
ABSTRACT
Growing evidence demonstrates that macroautophagy/autophagy in the host stroma influences the tumor microenvironment. We have uncovered that autophagy in host stromal fibroblasts is compulsory to initiate and maintain the desmoplastic fibrotic response that fosters mammary tumor progression. Genetic loss of fibroblast autophagy impedes COL1A/type 1 collagen secretion, which is required for the development of a stiff tissue matrix permissive for mammary tumor growth. As a result, stromal fibroblast autophagy deficiency impairs mammary tumor progression in vivo, even when the cancer cells themselves remain autophagy competent. Our results provide unique conceptual insight into how the autophagy pathway can be modulated to abolish the desmoplastic response required for cancer progression.
Autophagy functions as a survival and metabolic adaptation pathway in both normal and cancerous cells. These findings have led to tremendous interest in autophagy as a therapeutic target against advanced cancers. The effects of autophagy in the tumor microenvironment (TME) likely influence the anti-tumor effects of systemically administered autophagy inhibitors in cancer patients. Cancer-associated fibroblasts (CAFs) represent a principal component of the TME, modulating tumor cell behavior through diverse mechanisms, including the synthesis of growth, tissue remodeling and angiogenic factors, cytokines, and extracellular matrix (ECM) components. Indeed, many solid tumors exhibit striking histological evidence of fibroblast proliferation and activation, termed desmoplasia. Although desmoplasia in human cancers tightly correlates with poor prognosis, the molecular mechanisms that generate and maintain this desmoplastic response remain unclear. Accordingly, we scrutinized whether and how autophagy in host stromal fibroblasts affects tumor progression and desmoplasia.
We initially employed an autochthonous mammary tumor model driven by the polyoma middle T (PyMT) oncogene to assess how genetic autophagy deletion in stromal fibroblasts affects tumor progression [Citation1]. We generated PyMT compound transgenic mice bearing conditional alleles independently targeting essential Atg genes (Atg12 or Atg5) using Cre recombinase driven by the S100A4/fibroblast specific protein 1 promoter (S100A4-Cre). Importantly, in these models, the tumor cells themselves remain fully autophagy competent. S100A4-Cre-mediated deletion of either Atg gene profoundly attenuates primary tumor growth compared to autophagy-competent controls. We complemented these studies with two different transplantation approaches in order to dissect the specific functions of autophagy in mouse mammary fibroblasts (MMFs). First, we co-mixed autophagy-competent or -deficient MMFs with autophagy-competent PyMT tumor cells and implanted this mixture into the mammary glands of syngeneic host mice. Intriguingly, mice implanted with PyMT tumor cells co-mixed with autophagy-deficient MMFs have significantly longer tumor latency and reduced end-stage tumor volume compared to those implanted with PyMT tumor cells co-mixed with autophagy-competent MMFs.
Second, we utilized an established mammary fat pad clearing and transplantation technique, where the mammary glandular epithelium is removed and the cleared mammary fat pad (MFP) is engrafted with either autophagy-competent or autophagy-deficient MMFs. Masson’s Trichrome and picrosirius red staining demonstrated that the engrafted stroma derived from autophagy-deficient MMFs exhibits significantly reduced COL1A/type 1 collagen deposition compared to engrafting with autophagy-competent MMFs. COL1A deposition in the stroma is a key mediator of ECM stiffness, which facilitates both tumor cell proliferation and survival. Using atomic force microscopy, we verified that MFPs primed with autophagy-deficient MMFs exhibit significantly reduced tissue stiffness compared to controls. Importantly, these autophagy-dependent increases in collagen deposition and the resultant stiffening of the stroma represents a cardinal feature of tumor desmoplasia.
Due to these reductions in COL1A deposition and tissue stiffness, we hypothesized that MFPs primed with autophagy-incompetent MMFs would not support robust PyMT tumor growth. Three weeks following the engraftment of either autophagy-competent or -incompetent MMFs into the cleared MFPs, we introduced autophagy-competent PyMT tumor cells. Once again, mice primed with autophagy-deficient MMFs gave rise to profoundly reduced PyMT tumor burden versus cohorts primed with autophagy-competent MMFs. Based on these results, we propose that autophagy deficiency in stromal fibroblasts impairs COL1A deposition, which creates a TME inhospitable for mammary tumor progression.
In addition to collagen deposition, another key feature of tumor desmoplasia is increased immune and inflammatory cell infiltration, which partly arises from CAF-mediated secretion of pro-inflammatory cytokines. Recent studies demonstrate ATGs are required for efficient secretion of various pro-inflammatory cytokines, including IL6 (interleukin 6), a well-known pro-fibrotic, pro-angiogenic, and tumor-promoting cytokine secreted by CAFs. Cytokine array profiling corroborated that IL6 secretion in autophagy-incompetent MMFs is significantly attenuated compared to controls. Interestingly, MFPs primed with autophagy-deficient MMFs have significantly attenuated neo-angiogenesis compared to controls, but it is uncertain if these phenotypes are IL6 dependent. Furthermore, MFPs derived from autophagy-deficient MMFs show significantly reduced recruitment of macrophages (ADGRE1/F4/80+) and lymphocytes (CD3+) suggesting autophagy in stromal fibroblasts promotes both innate and adaptive immune cell recruitment. Further studies are needed to ascertain the precise role of autophagy-regulated cytokine secretion in modulating these aspects of the desmoplastic response.
Importantly, autophagy deficiency does not globally impair MMF function, including the induction of fibroblast differentiation factors and the activation of the fibrogenic TGFB/TGF-β pathway. Rather, autophagy deficiency leads to specific secretory defects critical for the desmoplastic response, including COL1A and pro-inflammatory cytokine secretion. Furthermore, our results using MMFs corroborate prior reports in other cell types that autophagy supports collagen secretion. Specifically, misfolded type 1 pro-collagen (PC1) is normally degraded in the lysosome via autophagy. Upon autophagy ablation, the resulting defects in PC1 proteostasis suppress collagen secretion via an ER quality control feedback mechanism. Our results confirmed the reduced delivery and degradation of PC1 in the lysosomes of autophagy-deficient MMFs, indicating that autophagy in MMFs supports pro-collagen proteostasis which is required for efficient COL1A secretion ().
Figure 1. Autophagy in stromal fibroblasts supports desmoplasia and tumor growth in trans. Enhanced COL1A deposition and inflammatory cell recruitment are hallmarks of tumor desmoplasia. Autophagy in stromal fibroblasts promotes the degradation of misfolded type 1 pro-collagen (PC1), which maintains cellular proteostasis and enables the efficient secretion of COL1A necessary for creating the stiff fibrotic matrix found in desmoplastic stroma. At the same time, stromal fibroblast autophagy facilitates the secretion of IL6 and other pro-angiogenic and immune-modulatory cytokines. This enables the recruitment of both innate and adaptive immune cells to the tumor microenvironment (TME). Collectively, these pathways create a hospitable TME for tumor cell proliferation and survival. As a result, stromal fibroblast autophagy promotes tumor growth
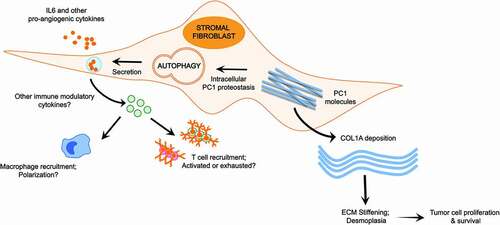
To date, studies of autophagy in the host stroma implicate the transfer of autophagy-derived amino acids from host cells to tumor cells as the principal tumor-promoting mechanism. Our study provides new mechanistic perspectives into host autophagy by demonstrating that in fibroblasts autophagy supports key aspects of the desmoplastic fibrotic response, including COL1A secretion, ECM stiffening, pro-inflammatory cytokine secretion and neo-angiogenesis, all of which are collectively required to support tumor growth (). Future studies should elucidate how fibroblast autophagy affects various innate and adaptive immune subsets within the TME, especially in the context of adjuvant immunotherapy.
Disclosure statement
JD is a member of the Scientific Advisory Board of Vescor Therapeutics.
Additional information
Funding
Reference
- Rudnick JA, Monkkonen T, Mar FA, et al. Autophagy in stromal fibroblasts promotes tumor desmoplasia and mammary tumorigenesis. Genes Dev. 2021;35(13–14):963–975. PMID: 34168038 PMCID: PMC8247603.