
ABSTRACT
Macroautophagy/autophagy, a highly conserved lysosome-dependent degradation pathway, has been intensively studied in regulating cell metabolism by degradation of intracellular components. In this study, we link autophagy to RNA metabolism by uncovering a regulatory role of autophagy in ribosomal RNA (rRNA) synthesis. Autophagy-deficient cells exhibit much higher 47S precursor rRNA level, which is caused by the accumulation of SQSTM1/p62 (sequestosome 1) but not other autophagy receptors. Mechanistically, SQSTM1 accumulation potentiates the activation of MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) signaling and promotes the assembly of RNA polymerase I pre-initiation complex at ribosomal DNA (rDNA) promoters, which leads to an increase of 47S rRNA transcribed from rDNA. Functionally, autophagy deficiency promotes protein synthesis, cell growth and cell proliferation, both of which are dependent on SQSTM1 accumulation. Taken together, our findings suggest that autophagy deficiency is involved in RNA metabolism by activating rDNA transcription and provide novel mechanisms for the reprogramming of cell metabolism in autophagy-related diseases including multiple types of cancers.
Abbreviations: 5-FUrd: 5-fluorouridine; AMPK: AMP-activated protein kinase; ATG: autophagy related; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; ChIP: chromatin immunoprecipitation; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MAPK/ERK: mitogen-activated protein kinase; MTOR: mechanistic target of rapamycin kinase; NBR1: NBR1 autophagy cargo receptor; NFKB/NF-κB: nuclear factor kappa B; NFE2L2/NRF2: nuclear factor, erythroid 2 like 2; OPTN: optineurin; PIC: pre-initiation complex; POLR1: RNA polymerase I; POLR1A/RPA194: RNA polymerase I subunit A; POLR2A: RNA polymerase II subunit A; rDNA: ribosomal DNA; RPS6KB1/S6K1: ribosomal protein S6 kinase B1; rRNA: ribosomal RNA; RUBCN/Rubicon: rubicon autophagy regulator; SQSTM1/p62: sequestosome 1; STX17: syntaxin 17; SUnSET: surface sensing of translation; TAX1BP1: Tax1 binding protein 1; UBTF/UBF1: upstream binding transcription factor; WIPI2: WD repeat domain, phosphoinositide interacting 2; WT: wild-type.
KEYWORDS:
Introduction
Macroautophagy/autophagy is a highly conserved intracellular degradation pathway by which cellular materials are engulfed by double-membraned vesicles and transported to lysosomes for digestion [Citation1]. Genetic studies from yeast to mammals have identified a number of ATG (autophagy related) proteins, which control different stages of autophagy, including autophagy initiation, autophagosome formation, autophagic cargo assembly, and autophagosome-lysosome fusion [Citation1]. Various autophagy-deficient models generated with knockout of ATG genes have been utilized to elucidate the regulatory role of autophagy in a plethora of physiological processes, including cellular adaptation to stresses, host defense against infection, and embryonic development [Citation2–5]. Recently, mutations in the ATG genes have been identified in multiple human diseases, such as cancers, neurodegenerative diseases, and metabolic disorders [Citation6]. However, the molecular links between the dysregulation of autophagy and the pathogenesis of human diseases remain largely unknown.
As one of best-known autophagy receptors, SQSTM1/p62 (sequestosome 1) mediates the autophagic degradation of ubiquitinated cargoes by targeting them to autophagosomes [Citation7]. Interestingly, SQSTM1 is initially identified as a regulator of cellular signal transduction by interacting with PRKC/atypical protein kinase C [Citation8]. So far, many lines of evidence have demonstrated the central role of SQSTM1 in the regulation of a number of cellular signaling pathways, including activation of NFKB/NF-κB (nuclear factor kappa B), inhibition of MAPK/ERK (mitogen-activated protein kinase), and stimulation of NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2)-dependent anti-oxidant response [Citation9–11], both of which support SQSTM1 as a critical player in a variety of cellular processes, such as inflammation, adipogenesis and cell death [Citation9–11]. Surprisingly, as the substrate of autophagy, SQSTM1 is also shown to be an upstream regulator of autophagy by controlling MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) signaling, one of the master regulators of autophagy initiation [Citation12]. Considering the central role of MTORC1 in regulating many fundamental cellular processes and deregulated MTORC1 signaling is implicated in various diseases [Citation13–15], SQSTM1 may also be involved in these MTORC1-regulated physiological or pathological processes by controlling MTORC1 activity.
Ribosome, the only organelle that provides framework on which protein synthesis takes place, is constituted with ribosomal proteins and ribosomal RNAs (rRNAs) [Citation16,Citation17]. The production of rRNAs is compromised with multiple steps, including at least transcription, processing and modifications, among which the transcription of precursor rRNA from thousands of ribosomal DNA (rDNA) copies by POLR1/RNA polymerase I appears to be the first pivotal rate-limiting step [Citation16,Citation18]. rDNA transcription consumes a lot of cellular energy and affects protein synthesis, cell growth and cell proliferation [Citation19,Citation20]. Many cellular nutrient- and energy-sensing pathways, such as MTORC1 and AMP-activated protein kinase (AMPK) signaling, have been utilized by the cell to link the availability of nutrients and energy to cell metabolism, such as rDNA transcription [Citation21,Citation22]. Interestingly, various cancer cells have been reported to show hyperactivation in rDNA transcription [Citation16,Citation23], suggesting that rDNA transcription activation may be involved in the pathogenesis of cancer.
In this study, we have investigated the role of autophagy in the regulation of rDNA transcription. Our results demonstrate that rDNA transcription is activated in autophagy-deficient cells. Accumulation of SQSTM1 but not other autophagy receptors potentiates the activation of MTORC1 signaling and promotes the assembly of POLR1 pre-initiation complex (PIC) at rDNA promoters, which leads to an increase in the synthesis of precursor RNA. Furthermore, we show that the accumulated SQSTM1 activates protein synthesis, cell growth and cell proliferation in autophagy-deficient cells.
Results
Autophagy deficiency upregulates 47S precursor rRNA level
To test whether autophagy is involved in the regulation of rRNA synthesis, we began by measuring the primary rRNA transcript 47S precursor rRNA level in autophagy-deficient cells. In HEK293 cells with knockout of ATG5 or ATG7, two essential genes for MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) lipidation, cellular 47S rRNA level was increased (). Moreover, when the serum-starved cells were stimulated with serum, cells with deletion of ATG5 or ATG7 exhibited a much higher increase in 47S rRNA level (). Consistent with this, the increase of 47S rRNA level in ATG5- or ATG7-deficient cells was abolished by re-introduction of ATG5 or ATG7 into the cells (). Besides autophagy, LC3 lipidation is also required for LC3-associated phagocytosis (LAP) [Citation24], we then tested whether LAP affects cellular 47S rRNA level. Interestingly, knockout of ATG16L1, another essential component for LC3 lipidation, but not RUBCN/Rubicon (rubicon autophagy regulator), a necessary component for LAP, significantly increased 47S rRNA level in cells (). Re-introduction of ATG16L1 totally abolished the increase of 47S rRNA in ATG16L1-deficient cells (). These results rule out the role of LAP in regulating cellular 47S rRNA level. In addition, we measured 47S rRNA level in cells with or without STX17 (syntaxin 17), functioning in autophagy initiation and autophagosome-lysosome fusion. A consistent result was also obtained with or without serum stimulation (). Of note, autophagy has been shown to be induced through an ATG5- and ATG7-independent manner [Citation25]. To clarify the link between autophagy and 47S rRNA in cells, we then measured 47S rRNA level in cells with or without ULK1, a core autophagy protein that is required for both ATG5- and ATG7-dependent and independent autophagy [Citation25,Citation26]. Clearly, ULK1 deletion also upregulated cellular 47S rRNA level and re-introduction of ULK1 reversed the increase ().
Figure 1. Autophagy deficiency upregulates 47S precursor rRNA level. (A-D) Cellular 47S rRNA level was measured by real-time PCR and normalized to ACTB mRNA. Cells with the deletion of the indicated gene were transfected with or without untagged plasmid that harboring the gene. Cells were cultured in nutrient-rich medium or treated with serum (6 h) after serum starvation (12 h). The statistical data are presented as mean ± SEM of three independent experiments. **P < 0.01. (E) Cellular ETS1 or GAPDH mRNA level was measured by real-time PCR and normalized to ACTB mRNA after incubation of the cells with 20 μg/ml actinomycin D (Act-D). GAPDH mRNA level was used as the internal control and did not change significantly in the course of the experiment. Act-D, a transcription inhibitor.
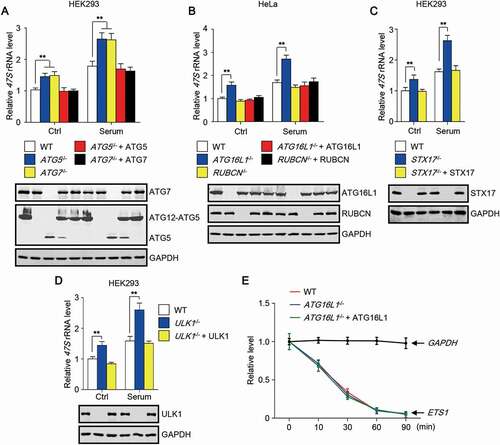
As autophagy is an intracellular degradation pathway, a simple hypothesis is that 47S rRNA may undergo autophagy-mediated degradation. We treated the cells with actinomycin D (Act-D) in high concentration, which has been shown to inhibit rDNA transcription in less than 30 s [Citation27], and checked the level of ETS1 [Citation27], derived from 47S rRNA, in wild-type (WT) and ATG16L1-deficient cells at different time points. GAPDH mRNA level was used as the internal control and remained nearly unchanged in the course of the experiment (). Consistent with the previous study [Citation27], the half-time of ETS1 was less than 30 min (). Obviously, the half-time of ETS1 remained unchanged in cells with or without ATG16L1 (), indicating that 47S rRNA may not be the direct substrate of autophagy.
These data together suggest that autophagy deficiency causes an increase in 47S rRNA level in the cell.
SQSTM1 mediates the increase of 47S rRNA in autophagy-deficient cells
As we have ruled out the possibility that 47S rRNA undergoes autophagic degradation, the player that mediates the increase of 47S rRNA in autophagy-deficient cells remains unknown. Autophagy receptors are required for the targeting of most identified autophagy cargoes, including protein aggregates, damaged organelles, and invading pathogens, to autophagosomes for degradation. Meanwhile, these autophagy receptors undergo autophagic degradation along with the substrates. Therefore, the protein levels of autophagy receptors are usually markedly upregulated in autophagy-deficient cells. A simple hypothesis is that the mediator, leading to the increase of 47S rRNA in autophagy-deficient cells, may be autophagy receptor(s). We began by measuring 47S rRNA level in HeLa cells with knockout of five major autophagy receptors (penta KO, pKO), including SQSTM1, NBR1 (NBR1 autophagy cargo receptor), TAX1BP1 (Tax1 binding protein 1), OPTN (optineurin) and CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2). Surprisingly, after serum stimulation, cellular 47S rRNA level was decreased in pKO HeLa cells (), suggesting that the five autophagy receptors may be responsible for the upregulation of 47S rRNA in autophagy-deficient cells. Of note, re-introduction of SQSTM1 but not other autophagy receptors reversed the decrease of 47S rRNA in pKO HeLa cells (). These data suggest that it is SQSTM1 but not other autophagy receptors mediating autophagy deficiency-induced upregulation of cellular 47S rRNA level.
Figure 2. SQSTM1 mediates the increase of 47S rRNA in autophagy-deficient cells. (A-B) Cellular 47S rRNA level was measured by real-time PCR. Cells with the deletion of the indicated gene(s) were transfected with or without untagged plasmid that harboring the gene. Cells were cultured in nutrient-rich medium or treated with serum (6 h) after serum starvation (12 h). pKO, knockout of SQSTM1, NBR1, TAX1BP1, OPTN and CALCOCO2. (C) 47S rRNA level was monitored by northern blot. Blotted RNA was hybridized to the biotin labeled probe comprising antisense sequences from human 47S rRNA or human ACTB mRNA. (D) Cellular 47S rRNA level of HeLa cells transfected with wild-type (WT) SQSTM1 or the indicated SQSTM1 mutant. K7A/D69A, unable to oligomerize; L343A, unable to bind to LC3; I431A, unable to interact with ubiquitinated substrates. (E) HeLa cells transfected with SQSTM1-MYC were treated with or without Act-D and incubated with 5-fluorouridine (5-FUrd) for 1 h. Then the cells were fixed and stained with anti-MYC and anti-BrdU. Scale bar: 10 µm. The statistical data are presented as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01.
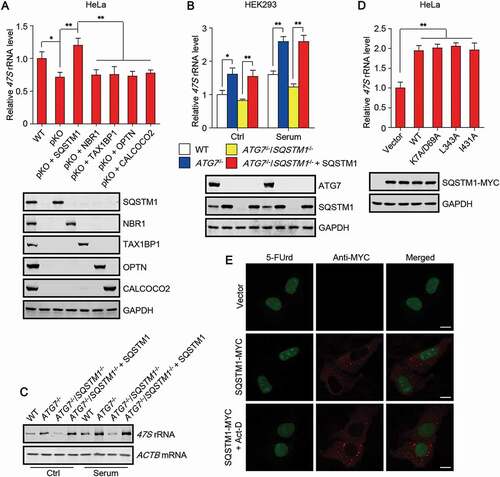
Moreover, deletion of SQSTM1 in ATG7 knockout HEK293 cells totally abolished the increase of 47S rRNA level with or without serum stimulation (). Re-introduction of SQSTM1 reversed the reduction of 47S rRNA level in ATG7 and SQSTM1 double knockout (DKO) cells (). In addition, we also monitored 47S rRNA level by northern blot. The blotted RNA was hybridized to biotin labeled probes comprising antisense sequences of human 47S rRNA or human ACTB mRNA, and detected by biotin antibody. Obviously, a consistent result was also obtained (). Notably, overexpression of WT SQSTM1 or SQSTM1 mutant, unable to oligomerize, to bind to LC3, or to interact with ubiquitinated substrates, upregulated cellular 47S rRNA level (), suggesting that the promoting effect of SQSTM1 on cellular 47S rRNA level does not rely on its function as autophagy receptor. We also utilized an in situ run-on assay, based on the incorporation of 5-fluorouridine (5-FUrd) into nascent RNA [Citation28], to assess rRNA synthesis activity directly. Clearly, SQSTM1 overexpression promoted 5-FUrd incorporation at the nucleolus, and the incorporation was totally abolished by Act-D treatment ().
These data suggest that SQSTM1 is sufficient and necessary to mediate the increase of 47S rRNA in autophagy-deficient cells and imply that the increase may be caused by rRNA synthesis activation at the nucleolus.
SQSTM1 activates rDNA transcription
We first examined the promoter activity of rDNA in autophagy-deficient cells using the pHrD-IRES-Luc (human rDNA promoter luciferase) reporter [Citation29]. The used promoter region is human rRNA promoter spanning 410 to 314 bp (accession number K01105) with respect to the transcription initiation site and the sequence was obtained from HeLa genomic DNA [Citation29]. Obviously, knockout of ATG16L1 but not RUBCN increased rDNA promoter activity, and re-introduction of ATG16L1 abolished the increase (). Moreover, deletion of SQSTM1 completely abolished the increase of rDNA promoter activity led by ATG7 knockout (). Re-introduction of SQSTM1 but not other autophagy receptor proteins rescued the reduction of rDNA promoter activity in pKO HeLa cells (). Of note, compared to WT SQSTM1, autophagy-deficient SQSTM1 mutant showed similar promoting effect on rDNA promoter activity (), confirming our observation that SQSTM1 upregulates cellular 47S rRNA level through an autophagy receptor-independent manner.
Figure 3. SQSTM1 activates rDNA transcription. (A-D) Cells with the deletion of the indicated gene(s) were transfected with or without untagged plasmid that harboring the gene. After 24 h, luciferase activity was measured. (E and F) The indicated cells were treated as indicated and subjected to chromatin immunoprecipitation (ChIP) assay using an anti-UBTF or an anti-POLR1A antibody. Anti-POLR2A was used as a negative control. The relative enrichment was determined by real-time PCR using primer set H42.9. (G) The indicated HEK293 cells were treated as indicated and subjected to immunoprecipitation by anti-UBTF, followed by western blot to detect POLR1A. Cells were cultured in nutrient-rich medium, treated with serum starvation (6 h), or stimulated with serum (3 h) after serum starvation. The statistical data are presented as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01.
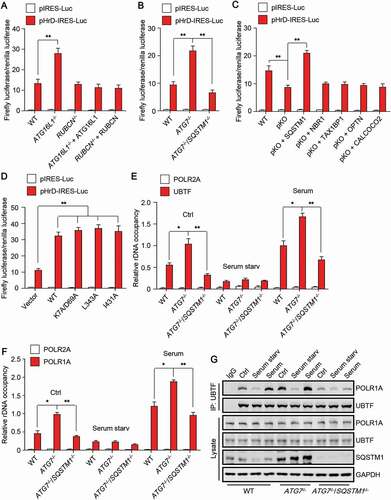
Because stimulation of rDNA promoter activity requires the formation of POLR1 PIC at rDNA promoter regions, we then carried out chromatin immunoprecipitation (ChIP) experiments to examine the rDNA promoter binding of UBTF/UBF1 (upstream binding transcription factor) and POLR1A/RPA194 (RNA polymerase I subunit A), two essential components of POLR1 PIC. Consistently, ATG7 knockout enhanced the binding of UBTF and POLR1A to the promoter regions of rDNA (H42.9), whereas SQSTM1 deletion totally revered the promoting effect (). Notably, serum starvation also abolished the binding of UBTF and POLR1A to the promoter regions in ATG7 knock out cells (), implying that nutrient-sensing pathway may be involved in autophagy deficiency-induced increase of rDNA promoter activity. POLR2A (RNA polymerase II subunit A) was used as a negative control to verify the specific binding of UBTF and POLR1A to the rDNA promoter regions (). Furthermore, we performed co-immunoprecipitation assay to check the influence of SQSTM1 on the assembly of POLR1 PIC by examining the interaction between UBTF and POLR1A. Notably, deletion of ATG7 promoted the binding of UBTF to POLR1A whereas deletion of SQSTM1 reversed the binding (). Interestingly, in ATG7-deficient cells, the protein level of SQSTM1 remained unchanged whereas the lysosomal distribution of SQSTM1 was significantly promoted upon serum stimulation (Fig. S1), indicating that lysosome-localized SQSTM1 may play an important role in regulating the assembly of POLR1 PIC.
These data together suggest that SQSTM1 activates rDNA transcription to upregulate cellular 47S rRNA level by promoting the assembly of POLR1 PIC at the rDNA promoter regions.
Autophagy deficiency potentiates the activation of MTORC1 signaling
Many lines of evidence have highlighted SQSTM1 as an important regulatory hub in various cellular signaling pathways, including MTORC1 signaling [Citation8–12]. Interestingly, MTORC1 signaling has been demonstrated to target a number of proteins to tightly control rDNA transcription [Citation19,Citation22,Citation30]. We then tested whether autophagy deficiency is involved in the regulation of MTORC1 signaling. By checking the phosphorylation of RPS6KB1/S6K1 (ribosomal protein S6 kinase B1) at Thr389, which site is specifically phosphorylated by MTORC1, we found that the phosphorylation of RPS6KB1 at Thr389 was significantly increased in cells with deletion of ATG5 or WIPI2 (WD repeat domain, phosphoinositide interacting 2) (). Meanwhile, knockout of ATG5 or WIPI2 led to the accumulation of SQSTM1 in cells (). Re-introduction of ATG5 or WIPI2 decreased the level of phosphorylated RPS6KB1 and SQSTM1 (). Of note, knockout of SQSTM1 was sufficient to abolish the increase of the phosphorylation of RPS6KB1 in cells with ATG7 deletion (). Moreover, re-expression of SQSTM1 rescued the phosphorylation of RPS6KB1 in ATG7 and SQSTM1 DKO cells (). Additionally, we found that overexpression of SQSTM1 but not other autophagy receptor significantly elevates the phosphorylation level of RPS6KB1 in cells upon serum stimulation (). The phosphorylation of RPS6KB1 induced by SQSTM1 overexpression in cells was completely abolished by the treatment of an MTOR inhibitor rapamycin (). In addition, Tsc2-deficient cells, in which MTORC1 was constitutively activated, displayed high basal phosphorylation of RPS6KB1, and SQSTM1 overexpression failed to further elevate the phosphorylation level (). These data together suggest that SQSTM1 accumulation is sufficient and necessary to potentiate MTORC1 activation in autophagy-deficient cells.
Figure 4. Autophagy deficiency potentiates the activation of MTORC1 signaling. (A-E) Phosphorylation of RPS6KB1 at Thr389 in the indicated cells treated as indicated. Cells were cultured in nutrient-rich medium, treated with serum starvation (6 h), or stimulated with serum (3 h) after serum starvation. Rapamycin (Rapa; 250 nM) were used along with serum stimulation. (F) Cellular 47S rRNA level of the cells treated as indicated in (E). Rapamycin, an MTOR inhibitor. The statistical data are presented as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01. (G) HEK293 cells transfected with or without SQSTM1-MYC were treated as indicated and subjected to immunoprecipitation by anti-UBTF, followed by western blot to detect POLR1A.
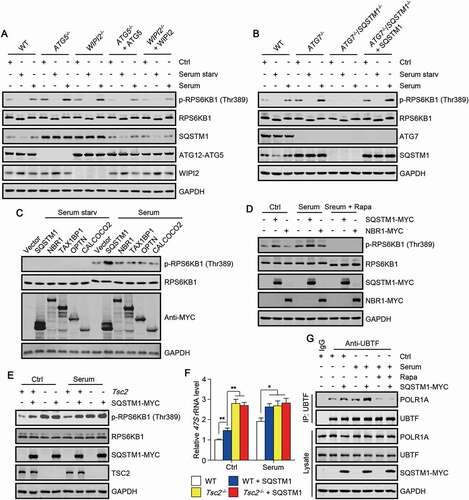
Then, we investigated whether SQSTM1-regualted MTORC1 activation is responsible for the increase of rDNA transcription in autophagy-deficient cells. By examining 47S rRNA level and rDNA promoter activity in cells, we found that the increase of 47S rRNA level and rDNA promoter activity caused by SQSTM1 overexpression could be totally abolished by the treatment of MTOR inhibitor rapamycin or Torin1 ((Fig. S2). Moreover, we found that SQSTM1 overexpression was unable to further increase 47S rRNA level in Tsc2-deficient cells (). We also checked the interaction between UBTF and POLR1A, and found that SQSTM1 overexpression enhanced UBTF-POLR1A interaction (). Interestingly, rapamycin treatment totally abolished the interaction between UBTF and POLR1A in cells with or without SQSTM1 overexpression ().
Taken together, these results suggest that MTORC1 signaling is required for SQSTM1 to mediate the activation of rDNA transcription in autophagy-deficient cells.
Autophagy deficiency stimulates protein synthesis, cell growth and cell proliferation
As a pivotal step for ribosome biogenesis, rDNA transcription has been suggested to play a promoting role in a number of cellular processes, including protein synthesis, cell growth and cell proliferation. To explore the physiological role of autophagy deficiency-induced rDNA transcription activation, we chose to examine protein synthesis, cell growth and cell proliferation in autophagy-deficient cells. The rate of protein synthesis was checked using the surface sensing of translation (SUnSET) assay, detecting the amount of puromycin incorporation into nascent proteins, which indicates the rate of nascent protein synthesis [Citation31,Citation32]. Obviously, knockout of ATG7 increased the puromycin incorporation in cells, and the increase was abolished by further knockout of SQSTM1 (). Of note, re-introduction of SQSTM1 rescued the puromycin incorporation in ATG7 and SQSTM1 DKO cells (). Puromycin incorporation has been shown to induce the formation of protein aggregates [Citation7], which can be degraded by autophagy. To further verify our observations, we also detected nascent protein synthesis using a nontoxic method [Citation33], by which an amino acid analog of methionine containing an alkyne moiety was added to the culture medium and incorporated into newly synthesized proteins. The alkyne-modified proteins were then detected by a click reaction between alkyne and Alexa Fluor 488 azide, whose fluorescence indicated nascent protein synthesis rate. A consistent result was also obtained, SQSTM1 knockout abolished ATG7 deletion-induced increase of protein synthesis rate, and the effect led by SQSTM1 deletion was rescued by SQSTM1 re-introduction (). Of note, the increase of protein synthesis rate, led by ATG7 knockout or SQSTM1 re-expression, was completely abolished by serum starvation or rapamycin treatment ().
Figure 5. Autophagy deficiency stimulates protein synthesis, cell growth and cell proliferation. (A) Global protein synthesis was detected using the surface sensing of translation (SUnSET) method in the indicated cells. Cells were cultured in nutrient-rich medium, treated with serum starvation (6 h), or stimulated with serum (3 h) after serum starvation. The specificity of the anti-puromycin (puro) antibody was demonstrated by a sample without puromycin treatment. Coomassie blue staining was used as the loading control. (B) Global protein synthesis was detected using a Click-iT HPG method in the indicated cells treated as indicated in (A) with or without rapamycin incubation. (C) Relative cell diameter of the indicated HEK293 cells treated with serum for 24 h after serum starvation for 12 h. (D) Relative cell number of the indicated HEK293 cells. (E) Relative cell diameter of the indicated HEK293 cells treated with serum for 24 h after serum starvation for 12 h. Cells were transfected with or without SQSTM1-MYC and treated with or without rapamycin. (F) Relative cell number of the indicated HEK293 cells. Rapa, rapamycin. The statistical data are presented as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
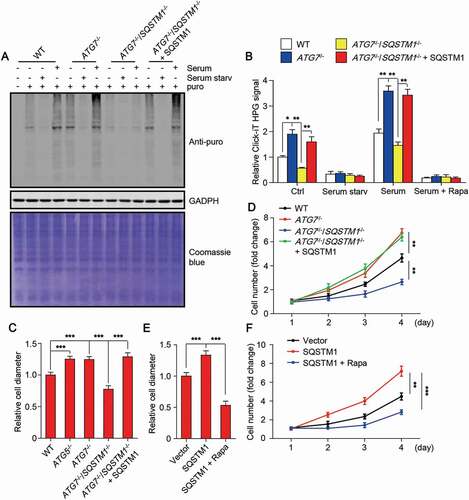
We also examined cell growth and cell proliferation, and found that knockout of ATG7 promotes cell growth, indicated by the average cell diameter, and cell proliferation, indicated by cell number (,). Interestingly, the increase of both cell growth and cell proliferation is reversed by the deletion of SQSTM1 (). Moreover, re-introduction of SQSTM1 rescued cell growth and cell proliferation in ATG7 and SQSTM1 DKO cells (). In addition, we found that SQSTM1 overexpression is sufficient to promote cell growth and cell proliferation, and the promoting effect is completely abolished by rapamycin treatment ().
Taken together, these data suggest that autophagy deficiency stimulates protein synthesis, cell growth and cell proliferation through a SQSTM1-dependent manner, which further confirm the promoting effect of autophagy deficiency on rDNA transcription in cells.
Discussion
In this study, we have uncovered the regulatory role of autophagy in rRNA metabolism. By elucidating the indispensable role of autophagy receptor SQSTM1 in autophagy deficiency-induced activation of MTORC1 signaling and rDNA transcription, our data suggest a promoting role of autophagy deficiency in rDNA transcription, by which autophagy deficiency stimulates protein synthesis, cell growth and cell proliferation. Our findings link autophagy, a catabolic process, to rDNA transcription, an anabolic pathway, and broaden the regulatory role of autophagy in cell metabolism and provide novel mechanisms for autophagy dysregulation-related diseases, such as cancer.
Autophagy deficiency led by deletion of different ATG genes has been demonstrated to render mice tumor prone [Citation34–36], suggesting that autophagy may suppress tumor initiation. Interestingly, the tumor suppressive property of autophagy is mainly attributed to the elimination of SQSTM1 [Citation37–39]. However, the pro-cancer properties of SQSTM1 remain largely unknown. Our study demonstrates that SQSTM1 is required for autophagy deficiency-induced rDNA transcription activation, which not only highlights the central role of SQSTM1 in autophagy deficiency-related cancers, but also implies that intervention of the activated rDNA transcription may be one more choice to combat these types of cancers.
Although our study suggests that autophagy deficiency-induced SQSTM1 accumulation potentiates MTORC1 signaling, the master regulator of rDNA transcription, we have not demonstrated that SQSTM1-induced MTORC1 activation is the only mechanism for rDNA transcription stimulation in autophagy-deficient cells. A recent study shows that SQSTM1 can target to the nucleolus through the nucleolar localization signal at the N-terminal domain of SQSTM1 [Citation40], it would be worthy to investigate whether SQSTM1 is directly involved in the regulation of rDNA transcription at the nucleolus.
Several previous studies have shown that ribosome can be targeted to autophagosomes for autophagic degradation upon nutrient starvation [Citation41–43]. Our study suggests that autophagy is also involved in rDNA transcription, one of the key steps of ribosome biogenesis. All these findings together suggest that autophagy emerges as a key coordinator that controls both the degradation and the production of ribosomes in response to intracellular or environmental changes. Under nutrient-rich conditions, autophagy occurs at a very low basal level, leading to the decrease of autophagic degradation of ribosomes and the increase of rDNA transcription, both of which may help maintain the huge amount of cellular ribosomes and support protein synthesis, cell growth and cell proliferation. By contrast, upon nutrient deprivation, autophagy is significantly induced, causing the activation of autophagic degradation of ribosomes and the inhibition of rDNA transcription, both of which may decrease the number of cellular ribosomes and suppress the anabolic processes to help the cell survive. Additionally, it would be interesting to explore whether there are some mediators, such as autophagy substrates, coordinating the autophagic degradation of ribosomes and the autophagy-regulated rDNA transcription. Moreover, because ribosome biogenesis is a multi-step process, whether autophagy is involved in the regulation of other steps, including the processing and modification of rRNAs, remains unknown and needs further investigations.
All kinds of rRNAs, accounting for the most nascent RNAs, are mainly derived from the 47S precursor rRNA, whose production consumes a tremendous amount of cellular energy. Intervention of rDNA transcription is supposed to significantly affect cellular energy status. Of note, autophagy is also tightly controlled by cellular energy- and nutrient-sensing pathways, such as AMPK and MTORC1 signaling [Citation44–46]. It would be interesting to explore whether intervention of cellular rDNA transcription activity affects autophagy induction, which may help us better understand the relationship between autophagy and rDNA transcription.
Materials and methods
Cell culture, transfection, reagents and treatment
HEK293, HeLa cells and MEFs were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, 11965092) supplemented with 10% fetal bovine serum (FBS) (Gibco, 10091148) at 37°C under an atmosphere of 5% CO2. Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) was used for plasmid transient transfection according to the manufacturer’s instructions. Cells were analyzed 16–24 h after transfection. Cells were incubated with DMEM referred to as serum starvation. Re-incubation of serum-starved cells with DMEM supplemented with 10% FBS referred to as serum stimulation.
HEK293 cells with deletion of WIPI2 were kindly provided by Dr. Han-Ming Shen (National University of Singapore Yong Loo Lin School of Medicine, Singapore). HeLa cells with deletion of ATG16L1 or RUBCN were kindly provided by Dr. Feng Shao (National Institute of Biological Sciences, Beijing, China). HeLa cells with deletion of SQSTM1, NBR1, TAX1BP1, OPTN and CALCOCO2 were kindly provided by Dr. Qiming Sun (Zhejiang University School of Medicine, Hangzhou, China). MEFs with or without deletion of Tsc2 and HEK293 cells with deletion of STX17 or SQSTM1 were described previously [Citation45,Citation47,Citation48]. HEK293 cells with deletion of ULK1, ATG5, ATG7 or both ATG7 and SQSTM1 were generated in our lab. Briefly, cells were transiently transfected with the corresponding pEP-KO plasmid and subjected to selection with puromycin (Selleckchem, S7417). Single clones were chosen and tested by western blot and DNA sequencing. The used sg (single-guide) RNA sequences were listed in Table S1.
Antibodies and plasmids
The following antibodies were used: anti-biotin (Abcam, ab53494), anti-CALCOCO2/NDP52 (Abcam, ab68588), anti-OPTN (Abcam, ab23666), anti-WIPI2 (Abcam, ab105459), anti-NBR1 (Cell Signaling Technology, 9891), anti-RPS6KB1/S6K1 (Cell Signaling Technology, 9202), anti-phospho-RPS6KB1/S6K1 (Thr389) (Cell Signaling Technology, 9205), anti-TAX1BP1 (Cell Signaling Technology, 5105S), anti-TSC2/Tuberin (Cell Signaling Technology, 3612), anti-ULK1 (Cell Signaling Technology, 8054S), anti-RUBCN/Rubicon (Cell Signaling Technology, 8465S), anti-ATG16L1 (MBL, PM040), anti-GFP (MBL, 598), anti-MYC (MBL, 562), anti-puromycin (Millipore, MABE343), anti-ATG5 (Novus Biologicals, NB110-53818), anti-SQSTM1/p62 (Proteintech, 18420-1-AP), anti-BrdU (Sigma-Aldrich, B8434), anti-LAMP1 (Santa Cruz Biotechnology, sc-17768), anti-UBTF/UBF1 (Santa Cruz Biotechnology, sc-13125), anti-POLR1A (Santa Cruz Biotechnology, sc-48385), anti-POLR2A (Santa Cruz Biotechnology, sc-9001), anti-GAPDH (Santa Cruz Biotechnology, sc-32233), anti-MYC (Santa Cruz Biotechnology, sc-40), anti-ATG7 (Sigma-Aldrich, A2856), anti-STX17 (Sigma-Aldrich, HPA001204), anti-TUBB (Sigma-Aldrich, T8328), donkey anti-rabbit IRDye800CW (LI-COR Biosciences, 926–32213), donkey anti-mouse IRDye680 (LI-COR Biosciences, 926–32222), goat anti-mouse IgG (H + L), Alexa Fluor 488 (Thermo Fisher Scientific, A-11001), donkey anti-rabbit IgG (H + L), Alexa Fluor 546 (Thermo Fisher Scientific, A10040). TMEM192-GFP was described previously [Citation46], ATG5, ATG7, ATG16L1, RUBCN, STX17, ULK1, WIPI2, SQSTM1, NBR1, TAX1BP1, OPTN and CALCOCO2 were constructed by inserting the corresponding human cDNA into a pCDNA3.1-MYC vector or untagged pCDNA3.1 vector.
Western blot and immunoprecipitation
Proteins from lysed cells were denatured and loaded on sodium dodecyl sulfate (SDS) polyacrylamide gels. Then the proteins were transferred to polyvinylidene difluoride membranes (Millipore, ISEQ00010), blocked in TBST (150 mM NaCl [Sangon Biotech, A610476], 10 mM TRIS-HCl, pH 7.5, and 0.1% Tween 20 [Sangon Biotech, A100777]) containing 5% (w:v) bovine serum albumin (Sangon Biotech, A602449), and incubated with the corresponding primary and secondary antibodies. The specific bands were analyzed by the western blot infrared imaging system (LI-COR Biosciences). For immunoprecipitation, cells were lysed with Triton X-100 lysis buffer (50 mM TRIS-HCl, pH 7.4, 150 mM NaCl [Sangon Biotech, A610476], 1% Triton X-100 [Sangon Biotech, A110694], 1% sodium deoxycholate [Sangon Biotech, A600150], 0.1% SDS [Sangon Biotech, A100227], 1 mM EDTA [Sangon Biotech, B540625]) containing protease inhibitors (Roche, 4693159001). After centrifugation at 12,000 x g for 15 min at 4°C, the supernatants were incubated with the indicated antibodies overnight and then protein A (Santa Cruz Biotechnology, sc-2001) or protein G (Santa Cruz Biotechnology, sc-2002) agarose for 4 h at 4°C. The immunocomplexes were washed 4 times and analyzed by western blot.
Lysosome isolation
Lysosomes were isolated as described previously [Citation46]. Briefly, the cells transiently transfected with TMEM192-GFP were harvested, washed and homogenized in extraction buffer (5 mM MOPS [Sangon Biotech, A620336], pH 7.65, 0.25 M sucrose [Sangon Biotech, A502792], 1 mM EDTA, 0.1% ethanol [Sangon Biotech, A500737]) containing protease inhibitors. The lysates were incubated with the specific GFP-Trap agarose (ChromoTek, gta) at 4°C for 2 h, then the bound lysosomes were washed using extraction buffer for 4 times and subjected to western blot.
Immunostaining and confocal microscopy
Cells were fixed in 4% formaldehyde (Sangon Biotech, A501912) followed by permeabilization with 0.1% Triton X-100. After washing twice in PBS (Sangon Biotech, E607008), cells were incubated in PBS with FBS (PBS, pH 7.4, 10% FBS) to block nonspecific sites of antibody adsorption. The cells were then incubated with appropriate primary and secondary antibodies. Cell images were acquired on a laser scanning confocal microscope LSM880 (Carl Zeiss) and analyzed with the LSM 880 software (Carl Zeiss).
RNA purification and real-time PCR
Total cellular RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, 15596018) and reverse transcribed using M-MLV reverse transcriptase (Promega, 9PIM170) according to the manufacturer’s protocol. To examine cellular 47S rRNA level, random hexamers were used for reverse transcription. Real-time PCR analysis was performed in 10 µl reaction mixture using the SYBR GREEN PCR Master Mix (Takara, DRR041A). The PCR reaction mixture includes the following components: 10 pmol of primer, 2 mM MgCl2, 200 μM dNTP mixture, 0.1 U of Taq DNA polymerase and universal buffer. Amplification specificity was checked by melting curve analysis. All of the reactions were performed in triplicate in the ABI7500 (Applied Biosystems). The sequences of primers used are listed in Table S2.
Northern blot
Total cellular RNA was separated on denaturing agarose gel and transferred to nylon membrane (Millipore, INYC00010). Blotted RNA was then crosslinked and hybridized to biotin labeled probes comprising antisense sequences of human 47S rRNA and human ACTB mRNA, and detected by biotin antibody. The probes were synthesized and the antisense sequences are listed in Table S3.
ChIP assay
Cells were cross-linked using formaldehyde, and lysed with SDS lysis buffer (50 mM TRIS-HCl, pH 8.1, 1% SDS, 10 mM EDTA) containing protease inhibitors, then followed by sonication. The crosslinked, sonicated chromatin was precleared before being incubated with 5 μg of the indicated antibodies and rotated at 4°C overnight. After extensive washes, immunocomplexes were treated with proteinase K (Roche, RPROTKSOL-RO) and decrosslinked. Bound DNA in the precipitates, as well as input DNA, was extracted, purified, and subjected to real-time PCR analysis using H42.9 primers corresponding to the promoter regions of the rDNA repeats. The sequences of primers used are listed in Table S2.
Luciferase reporter assay
Luciferase activity was measured using Dual-Luciferase assay [Citation29]. Briefly, pIRES-Luc or pHrD-IRES-Luc (expressing firefly luciferase) along with the internal control pRLTK (expressing Renilla luciferase) were co-transfected into cells. Cells were treated as indicated and lysed in passive lysis buffer, and assayed for luciferase activity using a luciferase assay kit (Promega, E1910) according to the manufacturer’s protocol. Reporter construct activity was normalized by comparison with activity from the Renilla luciferase construct.
Cell diameter determination
Cells were treated as indicated and harvested by trypsinization in 2 ml media and diluted 1:20 with PBS. Cell diameters were determined by flow cytometry using the Cytomic FC 500MCL (Beckman Coulter).
Protein synthesis and cell proliferation assays
For puromycin incorporation assay, cells were treated with 1 µM puromycin for 30 min and followed by extraction with digitonin supplemented permeabilization buffer (50 mM TRIS-HCl, pH 7.5, 5 mM MgCl2 [Sangon Biotech, A610328], 25 mM KCl [Sangon Biotech, A100395]) containing protease inhibitors to release free puromycin. Nascent proteins were detected with anti-puromycin antibody using western blot. For Click-iT HPG Alexa Fluor 488 protein synthesis assay, protein synthesis rate was assessed using a kit (Thermo Fisher Scientific, C10428) according to the manufacturer’s instructions. For cell proliferation assay, cells were counted using a cell counting kit (Bimake, B34302) according to the manufacturer’s instructions.
Statistical analysis
All the statistical data were presented as mean ± SEM. Statistical significance of the differences was determined using the Student’s t-test. P-value < 0.05 was considered to be statistically significant.
Supplemental Material
Download Zip (551.3 KB)Acknowledgments
We are grateful to the Imaging Center of Zhejiang University School of Medicine for their assistance in confocal microscopy. We thank Dr. Han-Ming Shen (National University of Singapore Yong Loo Lin School of Medicine, Singapore), Dr. Feng Shao (National Institute of Biological Sciences, Beijing, China), and Dr. Qiming Sun (Zhejiang University School of Medicine, Hangzhou, China) for kindly providing cell lines and reagents.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Feng YC, He D, Yao ZY, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41.
- Levine B, Mizushima N., Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741.
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830.
- Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348.
- Mizushima N, Levine B. Autophagy in human diseases. New Engl J Med. 2020;383:1564–1576.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145.
- Sanz L, Diaz-Meco MT, Nakano H, et al. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586.
- Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappa B mediator in tumorigenesis. Cancer Cell. 2008;13:343–354.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2020;12:213–U217.
- Lee SJ, Pfluger PT, Kim JY, et al. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 2020;11:226–232.
- Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146.
- Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–755.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Bio. 2020;21:183–203.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976.
- Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol. 2020;50:131–156.
- Grummt I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003;17:1691–1702.
- Russell J, Zomerdijk JC. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem Sci. 2005;30:87–96.
- Xu Y, Wan W, Shou X, et al. TP53INP2/DOR, a mediator of cell autophagy, promotes rDNA transcription via facilitating the assembly of the POLR1/RNA polymerase I preinitiation complex at rDNA promoters. Autophagy. 2016;12:1118–1128.
- Xu YF, Wu YS, Wang L, et al. Identification of curcumin as a novel natural inhibitor of rDNA transcription. Cell Cycle. 2020;19:3362–3374.
- Hoppe S, Bierhoff H, Cado I, et al. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci U S A. 2009;106:17781–17786.
- Mayer C, Zhao J, Yuan X, et al. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004;18:423–434.
- White RJ. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008;24:622–629.
- Heckmann BL, Green DR. LC3-associated phagocytosis at a glance. J Cell Sci. 2019;132:jcs222984.
- Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658.
- Chan EYW, Kir S, Tooze SA. SiRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474.
- Popov A, Smirnov E, Kovacik L, et al. Duration of the first steps of the human rRNA processing. Nucleus. 2013;4:134–141.
- Torrano V, Navascues J, Docquier F, et al. Targeting of CTCF to the nucleolus inhibits nucleolar transcription through a poly(ADP-ribosyl)ation-dependent mechanism. J Cell Sci. 2006;19:1746–1759.
- Ghoshal K, Majumder S, Datta J, et al. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J Biol Chem. 2004;279:6783–6793.
- Tsang CK, Liu H, Zheng XF. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle. 2010;9:953–957.
- Goodman CA, Mabrey DM, Frey JW, et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–1039.
- Schmidt EK, Clavarino G, Ceppi M, et al. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277.
- Chorghade S, Seimetz J, Emmons R, et al. Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. Elife. 2017;6:e24139.
- Jaber N, Dou Z, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–2008.
- Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434.
- Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800.
- Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284.
- Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075.
- Zhang J, Yang SZ, Xu B, et al. p62 functions as an oncogene in colorectal cancer through inhibiting apoptosis and promoting cell proliferation by interacting with the vitamin D receptor. Cell Prolif. 2019;52:e12585.
- Lobb IT, Morin P, Martin K, et al. A role for the autophagic receptor, SQSTM1/p62, in trafficking NF-kappaB/RelA to nucleolar aggresomes. Mol Cancer Res. 2021;19:274–287.
- An H, Harper JW. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol. 2018;20:135–143.
- Kraft C, Deplazes A, Sohrmann M, et al. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610.
- Wyant GA, Abu-Remaileh M, Frenkel EM, et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science. 2018;360:751–758.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141.
- Wan W, You Z, Xu Y, et al. mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell. 2017;68:323–335 e326.
- Wan W, You Z, Zhou L, et al. mTORC1-regulated and HUWE1-mediated WIPI2 degradation controls autophagy flux. Mol Cell. 2018;72:303–315 e306.
- Shen QH, Shi Y, Liu JQ, et al. Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy. 2021;17:1157–1169.
- You Z, Jiang WX, Qin LY, et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat Commun. 2019;10:5792.