2):e1006227.
- Hos NJ, Fischer J, Hos D, et al. TRIM21 is targeted for Chaperone-Mediated Autophagy during Salmonella typhimurium infection. J Immunol. 2020;205(9):2456–2467.
- Ashida H, Kim M, Sasakawa C. Manipulation of the host cell death pathway by Shigella. Cell Microbiol. 2014;16(12):1757–1766.
- Meunier E, Broz P. Quantification of cytosolic vs. vacuolar Salmonella in primary macrophages by differential permeabilization. J Vis Exp. 2015 Jul;28(101):e52960.
- Herb M, Schramm M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants (Basel). 2021 Feb 19;10(2).
- Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004 Oct;4(10):812–823.
- Kusnadi A, Park SH, Yuan R, et al. The cytokine TNF promotes transcription factor SREBP activity and binding to inflammatory genes to activate macrophages and limit tissue repair. Immunity. 2019 Aug 20;51(2):241–257 e9.
- Zhou QD, Chi X, Lee MS, et al. Interferon-mediated reprogramming of membrane cholesterol to evade bacterial toxins. Nat Immunol. 2020 Jul;21(7):746–755.
- Price JV, Russo D, Ji DX, et al. IRG1 and inducible nitric oxide synthase act redundantly with other interferon-gamma-induced factors to restrict intracellular replication of Legionella pneumophila. mBio. 2019 Nov 12;10(6). DOI:https://doi.org/10.1128/mBio.02629-19.
- Thurston TL, Wandel MP, von Muhlinen N, et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012 Jan 15;482(7385):414–418.
- Ravenhill BJ, Boyle KB, von Muhlinen N, et al. The Cargo Receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol Cell. 2019 Apr 18;74(2):320–329 e6.
- Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and Galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016 [ 2016 Oct];39(1):13–27.
- Hong M-H, Weng IC, Li F-Y, et al. Intracellular galectins sense cytosolically exposed glycans as danger and mediate cellular responses. J Biomed Sci. 2021 [ 2021 Mar 4];28(1):16.
- Kreibich S, Emmenlauer M, Fredlund J, et al. Autophagy proteins promote repair of endosomal membranes damaged by the Salmonella Type Three Secretion System 1. Cell Host Microbe. 2015 Nov 11;18(5):527–537.
- Weng I-C, Chen H-L, Lo T-H, et al. Cytosolic galectin-3 and −8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology. 2018;28(6):392–405.
- Birmingham CL, Canadien V, Kaniuk NA, et al. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature. 2008 Jan 17;451(7176):350–354.
- Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors.J Mol Biol. 2020 [ 2020 Jan 3];432(1):80–103.
- Florey O, Gammoh N, Kim SE, et al. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy. 2015;11(1):88–99.
- Heckmann BL, Teubner BJW, Tummers B, et al. LC3-Associated endocytosis facilitates β-Amyloid clearance and mitigates neurodegeneration in Murine Alzheimer’s disease. Cell. 2019 Jul 25;178(3):536–551. e14.
- Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007 Dec 20;450(7173):1253–1257.
- Lam GY, Cemma M, Muise AM, et al. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy. 2013 Jul;9(7):985–995.
- Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages [Review]. Front Immunol. 2019 [ 2019 May 24];10(1084):1084.
- Herb M, Farid A, Gluschko A, et al. Highly efficient transfection of primary macrophages with in vitro transcribed mRNA. J Vis Exp. 2019 Nov;9(153):e60143.
- Schramm M, Herz J, Haas A, et al. Acid sphingomyelinase is required for efficient phago-lysosomal fusion. Cell Microbiol. 2008 Sep;10(9):1839–1853.