
ABSTRACT
Owing to the dominant functions of mitochondria in multiple cellular metabolisms and distinct types of regulated cell death, maintaining a functional mitochondrial network is fundamental for the cellular homeostasis and body fitness in response to physiological adaptations and stressed conditions. The process of mitophagy, in which the dysfunctional or superfluous mitochondria are selectively engulfed by autophagosome and subsequently degraded in lysosome, has been well formulated as one of the major mechanisms for mitochondrial quality control. To date, the PINK1-PRKN-dependent and receptors (including proteins and lipids)-dependent pathways have been characterized to determine the mitophagy in mammalian cells. The mitophagy is highly responsive to the dynamics of endogenous metabolites, including iron-, calcium-, glycolysis-TCA-, NAD+-, amino acids-, fatty acids-, and cAMP-associated metabolites. Herein, we summarize the recent advances toward the molecular details of mitophagy regulation in mammalian cells. We also highlight the key regulations of mammalian mitophagy by endogenous metabolites, shed new light on the bidirectional interplay between mitophagy and cellular metabolisms, with attempting to provide a perspective insight into the nutritional intervention of metabolic disorders with mitophagy deficit.
Abbreviations: acetyl-CoA: acetyl-coenzyme A; ACO1: aconitase 1; ADCYs: adenylate cyclases; AMPK: AMP-activated protein kinase; ATM: ATM serine/threonine kinase; BCL2L1: BCL2 like 1; BCL2L13: BCL2 like 13; BNIP3: BCL2 interacting protein 3; BNIP3L: BCL2 interacting protein 3 like; Ca2+: calcium ion; CALCOCO2: calcium binding and coiled-coil domain 2; CANX: calnexin; CO: carbon monoxide; CYCS: cytochrome c, somatic; DFP: deferiprone; DNM1L: dynamin 1 like; ER: endoplasmic reticulum; FKBP8: FKBP prolyl isomerase 8; FOXO3: forkhead box O3; FTMT: ferritin mitochondrial; FUNDC1: FUN14 domain containing 1; GABA: γ-aminobutyric acid; GSH: glutathione; HIF1A: hypoxia inducible factor 1 subunit alpha; IMMT: inner membrane mitochondrial protein; IRP1: iron regulatory protein 1; ISC: iron-sulfur cluster; ITPR2: inositol 1,4,5-trisphosphate type 2 receptor; KMO: kynurenine 3-monooxygenase; LIR: LC3 interacting region; MAM: mitochondria-associated membrane; MAP1LC3: microtubule associated protein 1 light chain 3; MFNs: mitofusins; mitophagy: mitochondrial autophagy; mPTP: mitochondrial permeability transition pore; MTOR: mechanistic target of rapamycin kinase; NAD+: nicotinamide adenine dinucleotide; NAM: nicotinamide; NMN: nicotinamide mononucleotide; NO: nitric oxide; NPA: Niemann-Pick type A; NR: nicotinamide riboside; NR4A1: nuclear receptor subfamily 4 group A member 1; NRF1: nuclear respiratory factor 1; OPA1: OPA1 mitochondrial dynamin like GTPase; OPTN: optineurin; PARL: presenilin associated rhomboid like; PARPs: poly(ADP-ribose) polymerases; PC: phosphatidylcholine; PHB2: prohibitin 2; PINK1: PTEN induced kinase 1; PPARG: peroxisome proliferator activated receptor gamma; PPARGC1A: PPARG coactivator 1 alpha; PRKA: protein kinase AMP-activated; PRKDC: protein kinase, DNA-activated, catalytic subunit; PRKN: parkin RBR E3 ubiquitin protein ligase; RHOT: ras homolog family member T; ROS: reactive oxygen species; SIRTs: sirtuins; STK11: serine/threonine kinase 11; TCA: tricarboxylic acid; TP53: tumor protein p53; ULK1: unc-51 like autophagy activating kinase 1; VDAC1: voltage dependent anion channel 1
Introduction
Mitochondria are critical organelles determining a myriad of cellular metabolisms, including but not limited to the ATP production via electron transport coupled with oxidative phosphorylation, tricarboxylic acid (TCA) cycle, fatty acid β-oxidation, amino acid synthesis, calcium homeostasis and iron metabolism (bio-synthesis of heme and iron-sulfur cluster). Besides, from last two decades, it has been well understood that mitochondria serve as the signaling hubs to orchestrate intra-cellular and extra-cellular signals and communicate to other cellular compartments to fulfill the diverse functions under homeostatic and stressed conditions [Citation1,Citation2]. Moreover, mitochondria produce overwhelming majority of reactive oxygen species (ROS) as the inevitable by-product during the electron transport, while themself are particularly vulnerable to the ROS mediated oxidative damage [Citation3]. Mitochondria also dominate programmed cell death mainly by tuning the release of pro-apoptotic molecules, especially CYCS (cytochrome c, somatic) and DIABLO (diablo IAP-binding mitochondrial protein) [Citation4,Citation5]. The mitochondria associated BCL2 family, categorized into three sub-families including the anti-apoptotic proteins, the BH3-only pro-apoptotic proteins, and the pore-forming apoptotic proteins, coordinates the mitochondrial membrane permeabilization and the release of these apoptogenic factors [Citation6]. The compromised mitochondria would also drive other forms of regulated cell death, including necroptosis, ferroptosis and pyroptosis [Citation7]. Moreover, the accumulation of dysfunctional mitochondria has been widely documented to associate with the pathogenesis of multiple human diseases, especially the neurodegenerative diseases, autoimmune disorders and cancers [Citation8]. Therefore, maintenance of a functional mitochondria pool is fundamental for the cellular homeostasis and body fitness.
Mitochondrial quality control is precisely achieved by distinct mechanisms. Mitochondrially resident proteases and chaperons determine the integrity of mitochondrial proteome by hydrolyzing and unfolding-refolding the misfolded proteins, respectively [Citation9,Citation10]. Mitochondrial unfolded protein response (UPRmt), referring to the sufficient accumulation of misfolded mitochondrial proteins driving a retrograde signaling to activate the transcriptional response and facilitate the expression of mitochondrial chaperones and proteases, could reduce the mitochondrial unfolded proteins and promote recovery of mitochondrial proteome [Citation11]. Mitochondria are highly dynamic organelles, with constant fusion and fission, which is conferred by the fusion proteins MFNs (mitofusins), OPA1 (OPA1 mitochondrial dynamin like GTPase) and fission protein DNM1L (dynamin 1 like), as well as its mitochondrially localized receptors. Mitochondrial fusion neutralizes the damaged mitochondrial components, while mitochondrial fission could effectively segregate the damaged mitochondria from the whole mitochondrial architectural network for the subsequent degradation [Citation12]. Furthermore, mitochondria-derived vesicles (MDVs), generated through the selective incorporation of mitochondrial outer membrane, inner membrane, or matrix content, subsequently deliver specific mitochondrial contents to late endosome or multivesicular bodies for degradation, are recognized as a novel avenue for mitochondrial quality control at sub-organelle level [Citation13,Citation14]. Increasing evidence has emerged that mitophagy, a selective recognition and degradation of mitochondria by autophagic machinery, is regarded as one of the major mechanisms for mitochondrial quality control [Citation15,Citation16].
Pioneering studies reported the focal cytoplasmic degradation of mitochondria from 1950s to 1970s. Specifically, Hruban and colleagues observed that mitochondria are broken down within the cytoplasmic inclusions in 1963 [Citation17]. In 1979, Greene and colleagues observed a selective degeneration of mitochondria associates with electron-lucent mitochondrial inclusions in proximal renal tubular epithelium induced by hyperbaric oxygen exposure [Citation18]. These early studies based on electron microscopy groundbreakingly proposed the concepts of autophagosome maturation and selective autophagy. The early history of mitophagy research was comprehensively summarized [Citation19]. In 2002, the phenomenon was reemerged. A complete removal of mitochondria was observed in cultured primary sympathetic neurons during apoptosis induction in the presence of CASPs (caspases) inhibitors [Citation20]. In 2005, the term “mitophagy” was first proposed to describe the selective mitochondrial autophagy by John Lemasters [Citation21]. Since then, increasing studies focused on this booming field and several regulatory pathways including the PINK1 (PTEN induced kinase 1)-PRKN/PARKIN (parkin RBR E3 ubiquitin protein ligase)-dependent mitophagy and the receptors (including protein and lipid mitophagy receptors)-mediated mitophagy have been deciphered. Furthermore, defects in mitophagy have been evidenced to result in the accumulation of damaged mitochondria, which is implicated in several disease pathogenesis [Citation22]. Herein, we critically summarize the major regulatory circuits of mitophagy in mammalian cells, with a focus on dissecting the mitophagy regulation by endogenous metabolites, in an attempt to provide a perspective insight into the nutritional intervention of metabolic disorders with mitophagy deficit.
Major mitophagy pathways
Macroautophagy/autophagy refers to a cellular catabolic process, sequentially manifested by the formation of sequestering compartments, phagophores, that engulf the to-be-degraded cellular components, including proteins, protein aggregates, lipid, organelles, invading pathogens and even nuclear lamina. The phagophores then mature into double-membraned vesicles (autophagosomes) that fuse with an endosome and/or lysosome, resulting in subsequent degradation of the engulfed components by the lysosomal hydrolases [Citation23]. Mitophagy is a specialized form of autophagy to selectively recognize and remove the damaged or superfluous mitochondria, sharing fundamental features and core proteins with general autophagy. The specificity of the phagophore engulfing mitochondria is achieved by distinct mechanisms. To data, both the PINK1-PRKN-dependent mitophagy and receptor-mediated mitophagy have been elucidated in mammalian cells.
PINK1-PRKN axis-dependent mitophagy
Early studies using loss-of-function mutants of Drosophila suggested that both the mutations in Pink1 (encoding a homolog of the serine-threonine kinase PINK1) and park (encoding a homolog of the E3 ubiquitin ligase PRKN), two major genes associated with Parkinson disease, result in disturbed mitochondrial morphology and declined mitochondrial functionalities, accompanied with locomotor deficits, muscle degeneration, male sterility and neuronal loss [Citation24,Citation25]. The transgenic expression of park markedly ameliorates mitochondrial functionalities and restores the neuronal fitness at the genetic background with Pink1 loss of function, but not vice versa, suggesting that PRKN functions downstream of PINK1 to determine the mitochondrial integrity [Citation24,Citation26].
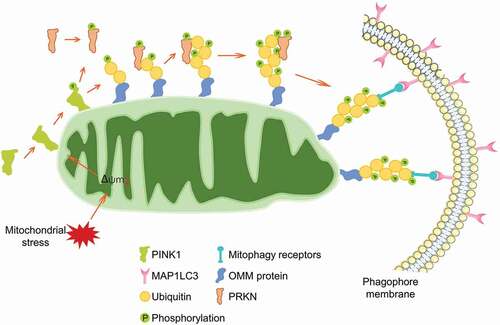
The initial understanding of the PINK1-PRKN axis-dependent mitophagy was originated by Dr. Richard Youle and colleagues. In 2008, they reported that PRKN is recruited to the outer membrane of depolarized mitochondria, where it facilitates the autophagic degradation of these damaged mitochondria [Citation27]. This groundbreaking study boosted up the mitophagy research. Since then, the regulatory circuit of the PINK1-PRKN axis-dependent mitophagy has been well understood (). Under basal condition, PINK1 is imported into the mitochondria, where it is cleaved by the mitochondrial rhomboid protease PARL (presenilin associated rhomboid like) [Citation28,Citation29]. The processed product is released into the cytosol and constitutively degraded by the E3 ubiquitin ligases UBR1, UBR2 and UBR4 [Citation30]. During mitochondrial depolarization, PINK1 escapes from PARL proteolytic cleavage and trans-localizes onto the mitochondrial outer membrane [Citation31,Citation32]. On the outer membrane, PINK1 forms a dimer and associates with the TOMM (translocase of outer mitochondrial membrane) complex to reduce the mitochondrial import of newly synthesized proteins [Citation31,Citation33]. The TOMM complex guides the correct positioning of dimeric PINK1 and facilitates its kinase activity through supporting its auto-phosphorylation, which is required for the PRKN recruitment and mitophagy execution [Citation32]. Besides, TOMM7 [Citation34,Citation35], IMMT/Mitofilin/MIC60 (inner membrane mitochondrial protein) [Citation36] and mitochondrial protease OMA1 [Citation34] are involved in regulating PINK1 stabilization on the outer membrane [Citation37]. On one hand, PINK1 phosphorylates the preexisting ubiquitin at Ser65 on the mitochondrial surface [Citation38,Citation39]. On the other hand, the phosphorylated ubiquitin could be captured by PRKN, inspiring PRKN phosphorylation at Ser65 in the ubiquitin-like domain by PINK1 [Citation40–42]. Phosphorylation at Ser65 and binding to the phosphorylated ubiquitin facilitate the conformational change of the ubiquitin-like domain and relieve of auto-inhibition [Citation43–45], leading to PRKN activation to ubiquitinate mitochondrial proteins on the outer membrane, which serves as a “eat-me” signaling for ultimate mitophagy [Citation46,Citation47]. Furthermore, phosphorylated MFN2 [Citation48] and VDAC1 (voltage dependent anion channel 1) [Citation49] are also important for PRKN translocation. PINK1 and the phosphorylated ubiquitin also drive the mitochondrial recruitment of autophagy receptor proteins including CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) and OPTN (optineurin), which facilitate mitochondria engulfed by autophagosome through direct interaction with MAP1LC3 (microtubule associated protein 1 light chain 3) [Citation50,Citation51].
Figure 1. The regulatory circuit of PINK1-PRKN-mediated mitophagy. PINK1 is imported into healthy mitochondria and cleaved by mitochondrial protease, leading to a quick degradation. During mitochondrial depolarization, PINK1 is stabilized and translocated onto the outer mitochondrial membrane, where it recruits and phosphorylates PRKN, phosphorylates the ubiquitin molecules, leading to the enzymatic activation of PRKN. PRKN ubiquitinates numerous mitochondrial outer membrane proteins. These signals lead to autophagy receptors OPTN and CALCOCO2 translocating onto mitochondria for the selective recognition of mitochondria by the phagophore membrane.
According to the mechanistic studies based on Drosophila, mammalian cells, mice and epidemiological analysis of Parkinson disease, genetic variations of PINK1 and PRKN are strongly associated with the pathogenesis of Parkinson disease, in which the mitochondrial dysfunction is primarily formulated [Citation52]. However, it is still not certain whether this is due to their functions in mitophagy or others, as basal mammalian mitophagy could occur independently of PINK1 or PRKN in a variety of tissues including brain [Citation53,Citation54]. Moreover, PINK1 and PRKN have been reported to engage in a wide range of mitochondrial behaviors. PINK1 could regulate mitochondrial mobility by phosphorylating and promoting the proteasomal degradation of RHOT/MIRO (ras homolog family member T) in a PRKN-dependent manner [Citation55,Citation56]. PRKN sustains PPARGC1A (PPARG coactivator 1 alpha) expression and supports mitochondrial biogenesis by ubiquitinating and promoting the proteasomal degradation of ZNF746/PARIS (zinc finger protein 746), a transcriptional repressor of PPARGC1A [Citation57,Citation58]. Knockdown of PINK1 reduces the enzymatic activities of several respiratory chain complexes [Citation59,Citation60]. Additionally, both Pink1 and Prkn genetically interact with multiple mitochondrial dynamics associated genes, including Dnm1l, Mfn and Opa1 [Citation61–63]. Recently, Han et al. reported that PINK1 could phosphorylate DNM1L at S616 to regulate mitochondrial fission [Citation64]. Other studies elaborated that PINK1 and PRKN could modulate mitochondrial PRKA/PKA (protein kinase AMP-activated) signaling [Citation65,Citation66], mitochondrial calcium homeostasis [Citation67,Citation68] and mitochondrial chaperone [Citation69]. Therefore, further studies are needed to dissect the exact physio-pathological functions of the PINK1-PRKN-mediated mitophagy in Parkinson disease.
Receptor-mediated mitophagy
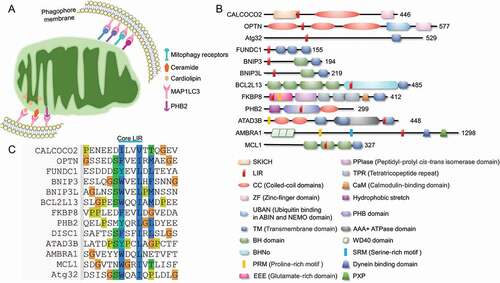
The specific sequestration of the certain cargo for selective autophagy is mainly achieved by autophagy receptors. The autophagy receptors always target the to-be-degraded cargo (damaged organelles, protein aggregates or invading pathogens) and tether them to the phagophore membrane by interacting with MAP1LC3 members (including MAP1LC3A, MAP1LC3B, MAP1LC3B2 and MAP1LC3C) or the homologs (GABARAP, GABARAPL1 and GABARAPL2) via an LC3-interacting region (LIR) motif [Citation70]. The canonical LIR is composed of a (W/F/Y)XX(L/I/V) core motif that interacts with two hydrophobic pockets of the LIR docking site in MAP1LC3 and the homologs anchored in the phagophore membrane () [Citation71]. In yeast, Atg32 was identified as a selective mitophagy receptor by two independent groups [Citation72,Citation73]. In mammalian cells, more than 10 mitophagy receptors have been identified for the specific sequestration of mitochondria for mitophagy program. Emerging evidence shows that these different receptors sense distinct stress signals to coordinate the mitochondrial quality control.
Figure 2. Major receptors mediated mitophagy. (A) Mitophagy receptors are mainly localized on mitochondrial outer membrane. The mitochondrial inner membrane resident cardiolipin could transfer to the outer membrane and bind to MAP1LC3 directly, while the rupture of outer membrane leads to the exposure of PHB2, leading to the PHB2-MAP1LC3 interaction to promote the PINK1-PRKN-mediated mitophagy. (B) Domain architectures of major protein mitophagy receptors. (C) Protein mitophagy receptors harbor LIR regions and directly bind to MAP1LC3 and homologs to initiate mitophagy.
FUNDC1-mediated mitophagy
FUNDC1 (FUN14 domain containing 1) is a mitochondrial outer membrane protein, with its N terminus exposed to the cytosol and C terminus inserted into the mitochondrial outer membrane. In the N terminus, FUNDC1 possesses a canonical LIR motif (Y18XXL21) (). Dependent on this LIR, FUNDC1 could directly interact with MAP1LC3 and homologs, and thus drive mitophagy. Mutants in the LIR disrupt the FUNDC1-MAP1LC3 interaction and significantly prevent hypoxia-induced mitophagy [Citation74]. The mitophagy-driving activity of FUNDC1 is determined by several dynamic phosphorylation and dephosphorylation. Specifically, SRC (SRC proto-oncogene, non-receptor tyrosine kinase) kinase and CSNK2 (casein kinase 2) phosphorylate FUNDC1 at Tyr18 (exactly in the LIR motif) and Ser13, respectively, which restrain the FUNDC1-MAP1LC3 interaction. Dephosphorylation of FUNDC1 at Ser13 by the mitochondrial phosphatase PGAM5 strengthens the FUNDC1-MAP1LC3 interaction and facilitates the FUNDC1-dependent mitophagy [Citation74–76]. BCL2L1 (BCL2 like 1), an anti-apoptotic member in BCL2 family, directly binds to PGAM5 phosphatase, limits the phosphatase activity, and thus maintains the FUNDC1 phosphorylation at Ser13 to antagonize the FUNDC1-dependent mitophagy [Citation77,Citation78]. Besides, ULK1 (unc-51 like autophagy activating kinase 1), an upstream kinase for autophagy initiation, phosphorylates FUNDC1 at Ser17, which expedites the FUNDC1-MAP1LC3 interaction and is required for the FUNDC1-dependent mitophagy [Citation79]. It is still elusive but of great interests that why FUNDC1 needs these distinct phosphorylated regulations, and why phosphorylation at different (but adjacent) sites by the corresponding kinases or phosphatases leads to completely opposite MAP1LC3-binding capacity. Furthermore, it is also unclear which phosphatases are involved in the dephosphorylation of FUNDC1 at Tyr18 and Ser17. Additionally, the mitochondrially resident E3 ligase MARCHF5 (membrane associated ring-CH-type finger 5) could directly interact with FUNDC1 and catalyze its ubiquitination at Lys119 for the subsequent proteasomal degradation, thus desensitize hypoxia-induced mitophagy () [Citation80]. FUNDC1 is evolutionally conserved. Recently, it has been reported that the FUNDC1 ortholog, FNDC-1 and Fundc1, similarly serve as mitophagy receptor and mediate mitophagy in C. elegans and D. rerio, respectively [Citation81–83].
Figure 3. Regulatory circuit of FUNDC1-mediated mitophagy. FUNDC1 is phosphorylated by CSNK2, ULK1 and SRC kinases at Ser13, Ser17 and Tyr18, respectively. The Ser13 is dephosphorylated by mitochondrial phosphatase PGAM5, which is finely tuned by BCL2L1. The phosphorylation at Ser17 while dephosphorylation at Ser13 and Tyr18 under mitochondrial stresses (especially hypoxia) enhances the FUNDC1-MAP1LC3 interaction and stimulates mitophagy induction. The mitochondrial E3 ligase MARCHF5 could ubiquitinate FUNDC1 at Lys118 for its proteasomal degradation and thus desensitize hypoxia induced mitophagy. FUNDC1 is also tuned by transcriptional factor NRF1-PPARGC1A and MIR137 at transcriptional and post-transcriptional level, respectively. FUNDC1 is enriched in MAM structure by interacting with ER resident CANX and ITPR2, where it promotes the release of ER Ca2+ to mitochondria and cytosol. Under mitochondrial stresses, FUNDC1 disassociates with OPA1 and CANX, while associates with DNM1L at MAMs to facilitate mitochondrial fission and mitophagy.
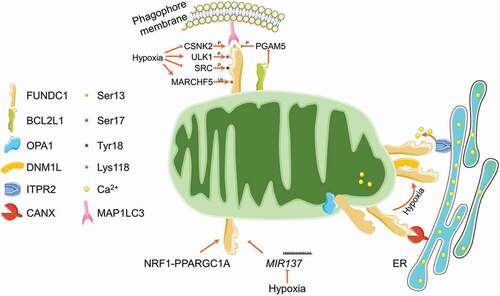
FUNDC1-mediated mitophagy is also finely tuned at transcriptional and post-transcriptional levels (). Specifically, our recent study reported that NRF1 (nuclear respiratory factor 1) and PPARGC1A, the key transcriptional factor and co-factor involved in mitochondrial biogenesis, transcriptionally up-regulate the FUNDC1 expression and expedite mitophagy [Citation84], which highlights the coordination of mitochondrial integrity by coupling mitophagy and mitochondrial biogenesis via PPARGC1A-NRF1-FUNDC1 axis [Citation85]. MIR137 (microRNA 137), a hypoxia down-regulated microRNA, could target 3′ UTR of FUNDC1 mRNA and significantly reduce the FUNDC1 protein level, leading to impaired mitophagy without affecting general autophagy [Citation86].
Chen and colleagues reported that FUNDC1 could interact with the mitochondrial dynamics proteins OPA1 and DNM1L, thus coordinate both mitochondrial fusion and fission. Mitochondrial stress drives the disassembly of the FUNDC1-OPA1 complex while strengthens the DNM1L recruitment onto mitochondria by enhancing the FUNDC1-DNM1L interaction. The dephosphorylation at Ser13 is important for this switch from FUNDC1-OPA1 interaction to FUNDC1-DNM1L interaction, thus triggers the mitochondrial network disruption and mitophagy initiation [Citation87]. Mitochondria always connect with endoplasmic reticulum (ER) by the so-called mitochondria-associated membrane (MAM) structure. MAM is important for rapid exchange of biological molecules between mitochondria and ER to fulfill the diverse signaling transduction [Citation88]. Moreover, MAM provides a unique location for DNM1L recruitment and mitochondrial fission [Citation89,Citation90]. It has been reported that MAM involves in the autophagosome and specific mitophagosome formation [Citation91,Citation92]. Two independent studies reported that FUNDC1 is enriched in MAM structure, and binds to the MAM components CANX (calnexin) and ITPR2 (inositol 1,4,5-trisphosphate receptor type 2) [Citation93,Citation94], which is functionally important for mitochondrial fragmentation and mitophagy initiation ().
By using fundc1 knockout mice, increasing evidence is emerging that the FUNDC1-dependent mitophagy is essential for mitochondrial quality control, while defect in this pathway leads to diverse disease pathogenesis. On one hand, FUNDC1-mediated mitophagy in brown fat tissue is essential for adaptive thermogenesis by coordinating mitophagy and mitochondrial biogenesis [Citation84]. FUNDC1-mediated mitophagy in white adipose tissue is important for restraining high-fat diet induced obesity and insulin resistance. Deficiency in this mitophagy by specific deletion of Fundc1 in adipose tissue exacerbates the accumulation of dysfunctional mitochondria in adipocytes, leading to aggravated oxidative stress, apparent macrophages infiltration and abominable metabolic disorder [Citation95]. Muscle-specific deletion of Fundc1 also impairs the mitochondrial function, however antagonizes the high-fat diet-induced obesity and insulin resistance [Citation96]. It is still unknown why FUNDC1 in adipose tissue and skeletal muscle possesses completely opposite impact on metabolic homeostasis in mice, although in both tissues FUNDC1-mediated mitophagy is important for the removal of damaged mitochondria. On the other hand, it has been revealed that the FUNDC1-mediated mitophagy is indispensable for tissue protection from injury and toxic insults (especially the ischemia-reperfusion injury) in brain [Citation97], heart [Citation93,Citation98–100], liver [Citation101], kidney [Citation102], and intestine [Citation103]. Furthermore, FUNDC1 is regarded as a tumor suppressor and defect in this mitophagy accelerates the tumorigenesis of hepatocellular carcinoma by hyperactivating inflammasome and excessively secreting pro-inflammatory cytokines [Citation104].
BNIP3- and BNIP3L-mediated mitophagy
Both BNIP3 (BCL2 interacting protein 3) and BNIP3L/NIX (BCL2 interacting protein 3 like) were originally identified as BH3-only pro-apoptotic proteins integrated in the mitochondrial outer membrane via C-terminal transmembrane domain, although their BH3 domains are poorly conserved [Citation105]. BNIP3 harbors a canonical LIR motif essential for the interaction with MAP1LC3 and mitophagy induction () [Citation106–108]. The phosphorylation of BNIP3 at Ser17 and Ser24 flanking the LIR motif enhances the BNIP3-MAP1LC3 interaction and facilitates mitophagy [Citation109]. However, the detailed regulatory mechanism is still vague.
BNIP3L is essential for the maturation of red blood cells. Bnip3l ablation results in the retardation of erythroid cells differentiation, leading to anemia and splenomegaly in mice [Citation110]. The following studies revealed that mitochondria are retained during the maturation of reticulocytes in bnip3l null mice [Citation111,Citation112]. Structurally, BNIP3L contains a LIR motif in the N terminus toward cytoplasm. Through this LIR, BNIP3L could directly interact with GABARAP and MAP1LC3 to initiate mitophagy () [Citation113,Citation114]. Similarly, BNIP3L-mediated mitophagy can be regulated by dynamic phosphorylated modifications. PRKA kinase phosphorylates BNIP3L at Ser212 leading to the translocation of BNIP3L from mitochondria and sarcoplasmic reticulum to cytosol, subsequently impairing mitophagy program [Citation115]. The phosphorylation of Ser34 and Ser35 adjacent to the LIR motif strongly enhances the interaction between BNIP3L and MAP1LC3 homologs, thus facilitates the phagophore sequestration of mitochondria [Citation116]. Ser81 was reported as another phosphorylable residue. Phosphorylation at this site reinforces the binding affinity to MAP1LC3 family members [Citation117]. Additionally, BNIP3L dimerization robustly strengthens the MAP1LC3-BNIP3L interaction and subsequently boosts the mitophagy induction [Citation118].
Both BNIP3 and BNIP3L are transcriptionally dominated by the transcriptional factor HIF1A (hypoxia inducible factor 1 subunit alpha) [Citation119–121]. The hypoxia-responsive element in the promoter region encoding by Bnip3 confers it hypoxia-responsive [Citation122]. Alternatively, FOXO3 (forkhead box O3) was reported to transcriptionally regulate the expression of BNIP3 and BNIP3L [Citation123]. Circadian gene ARNTL (aryl hydrocarbon receptor nuclear translocator like) also regulates the transcription of BNIP3 via binding to the E-Box element within its promoter region, which is important for mitophagy induction and sustains the cardiac function [Citation124]. Additionally, BNIP3 is also transcriptionally regulated by TP53 (tumor protein p53), NFKB1 (nuclear factor kappa B subunit 1), and other tumor-relevant transcriptional factors in the corresponding circumstances [Citation125]. Furthermore, the expression of BNIP3 could be epigenetically suppressed by hypermethylation of the promoter region [Citation126,Citation127]. These complicated regulations of BNIP3 and BNIP3L suggest a regulatory necessity of the BNIP3- and BNIP3L-mediated mitophagy in response to distinct physiological adaptions and stressed conditions.
Similar to FUNDC1, BNIP3- and BNIP3L-mediated mitophagy is important for maintaining the mitochondrial integrity and tissue-protective. Specifically, the HIF1A-BNIP3 and HIF1A-BNIP3L axes are critical to protect brain [Citation117,Citation128] and kidney [Citation129,Citation130] against ischemia-reperfusion injury. On the contrary, Jin and colleagues reported that the BNIP3-mediated mitophagy overtly depletes the mitochondria resulting in metabolic disorder and aggregating cardiac ischemia-reperfusion injury [Citation131]. BNIP3 is proved as a tumor suppressor [Citation125], and loss of BNIP3-mediated mitophagy promotes mammary tumor growth and increases lung metastasis by remodeling the cellular metabolism toward the aerobic glycolysis [Citation132]. BNIP3 expression also restrains the pancreatic cancer [Citation127] and colorectal cancer [Citation126,Citation133]. However, whether the mitophagy-driving activity of BNIP3 is involved in these tumor suppressions was not studied. On the contrary, BNIP3L-mediated mitophagy may function as a tumor promoter in pancreatic ductal adenocarcinoma through limiting the mitochondrial glucose flux and supporting the redox regulation. Bnip3l deletion markedly delays the progression of pancreatic cancer and improves survival in a murine model of pancreatic ductal adenocarcinoma [Citation134]. Hence, it is of great interest to further dissect the exact roles of BNIP3- and BNIP3L-mediated mitophagy in tissue injury and tumorigenesis.
It should not be ignored that both BNIP3 and BNIP3L are originally identified as pro-apoptotic proteins. They interact with BCL2 and BCL2L1 via the transmembrane domain, release BH3-only proteins to interact with BAX or BAK1 and induce CYCS release, CASPs activation, and cell death. It has been assumed that the BNIP3- and BNIP3L-mediated mitophagy is cell-protective, while loss of this mitophagy would accelerate the cell death [Citation121]. Moreover, BNIP3- and BNIP3L-mediated mitophagy and metabolic reprogramming regulate the cell fate determination during the differentiation of stem cells and de-differentiation of the induced pluripotent stem cells [Citation135]. However, it is still unclear whether any endogenous metabolites could dominate the cellular fates decision toward protective mitophagy, apoptosis, differentiation or de-differentiation by manipulating BNIP3 and BNIP3L.
BCL2L13-mediated mitophagy
BCL2L13/BCL-Rambo (BCL2 like 13) was originally identified as a pro-apoptotic protein anchored on mitochondrial outer membrane with its N terminus exposed to the cytosol and C terminus in the intermembrane space [Citation136]. Previous studies reported that BCL2L13 interacts with mitochondrial permeability transition pore (mPTP) components SLC25A4/ANT (solute carrier family 25 member 4) and VDAC, thus regulates the conformational transformation of mPTP for the release of CYCS [Citation137,Citation138]. In 2015, BCL2L13 was nominated as a mammalian Atg32 homolog as evidenced by its compensation of Atg32 to initiate mitophagy in yeast [Citation139]. The residues 273–276 constitute a functional LIR to selectively interact with MAP1LC3C, GABARAP and GABARAPL1 to promote mitophagy in response to mitochondrial depolarization () [Citation139,Citation140]. Murakawa and colleagues recently reported that BCL2L13 recruits the ULK1 complex to drive mitophagy [Citation141]. It has been well documented that BCL2L13 functions in multiple physiological processes including development and energy metabolism, while its dysregulation is associated with several pathological processes including tumorigenesis, bacterial infection, cardiovascular diseases and degenerative diseases [Citation142]. However, whether BCL2L13-mediated mitophagy is engaged in these processes is still unclear.
FKBP8-mediated mitophagy
FKBP8 (FKBP prolyl isomerase 8), a unique member of the FK506‐binding protein family, is predominantly localized in mitochondrial outer membrane. Early studies indicated that FKBP8 suppresses apoptosis by recruiting the anti-apoptotic proteins BCL2 and BCL2L1 to mitochondria [Citation143]. In addition, FKBP8 serves as a multifunctional chaperone important in hypoxia signaling and MTOR (mechanistic target of rapamycin kinase) pathway [Citation144]. FKBP8 was reported as a MAP1LC3‐interacting protein. Dependent on its LIR motif in the N terminus, FKBP8 can directly bind to MAP1LC3A and profoundly induce PRKN‐independent mitophagy () [Citation145]. A following study revealed that FKBP8 is critical for the mitochondrial fragmentation and mitophagy induction in response to iron depletion and hypoxia [Citation146]. Interestingly, FKBP8 itself escapes from the to-be-degraded mitochondria to ER during both the PRKN-mediated mitophagy and the FKBP8 ectopic-expression-induced mitophagy. This translocation is highly dependent on the basic amino acids in its C-terminal signal sequences [Citation145,Citation147]. Nevertheless, the detailed regulatory mechanisms and the physiological relevance of the FKBP8-mediated mitophagy are far from understood.
PHB2-mediated mitophagy
PHB2 (prohibitin 2) is a multifunctional protein possessing multiple localizations. Notably, PHB2 in mitochondrial inner membrane functions as a chaperone to stabilize the mitochondrial respiratory complexes and support the mitochondrial bioenergetics. In the nucleus, PHB2 was reported to modulate several transcriptional factors involved in the cell cycle regulation. PHB2 also localizes on the plasma membrane to modulate the cell signaling [Citation148]. In 2017, Wei and colleagues reported that PHB2 harbors a functional LIR motif and could interact with MAP1LC3, which is functionally important for the PRKN-mediated mitophagy in mammalian cells and C. elegans when the mitochondrial outer membrane is ruptured (). Thus, PHB2 is the first protein mitophagy receptor identified in the mitochondrial inner membrane. This enlightening study revealed a novel mitophagy paradigm, in which mitochondrial inner membrane performs a key role in cargo recognition for mitophagy [Citation149]. However, a recent study challenged this hypothesis and put forward an alternative proposal. Yan et. al found that the LIR motif is dispensable for PHB2-mediated mitophagy. PHB2 destabilizes and negatively regulates the enzymatic activity of PARL, the mitochondrial protease resident in the inner membrane essential for PINK1 hydrolysis. In this regard, PHB2 enhances PINK1 recruiting onto mitochondrial outer membrane and facilitates the PINK1-PRKN-mediated mitophagy [Citation150]. Therefore, further investigations are needed to explore the detailed regulatory circuit and physiological significance of PHB2-mediated mitophagy.
Mitochondrial lipid as mitophagy receptors
Besides to these mitochondrial proteins functioning as mitophagy receptors, mitochondrial lipid molecules, especially cardiolipin and ceramide, similarly serve as mitophagy receptors to mediate the specific sequestration of damaged mitochondria by autophagosome (). Cardiolipin is a unique phospholipid exclusively located in the mitochondrial inner membrane, where it supports proper cristae folding, respiratory chain and ATP synthesis [Citation151]. Upon pro-mitophagy stresses, cardiolipin externalizes to the mitochondrial surface, where it binds to MAP1LC3 for the subsequent phagophore recognition of mitochondria. Mechanistically, Arg10 and Arg11 residues in MAP1LC3 constitute the cardiolipin-binding pocket responsible for the MAP1LC3-cardiolipin interaction [Citation152,Citation153]. NME4 (NME/NM23 nucleoside diphosphate kinase 4), a hexameric protein resident in the mitochondrial intermembrane space, enables the cardiolipin externalization and is important for mitophagy initiation [Citation154]. A recent study reported that pro-IL1A (interleukin 1 alpha) is translocating onto mitochondria, where it directly interacts with cardiolipin through a similar motif found in MAP1LC3 for interaction with cardiolipin. This pro-IL1A-cardiolipin interaction competitively disrupts the MAP1LC3-cardiolipin interaction, leading to mitophagy defect, damaged mitochondria accumulation, and NLRP3 inflammasome hyper-activation. This study confirms the mitophagy-driving activity of cardiolipin [Citation155]. In addition, ceramide, a group of bioactive sphingolipids present in the bio-membrane, was also reported as a novel mitophagy receptor. Supplementation of a ceramide analog, or enhancing the generation of endogenous C18-ceramide by ectopic expression of CERS1 (ceramide synthase 1), triggers lethal mitophagy in cancer cells. The C18-ceramide can localize on mitochondria, where it binds to MAP1LC3 and mediates the phagophore engulfing mitochondria for mitophagy [Citation156,Citation157]. These findings collectively define novel mitophagy pathways mediated by the phospholipid components of mitochondrial membrane.
Other mitophagy receptors
Various studies characterized some other mitochondrial proteins as selective receptors for mitophagy in response to diverse stresses, including DISC1 (DISC1 scaffold protein) [Citation158], MCL1 [Citation159], AMBRA1 (autophagy and beclin 1 regulator 1) [Citation160] and ATAD3B (ATPase family AAA domain containing 3B) [Citation161]. The canonical xenophagy receptors CALCOCO2 and OPTN are also engaged in the PINK1-PRKN-mediated mitophagy as discussed above ().
It has been widely documented that different mitophagy pathways sense different mitochondrial stresses to initiate mitophagy for mitochondrial quality and quantity control in distinct circumstances [Citation15]. Recent studies have revealed the cross-talk between different mitophagy pathways for the coordination of mitophagy. The complicated but precise regulations ensure a proper mitophagy activity to maintain the cellular homeostasis.
Endogenous metabolites determine mitophagy
As the fundamental compartments for cellular metabolisms, mitochondria are highly sensitive to the intracellular metabolic programs and extracellular nutrients availabilities. Mitochondrial behaviors including morphological dynamics, mitochondrial metabolism and mitochondrial retrograde signaling are finely tuned by the physiological and pathological programs. Additionally, mitophagy, the major mechanism for mitochondrial quality and quantity control, is tightly manipulated by certain endogenous metabolites. It has been well characterized that the mitochondrial inevitable by-product ROS could facilitate mitophagy via distinct mechanisms, which has been comprehensively summarized in a recent review article [Citation162]. Specifically, Dagda and colleagues reported that the loss of PINK1 increases the mitochondrial superoxide production and elicits oxidative stress, which are critically important for the coordination of mitophagy and mitochondrial dynamics [Citation163]. Other studies suggested that the generation of mitochondrial ROS also initiates the PINK1-PRKN-dependent mitophagy, and this mitophagy could be abolished by expression of the mitochondrial antioxidant proteins [Citation164]. The decline in ATP production due to certain mitochondrial stresses could drive mitophagy through activation of AMP-activated protein kinase (AMPK) signaling pathway [Citation165]. Furthermore, other cellular metabolites associated with iron-, calcium-, glycolysis-TCA-, nicotinamide adenine dinucleotide (NAD)+-, amino acids-, fatty acids-, and cAMP-metabolism, also modulate mitophagy in corresponding circumstances.
Iron metabolites and mitophagy
Mitochondria provide the center compartment for cellular iron metabolism. The cytosolic iron is imported into mitochondrial matrix via the iron transporters SLC25A37/MFRN1 in erythroid cells and SLC25A28/MFRN2 in non-erythroid cells [Citation166]. Mitochondrial iron could be safely sequestered by FTMT (ferritin mitochondrial), or directly utilized for synthesis of heme and iron-sulfur cluster (ISC), two major iron-containing molecules required for a number of enzymes [Citation167]. Iron deprivation has been well documented to trigger mitophagy in different models. By utilizing a chemical screen, iron chelator deferiprone (DFP) was nominated as a potent mitophagy inducer in U2OS osteosarcoma and SH-SY5Y neuroblastoma cells. The exposure of DFP declines the mitochondrial respiration and drives mitophagy induction independent on PINK1 and PRKN, leading to the metabolic reprogramming toward glycolysis [Citation168]. DFP also elevates the expression of FTMT via HIF1A-SP1 (Sp1 transcription factor) axis and results in the translocation of FTMT precursor on the mitochondrial outer membrane, where FTMT precursor specifically interacts with NCOA4 (nuclear receptor coactivator 4, an autophagic receptor for ferritinophagy selectively degrading ferritin for iron turnover [Citation169]) and initiates mitophagy [Citation170]. Additionally, iron depletion was also reported to trigger mitophagy in pathogenic yeast Candida glabrata dependent on Atg32 homolog [Citation171], and in C. elegans dependent on pdr-1/PRKN and dct-1/BNIP3 [Citation172]. Specifically, pyoverdine, an iron-cheatable siderophore derived from bacterium Pseudomonas aeruginosa, disrupts the mitochondrial iron homeostasis and triggers mitophagy in the host C. elegans [Citation173]. Taken together, these studies enlighten a crosstalk between mitophagy and iron homeostasis.
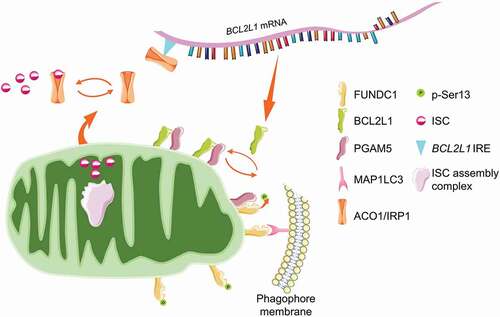
Recently, our group revealed a fundamental role of mitochondrial ISC assembly in determining mitophagy (). RNAi screen identified that the silence of mitochondrial genes involved in ISC assembly triggers mitophagy in a FUNDC1-dependent manner. Mechanistically, ISC supports the aconitase activity of ACO1 (aconitase 1), while defective ISC bio-synthesis switches ACO1 to ISC-free IRP1 (iron regulatory protein 1) apoprotein, which is then binding to the newly identified iron regulatory element (IRE) in the 5ʹUTR of BCL2L1 mRNA. IRP1 apoprotein binding leads to the translational repression of BCL2L1, triggering FUNDC1 dephosphorylation and mitophagy initiation by releasing the PGAM5 phosphatase [Citation174]. This study enlightens a novel regulatory circuit comprising ACO1/IRP1-BCL2L1-PGAM5-FUNDC1 axis in determining mitophagy in response to iron stress and sheds new light on the mitophagy-restraining activity of ISC by supporting BCL2L1 translation. Similarly, a previous study revealed that the silence of FXN (frataxin), one of the most significant mitochondrial proteins involved in ISC bio-synthesis, also drives mitophagy program both in mammalian cells and C. elegans [Citation172].
Figure 4. ISC antagonizes FUNDC1-mediated mitophagy. ISC is mainly synthesized in mitochondria by the ISC assembly complex. The newly assembled ISC is transported into the cytosol to support the aconitase activity of ACO1/IRP1 for the TCA cycle. During iron deficiency or genetic susceptibility, defective ISC assembly enables ACO1/IRP1 unbound. The free ACO1/IRP1 targets to the IRE region of BCL2L1 mRNA, leading to the translational suppression and protein downregulation. The mitochondrial phosphatase PGAM5 is liberated to dephosphorylate FUNDC1 and facilitates mitophagy.
Hemin facilitates mitophagy in a BNIP3L-dependent manner in human chronic myeloid leukemia cell line K562 [Citation175]. Knockdown of ALAS2 (5ʹ-aminolevulinate synthase 2), the rate-limiting enzyme catalyzing heme synthesis, downregulates the expression of BNIP3L [Citation176]. HMOX1 (heme oxygenase 1) is the stress-responsive enzyme catalyzing the degradation of heme and yielding biliverdin, carbon monoxide (CO) and ferrous iron. It was reported that HMOX1 is mitochondria-protective by maintaining the expression of PINK1 and PRKN [Citation177,Citation178]. CO is an endogenous gaseous molecule conferring protection against cellular stress at distinct aspects [Citation179]. CO inhalation increases the expression of PINK1 and PRKN, leading to both mitophagy and mitochondrial biogenesis to maintain the mitochondrial homeostasis in cultured hepatocytes in vitro and in liver tissue in vivo [Citation180]. However, the physiological role of endogenous CO in mitophagy is still unclear.
Collectively, the emerging evidence has revealed the tight communication between iron metabolism and mitophagy. As dozens of mitochondrial proteins possess heme or ISC (or both) as the essential co-factors, it is not unexpected that iron deprivation and defective ISC assembly could disrupt the mitochondrial architecture, compromise mitochondrial functionalities and drive mitophagy. Furthermore, disrupted iron homeostasis (both iron deposition and iron deficiency) and abnormal iron metabolism have been widely reported to strongly associate with diverse disease pathogenesis, during which mitochondrial dysfunction is primarily formulated [Citation181]. Therefore, it is of great clinical importance to explore the exact relevance of mitophagy and iron metabolism in the pathogenesis of these human diseases.
Ca2+ and mitophagy
Calcium ion (Ca2+) is one of the most versatile signal molecules. Mitochondria serve as important regulators of cellular Ca2+. The appropriate mitochondrial Ca2+ facilitates mitochondrial bioenergetics by sustaining the TCA activity through supporting several fundamental enzymes, including pyruvate dehydrogenase, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase. However, the uncontrolled increase in cytoplasmic Ca2+ and mitochondrial Ca2+ could drive cell death. Specifically, prolonged mitochondrial Ca2+ overload triggers the opening of mPTP, leading to the release of pro-apoptotic molecules and activation of downstream CASPs [Citation182]. Mitochondrial outer membrane is permeable to solutes and ions, while Ca2+ across the inner membrane is highly regulated. Through the VDACs in the outer membrane, MCU (mitochondrial Ca2+ uniporter) mediates the Ca2+ transfer through the inner membrane. The close contacts between mitochondria and ER render the rapid Ca2+ uptake by mitochondria from ER through ITPRs-VDACs-MCU. Additionally, mitochondria could also export excessive Ca2+ and thus ensure the mitochondrial Ca2+ homeostasis. SLC8B1/NCLX (solute carrier family 8 member B1) is the mitochondrial Na+-Ca2+ exchanger, mediates mitochondrial Ca2+ efflux by catalyzing Na+- or Li+-dependent Ca2+ export [Citation183].
As a vital signal molecule, Ca2+ could regulate the autophagic flux, although the mechanisms seem complicated and the effects are context-dependent [Citation184]. Along with the downstream kinases, including PRKC/PKC (protein kinase C), CAMKK2 (calcium/calmodulin dependent protein kinase kinase 2), CAMK (calcium/calmodulin-dependent kinase), MAPK1/ERK2 (mitogen-activated protein kinase 1)-MAPK3/ERK1 and PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), Ca2+ could stimulate the autophagy induction [Citation184]. However, several studies reported that Ca2+ could antagonize the autophagic flux. Specifically, Ca2+ is indispensable for CAPN1 (calpain 1)-mediated cleavage of ATG5, and this ATG5 cleavage inhibits ATG12-ATG5 conjugation and prevents autophagy induction [Citation185]. By sustaining the mitochondrial bioenergetics, it is not unexpected that mitochondrial Ca2+ could inactivate AMPK pathway and hence suppress the autophagy by supporting ATP production [Citation186,Citation187].
Ca2+ could also modulate the selective mitophagy. The sequestering capacity of Ca2+ by mitochondria is highly dependent on the membrane potential. During mitochondrial depolarization, Ca2+ released from mitochondria is captured by RHOT2, a key component of the adaptor complex anchoring mitochondria to motor proteins. The phosphorylated (by PINK1) and Ca2+-binding RHOT2 realigns from tetramers to monomers on the mitochondrial outer membrane, and functions as a platform for the subsequent PRKN translocation [Citation188]. Besides, there is a similar mechanism for RHOT1. Ca2+-binding RHOT1 promotes PRKN translocation, while RHOT1 mutations in the EF-hands essential for Ca2+ binding decrease glutamate-induced calcium-provoked mitophagy in neurons [Citation189]. It has been widely recognized that the Ca2+-binding of RHOT1 and RHOT2 arrests mitochondrial mobility [Citation190]. Therefore, Ca2+ sensors RHOT1 and RHOT2 act as key modulators for the coordination of mitochondrial mobility and the PINK1-PRKN-dependent mitophagy. Due to a significant implication of mitochondrial Ca2+ in determining mitochondrial integrity and programmed cell death, it is of great value to further elucidate the relationship between mitochondrial Ca2+ and mitophagy, especially in related human disease pathogenesis.
Glycolysis-TCA cycle associated metabolites and mitophagy
Mitochondria are located at the center for both catabolism and anabolism of bio-molecules. The cytosolic glycolysis breaks down glucose into pyruvate to fuel the TCA cycle. The following TCA cycle enzymatically catalyzes nutrients such as lipids, carbohydrates and amino acids to generate smaller units and metabolites, supporting the oxidative phosphorylation for ATP production. Normal mitochondrial functionalities are important for these cellular metabolisms. Reversely, the related metabolites are potent modulators of mitophagy ().
Table 1. Glycolysis-TCA cycle associated metabolites and mitophagy
Glucose provides the major carbon source for cellular bioenergetics. However, high plasma glucose (or hyperglycemia) is one of the most causative factors of diabetes, and is strongly correlated with the diabetes-related complications by damaging a group of kidney cells, neurons and endothelial cells [Citation191]. It was firstly reported by H. Crabtree in 1929 that high level of glucose could suppress the cellular respiratory flux, known as Crabtree effect [Citation192]. A number of hypotheses on the mechanisms underlying the Crabtree effect have been formulated. Recently, it has been reported that high level of glucose could induce mitophagy in diabetic platelets [Citation193] and neuronal cells [Citation194]. Mechanistically, hyper-production of ROS due to the mitochondrial fission may trigger this mitophagy [Citation195]. Furthermore, high level of glucose upregulates the HIF1A activity [Citation196,Citation197], and thus induces the expression of PINK1 [Citation198] or BNIP3 [Citation199] for mitophagy induction, leading to the decrease in oxidative phosphorylation and increase in glycolysis. Therefore, the mitophagy-driving capacity of glucose may partially contribute to the Crabtree effect, although some other studies suggested that high level of glucose could inhibit mitophagy in other types of cells. Furthermore, several glucose metabolites as discussed below could also modulate mitophagy in corresponding circumstances.
Lactate, previously regarded as the waste product of glycolysis, is gradually proved to serve as a major circulating carbohydrate fuel and a circulating redox buffer that equilibrates the NADH:NAD+ ratio across cells and tissues in mammals [Citation200]. Additionally, lactate could facilitate mitochondrial biogenesis via upregulation of PPARGC1A [Citation201]. Lactate could block the autophagic flux and suppress the BNIP3-related mitophagy by blunting the expression of BNIP3 through the NR4A1 (nuclear receptor subfamily 4 group A member 1)-PRKDC (protein kinase, DNA-activated, catalytic subunit)-TP53 axis in vascular smooth muscle cells. This mitophagy inhibition aggregates the calcification of vascular smooth muscle cells in the presence of lactate [Citation202,Citation203].
Pyruvate, the C3 product of glycolysis, fuels the TCA cycle through decarboxylation and generation of acetyl-coenzyme A (acetyl-CoA) inside mitochondria. Pyruvate can be reduced to lactate and NAD+ in the cytosol, thereby supporting the NAD+ pool [Citation204]. Early studies elucidated that both pyruvate administration and enhanced MPC (mitochondrial pyruvate carrier)-mediated mitochondrial pyruvate utilization facilitate mitochondrial biogenesis [Citation205,Citation206]. Recently, two independent groups reported that pyruvate could accelerate mitophagy by stabilizing PINK1. Ectopic expression of PDK4, a pyruvate dehydrogenase kinase phosphorylating and negatively determining the enzymatic activity of pyruvate dehydrogenase complex, leads to pyruvate accumulation and expedites mitophagy in response to mitochondrial depolarization. Supplementation of pyruvate alone facilitates PINK1 stabilization, drives PINK1 and MAP1LC3 recruitment onto mitochondrial surface and stimulates mitophagy induction [Citation207]. The underlying mechanism was elaborated by Kim and colleagues. Pyruvate deprivation restrains the generation of NAD+ (an important mitophagy inducer, which will be discussed below), leading to the inhibition of lysosomal acidification and mitophagy defect by increasing the acetylation of v-ATPase ATP6V0A1 [Citation208].
Acetyl-CoA, the central cellular intermediate, is not only the catabolic product of the glycolytic pyruvate, fatty acids and branched-chain amino acids, but also the anabolic precursor for fatty acids, steroids and certain amino acids. Acetyl-CoA is the sole donor of acetyl groups for protein acetylation and thus extensively dominates the gene expression and cellular signaling. Furthermore, acetyl-CoA metabolism is regulated by compartmentalization, and mitochondria locate at the center [Citation209]. It has been well understood that acetyl-CoA is a master autophagy regulator. Nutrient starvation causes a rapid depletion of acetyl-CoA and initiates the autophagy program, while replenishment of acetyl-CoA or dimethyl-α-ketoglutarate to complement sufficient amount of acetyl-CoA, could strongly suppress the starvation-induced autophagy. Mechanistically, acetyl-CoA determines the acetylated status of several core autophagy components by controlling the enzymatic activity of acetyltransferase EP300 [Citation210]. Alternatively, acetyl-CoA also dominates the acetylation of RPTOR (regulatory associated protein of MTOR complex 1) and manipulates MTOR activation [Citation211,Citation212]. Given the significant role of acetyl-CoA in metabolism and signaling transduction, it is not unexpected that acetyl-CoA could manipulate mitophagy. During starvation, the mitochondrial acetyltransferase BLOC1S1/GCN5L1 (biogenesis of lysosomal organelles complex 1 subunit 1) is inactivated due to the rapid exhaustion of acetyl-CoA. The inactivation of BLOC1S1 leads to an extensive reduction in the acetylated modifications of mitochondrial protein, leading to a PRKN-independent mitophagy [Citation213]. However, the exact mechanism by which mitochondrial acetylation manipulates mitophagy is still unknown. It is assumed that the deacetylations of mitochondrial proteins enable or facilitate the ubiquitination in an allosteric manner, which may signal to mitophagy induction [Citation214].
Additionally, the TCA intermediate succinate could modulate mitophagy in certain circumstances [Citation215]. Early studies revealed that succinate stabilizes the transcription factor HIF1A [Citation216], boosts ROS generation [Citation217], and modifies certain substrate proteins through succinylation [Citation218]. It is still elusive whether HIF1A, ROS or succinylation is involved in succinate-induced mitophagy or not.
In conclusion, the glycolysis-TCA cycle associated intermediates are important to fuel oxidative phosphorylation for ATP production and supply certain intermediates for anabolism and redox determination. The anaerobically glycolytic lactate and the aerobically glycolytic pyruvate, as well as its downstream product acetyl-CoA could manipulate mitophagy as discussed above. It is assumed that the mitophagy-mediated metabolic remodeling confers the cellular adaption to the metabolic dynamics for maintaining the cellular homeostasis.
NAD+ and mitophagy
As a crucial co-enzyme important for hundreds of redox reactions and an essential co-factor for non-redox enzymes (including SIRTs [sirtuins] and PARPs [poly(ADP-ribose) polymerases]), NAD+ supports a group of metabolism pathways including glycolysis, TCA cycle, oxidative phosphorylation, and fatty acid oxidation, maintains the redox homeostasis by coordinating the generation and detoxification of ROS, sustains genomic stability, and manipulates gene expression [Citation219,Citation220]. Furthermore, a gradual decline in the cellular NAD+ and prolonged disequilibrium of NAD+ metabolism have been widely formulated in series of human diseases, especially aging associated metabolic disorders, cancers and neurodegenerative diseases, suggesting NAD+ metabolism as a potential therapeutic target for these human diseases [Citation220,Citation221].
Although previous study implied that mitochondria could uptake cytosolic NAD+, it was just recently identified that SLC25A51 functions as the mitochondrial NAD+ transporter and dictates the mitochondrial NAD+ pool. SLC25A51 deficiency leads to increase in the mitochondrial mass but significant decrease in mitochondrial oxygen consumption rate, demonstrating a disturbed mitochondrial turnover during mitochondrial NAD+ depletion [Citation222]. NAD+ repletion has been widely documented to ameliorate mitochondrial functionalities and protect against certain metabolic disorders [Citation223–226]. Although the UPRmt conferring the retrograde signal from mitochondria to nucleus contributes to the beneficial effect of NAD+ [Citation223,Citation226,Citation227], it is not surprised that NAD+ could modulate mitophagy to maintain the mitochondrial homeostasis (). The challenge of nicotinamide (NAM), a metabolic precursor of NAD+, results in disturbed mitochondrial architecture and initiates mitophagy in a SIRT1-dependent manner. This mitophagy could be blocked by inhibition of NAM conversion to NAD+ [Citation228,Citation229]. Xeroderma pigmentosum is a rare autosomal-recessive disorder with severe ultraviolet sensitivity and an increased risk of skin cancers due to the defective DNA repair. Xeroderma pigmentosum patient-derived cells present a clinical mitochondrial pathology, with mitochondrial dysfunction and mitophagy deficit. Mechanistically, the PARP1 hyper-activation due to the DNA damage leads to NAD+ exhaustion, which results in mitochondrial hyper-polarization, excessive cleavage of PINK1, and disorder in the PINK1-PRKN-dependent mitophagy. Administration of NAD+ precursors nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are able to restore the mitophagy and improve the mitochondrial functionalities in patient derived cells [Citation230]. Furthermore, defects in ATM (ATM serine/threonine kinase), the master sensor and repairing regulator of DNA double-strand breaks, leads to a similar decline in NAD+. Damaged mitochondria accumulate in ATM-deficient neurons, mice, and worms. Treatment of NR or NMN complements NAD+ levels and restores mitophagy in ATM-deficient cells through reestablishing SIRT1 activity. dct-1/BNIP3 and pink-1/PINK1 engage in this mitophagy pathway [Citation231]. Additionally, it was also reported that NAD+ replenishment could restore mitophagy, which is crashed in Alzheimer disease and Werner syndrome due to the depletion of NAD+ [Citation232,Citation233]. Overall, these impressive studies collectively elaborated NAD+ as an endogenous mitophagy inducer.
Figure 5. NAD+ boosts mitophagy. Cellular NAD+ could be complemented by food supplementation of the metabolic precursors including NAM, NR and NMN, or synthesized de novo. SIRTs and PARPs are two major NAD+-dependent enzymes catalyzing the NAD+ to yield NAM, which is gradually elevated during aging, DNA damage and other genetic or environmental insults. NAD+ is imported into mitochondria to sustain several mitochondrial metabolisms. It was reported that NAD+ facilitates mitochondrial biogenesis via SIRTs-PPARGC1A axis. Additionally, NAD+ activates AMPK kinase through supporting SIRTs deacetylation of STK11, or through ATM-STK11-AMPK or ATM-AMPK axes. Alternatively, the activated SIRTs could mediate deacetylation of autophagy core components MAP1LC3, ATG5 and ATG7, as well as deacetylation of FOXOs to promote the expression of BNIP3, MAP1LC3 and ATGs, or enhance PINK1 stabilization, which collaboratively strengthen the mitophagy program.
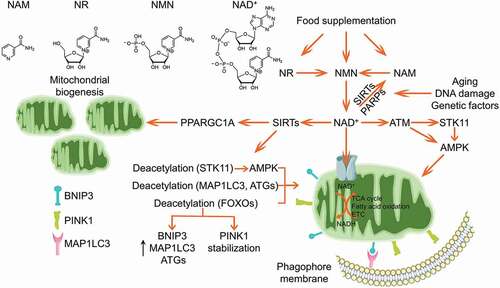
AMPK locates at the center of NAD+-induced mitophagy. As a major metabolic regulator, AMPK is one of the most important guardians for mitochondrial homeostasis, including dynamics, biogenesis and mitophagy [Citation165]. Specifically, AMPK phosphorylates MFF, the primary receptor for DNM1L translocation onto mitochondrial outer membrane, leading to mitochondrial fission [Citation234]. Furthermore, AMPK phosphorylates ULK1 to stimulate mitophagy induction [Citation235]. AMPK activity is suppressed in NAD+-deficient cells, while NAD+ replenishment could restore AMPK activation [Citation232]. Mechanistically, ATM-mediated activation of STK11/LKB1 (serine/threonine kinase 11), the upstream kinase responsible for AMPK phosphorylation, is involved in NAD+-mediated AMPK activation and mitophagy induction. ATM could also directly phosphorylate AMPK [Citation236]. Furthermore, NAD+ could activate AMPK by sustaining SIRT1 deacetylation of STK11 [Citation237]. However, other pathways are likely involved in the NAD+-induced mitophagy, as knockdown of AMPK is unable to completely ablate the NAD+-induced mitophagy [Citation232]. The elevation of NAD+ supports the enzymatic activity of SIRT1, a master mitophagy regulator by finely tuning the acetylated status of MAP1LC3 [Citation238], ATG5 and ATG7 [Citation239], or activating the daf-16/FOXO-dct-1/BNIP3 pathway [Citation231]. NAD+ also shapes mitochondrial morphology by manipulating the dynamics proteins [Citation226]. Moreover, this NAD+-mediated mitophagy is mitochondria- and cell-protective. As a group of aging-related diseases are manifested by NAD+ exhaustion due to the elevated DNA damage, genetic or epigenetic insults and environmental toxins, replenishment of NAD+ by supplementation of the metabolic precursors provides a highly potent therapeutic design for these diseases.
Amino acids associated metabolites regulating mitophagy
Amino acids are the metabolic blocks for protein synthesis. They could also fuel cellular bioenergetics as carbon source, and certain amino acids are dedicating to signaling transduction as signal molecules. Specifically, amino acids are sensed by the corresponding sensors, leading to the activation of MTOR and suppression of autophagic flux [Citation240]. It has been reported some amino acids associated metabolites as mitophagy regulators in corresponding circumstances ().
Table 2. Amino acids associated metabolites regulating mitophagy
Glutamine, the most abundant amino acid, functions as the nitrogen source for the bio-synthesis of nucleotides, amino acids, and hexamine, as well as carbon source to fuel the TCA cycle for bioenergetics through glutaminolysis [Citation241]. During glutaminolysis, glutamine is converted to α-ketoglutaric acid (α-KG) and ammonia, which is catalyzed by mitochondrial GLS (glutaminase). Ammonia was previously identified as a diffusible autophagy-inducing molecule in cancer cells [Citation242]. Ammonia supplementation increases the expression of BNIP3, PINK1, and PRKN, leading to the increased mitophagy in mammalian cells. Inhibition of GLS activity and glutamine withdrawal reduce the expression of BNIP3 [Citation243]. Mechanistically, ammonia exposure facilitates HIF1A stabilization and elevates the transcription of the target genes (including BNIP3) under normoxic conditions [Citation244]. Glutamate, the deamidated product of glutamine during glutaminolysis, was reported to facilitate PRKN translocation onto mitochondria. Excessive glutamate could trigger mitochondrial Ca2+ entry and Ca2+ binding to RHOT1, which leads to the arrest of mitochondrial mobility and PRKN recruitment onto mitochondria for the initiation of mitophagy, as discussed above [Citation189,Citation245].
γ-Aminobutyric acid (GABA), a well-recognized inhibitory neurotransmitter, is a four-carbon non-protein amino acid, which is synthesized primarily from glutamate catalyzed by GAD (glutamate decarboxylase). The disorders in GABA metabolism are manifested in diverse human neurological diseases [Citation246]. It was reported that the increase in endogenous GABA level could inhibit the pexophagy (a selective type of autophagy degrading peroxisomes) and mitophagy, but not other autophagy related pathways in yeast. This deficit in pexophagy and mitophagy could be restored by supplementation of rapamycin, indicating a TOR activation-dependent manner of GABA restraining pexophagy and mitophagy. Furthermore, supplementation of GABA, or genetic ablation of ALDH5A1 (aldehyde dehydrogenase 5 family member A1; which catalyzes GABA to succinate to fuel the TCA cycle, leading to the decrease in endogenous GABA) leads to the accumulation of morphologically abnormal mitochondria and defective mitophagy in mammalian cells. This study elaborates the mitophagy-restraining activity of GABA, which may implicate in the pathogenesis of GABA disorder associated diseases [Citation247].
Nitric oxide (NO) is a lipophilic, highly diffusible, and short-lived metabolite of NOS (nitric oxide synthase) that catalyzes the conversion of arginine into citrulline. It has been well understood that NO acts as a signaling molecule and plays the pleiotropic patho-physiological roles both in normal and diseased circumstances. Specifically, NO dictates S-nitrosylation of the target substrates and engages in the post-translational regulation and signaling transduction [Citation248]. Excess NO is converted to the toxic reactive nitrogen species, which could bind to the electron transport chain components, leading to the disruption of mitochondrial functionalities [Citation249]. NO could initiate the PINK1-PRKN-mediated mitophagy. The exposure of NO donor triggers PRKN translocation onto mitochondria even in PINK1 depleted cells. This NO-induced mitophagy facilitates PINK1-null dopaminergic neuronal cells to recover from mitochondrial damage [Citation250]. It is assumed that S-nitrosylation of PRKN at Cys323 enhances the E3 ligase activity and accelerates mitophagy [Citation251]. However, other studies reported that S-nitrosylation of PINK1 or PRKN leads to significantly reduced PRKN translocation onto mitochondria [Citation252,Citation253]. Besides, NO was proved to disturb autophagosome formation through distinct mechanisms [Citation254]. Therefore, it seems more complicated for NO manipulating mitophagy. Further mechanistic studies are needed.
Spermidine is another arginine derived polyamine metabolite. For mammals, spermidine could be obtained via autonomous synthesis by using arginine as the precursor, synthesis by the gut microbiota and uptake from dietary supplementation. Spermidine was previously identified as an anti-aging molecule in yeast, worms, flies, mice, and human cells through an autophagy-dependent manner [Citation255–257]. Specifically, spermidine performs highly cardiac-protective properties. Administration of spermidine stimulates mitophagy in cardiomyocytes, maintains the mitochondrial functionalities and delays cardiac aging in mice [Citation258,Citation259]. Furthermore, exposure of spermidine also reestablishes mitophagy and leads to the reverse of senescence in muscle stem cell [Citation260]. Mitophagy induction by spermidine has been linked to ATM kinase-dependent activation of PINK1-PRKN pathway [Citation261]. Additionally, spermidine elevates autophagy- and mitophagy-related components including BECN1 (beclin 1), MAP1LC3, PINK1, PRKN, ULK1, and ATG proteins, and promotes AMPK activation and MTOR inhibition [Citation262]. However, these studies mainly utilized exogenous administration of spermidine as models. The exact function and mechanism of endogenous spermidine in mitophagy are less characterized.
N-Acetyl-5-methoxytryptamine (melatonin) is a tryptophan-derived pleiotropic endogenous hormone exclusively synthesized in pineal gland. Melatonin is mainly supporting the optimal coordination of daily and seasonal rhythms. Harboring a prominent mitochondria-protective property, melatonin could improve mitochondrial integrity and protect against a wide variety of cardiovascular and neurodegenerative diseases. Melatonin could detoxify ROS, suppress ROS-mediated EIF2AK4/GCN2 (eukaryotic translation initiation factor 2 alpha kinase 4)-ATF4 (activating transcription factor 4)-BNIP3 axis-dependent mitophagy in placental trophoblasts and relieve cadmium-induced fetal growth restriction [Citation263]. Furthermore, emerging evidence indicated that melatonin antagonizes mitophagy in platelet via PPARG (peroxisome proliferator activated receptor gamma)-FUNDC1 axis, which could inhibit mitochondrial energy production, suppress platelet hyperactivity and alleviate cardiac ischemia-reperfusion injury [Citation264]. However, how the PPARG signaling regulates FUNDC1-dependent mitophagy is elusive. Melatonin also antagonizes mitophagy in cardiac microvasculature via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. Specifically, melatonin activates AMPK, facilitates DNM1L phosphorylation at Ser37 but dephosphorylation at Ser616, blunting the DNM1L-dependent mitochondrial fission. The maintenance of mitochondrial architecture restores VDAC1-HK2 interaction to prevent mPTP opening and the PINK1-PRKN activation, eventually block mitophagy and alleviate ischemia-reperfusion-induced cardiac injury [Citation265]. However, the same group also reported that melatonin activates ALDH2 (aldehyde dehydrogenase 2)-CGAS (cyclic GMP-AMP synthase)-STING1 (stimulator of interferon response cGAMP interactor 1)-TBK1 (TANK binding kinase 1) signaling axis and restores mitophagy in cardiomyocytes in mice model with Alzheimer disease [Citation266]. Melatonin could negate the increased expression of NR4A1, therefore block the activation of PRKDC and TP53, leading to BNIP3 upregulation, mitophagy reconstruction and improving mitochondrial functionalities in hepatocytes during the pathogenesis of nonalcoholic fatty liver disease [Citation267]. Therefore, the effect of melatonin on mitophagy seems complicated and context-dependent.
Besides supporting the biosynthesis of melatonin, tryptophan could also be mobilized to fuel the kynurenine pathway, the sole metabolism for de novo NAD+ synthesis. Disorder in kynurenine pathway is a potential causative factor for numerous aging diseases. The enzymes involved in this pathway are gradually inactivated with aging and mitochondrial dysfunction. The endpoint products of the kynurenine metabolism, kynurenic acid and NAD+, as well as several intermediate metabolites in the metabolic branches, are potential intervention targets of these diseases [Citation268,Citation269]. As a potent mitophagy inducer as discussed above, NAD+ de novo synthesis through the kynurenine pathway is critically important for the maintenance of mitochondrial integrity. Increase in NAD+ production by tryptophan supplementation or enhanced metabolic flux enhances mitochondrial function, leading to the increased lifespan and delayed pathology in C. elegans [Citation270], while loss of function of the kynurenine pathway associated critical enzymes results in congenital organ malformations [Citation271]. Moreover, it is worth noting that the kynurenine pathway also modulates the mitochondrial integrity independent on metabolites. KMO (kynurenine 3-monooxygenase), one of the rate-limiting enzymes in the kynurenine pathway, localizes in the mitochondrial outer membrane. A genome-wide RNAi screen identified KMO as a modulator of mitochondrial morphology and PRKN-dependent mitophagy [Citation272]. The increase in mitochondrial mass but decrease in respiratory capacity in cn/KMO-deficient flies supports the notion that KMO may regulate mitophagy. Drosophila studies showed that cn/KMO genetically interacts with Pink1, park and Drp1/DNM1L in a manner independent on kynurenine metabolism [Citation273]. However, the underlying mechanism is elusive.
Cysteine and methionine are primary sulfur-containing amino acids, which substantially maintain the cellular redox homeostasis by detoxifying free radicals and ROS [Citation274]. Therefore, it is not unexpected that both cysteine and methionine could impinge on the mitophagy activity. Hydrogen sulfide (H2S), derived from cysteine by CTH (cystathionine gamma-lyase) and CBS (cystathionine beta-synthase), is an important gasotransmitter and plays a critical role in regulating cardiovascular functions [Citation275]. Although lower concentration of H2S is beneficial for body fitness, higher level of H2S is toxic, especially to mitochondria by interfering the oxidative phosphorylation or sulfhydrating certain mitochondrial proteins [Citation276]. It was reported that exogenous H2S facilitates PRKN recruitment onto mitochondria and enhances the PINK1-PRKN-mediated mitophagy [Citation277]. H2S-mediated sulfhydration of PRKN and mitophagy associated deubiquitinase USP8 enhances the corresponding enzymatic activity and facilitates the PINK1-PRKN-mediated mitophagy [Citation278,Citation279]. However, other study demonstrated that the elevation of the endogenous H2S by CBS, CTH, and MPST triple gene therapy mitigates mitophagy in endothelial cells [Citation280].
Glutathione (GSH), the γ-L-glutamyl-L-cysteinyl-glycine tripeptide as a key determinant of cellular redox homeostasis, is assembled by GCL (glutamate cysteine ligase) ligating cysteine to glutamate to produce γ-glutamylcysteine, and following by GSS (glutathione synthetase) catalyzing glycine and γ-glutamylcysteine to yield GSH [Citation281]. GSH is converted into GSH disulfide (GSSG) and detoxifies oxidative stress by GSH-dependent peroxidases. Although GSH is not de novo synthesized in mitochondria, a line of evidence has been raised that multiple mitochondria associated GSH carrier proteins could mediate the mitochondrial transport of cytosolic GSH and determine the redox homeostasis inside the mitochondria [Citation282]. In yeast, decrease in endogenous GSH induced by chemical or genetic manipulation initiates selective mitophagy but not general autophagy, and this mitophagy could be suppressed by supplementation of a cell-permeable form of GSH or antioxidant N-acetyl-L-cysteine to normalize the cellular GSH abundance [Citation283]. Mechanistically, suppression of Atg32 expression by GSH leads to the mitophagy deficit [Citation284]. However, it is still elusive whether GSH could modulate mitophagy in mammalian cells.
Besides cysteine, methionine is another sulfur-containing amino acid. The methionine residues in proteins are susceptible to oxidation. Therefore, it was assumed that the methionine constitutes an important antioxidant defense mechanism and substantially contributes to the cellular redox homeostasis [Citation274,Citation285]. The methionine-restricted diet could dramatically extend the healthspan of a variety of model organisms. Mitophagy is required for this lifespan extension [Citation286]. Significant increases in PRKN and phosphorylated ubiquitin are observed during methionine restriction, suggesting that the PINK1-PRKN axis is involved in this mitophagy [Citation287]. Mechanistically, S-adenosylmethionine, the most important methyl donor derived from methionine metabolism, facilitates the methylation of the catalytic subunit of PPP2/PP2A (protein phosphatase 2), leading to the deficiency in autophagy and selective mitophagy [Citation288].
Fatty acids associated metabolites regulating mitophagy
Fatty acids are major energy source and important constituents of membrane lipids. Mitochondria play a dominative role in fatty acid metabolism, especially β-oxidation. Furthermore, the mitochondria derived citrate could shuttle to cytosol, where it is used to synthesize acetyl-CoA for lipogenesis. Mitochondria can also synthesize several phospholipids autonomously. Reversely, several fatty acids and the associated metabolites have been characterized to impinge on mitophagy activity to subsequently regulate the mitochondrial metabolism. Specifically, cardiolipin and ceramide are two well-characterized mitophagy receptors as discussed above. Furthermore, hyperlipemia could suppress mitophagy in a number of tissues via multiple mechanisms, suggesting a pathological role of the mitophagy suppression in obesity associated metabolic disorders [Citation289,Citation290].
Cholesterol, a key lipid molecule, is a metabolic precursor for the bio-synthesis of the steroid hormones, bile acids, and vitamin D. As mentioned above, mitochondria derived citrate could transport to cytosol and convert to acetyl-CoA for cholesterol de novo synthesis through the mevalonate pathway. Lysosomal storage disease Niemann-Pick type A (NPA) is characterized by the lysosomal cholesterol accumulation in diseased tissues including brain, spleen and liver, due to the genetic susceptibility or chemical interference. It has been reported that the fusion of mitochondria-containing autophagosomes with lysosomes is restrained in NPA hepatocytes, leading to the impaired mitophagy and aggregating acetaminophen induced liver injury [Citation291]. The similar phenomenon is also observed in NPA neuron where lysosomal cholesterol accumulates [Citation292]. Lysosomal cholesterol accumulation facilitates the hyper-activation of MTORC1 signaling, leading to the disruption of mitophagy program. Genetic or pharmacologic inhibition of MTORC1 could restore the lysosomal degradation of mitochondria, and ameliorate mitochondrial integrity in NPA neurons [Citation293]. Alternatively, cholesterol overload aggravates the oxidative damage to promote the formation of OPTN aggregates, thereby blocking the OPTN translocation onto mitochondria for the PINK1-PRKN-mediated mitophagy [Citation294]. These studies thus collectively highlight the mitophagy-restraining capacity of cholesterol.
Phosphatidylcholine (PC) is the predominant phospholipid class and required for the buildup of cell membrane. An overwhelming majority of PC is synthesized via the Kennedy pathway by using choline as the substrate. Deacceleration of PC de novo bio-synthesis by genetic ablation of choline kinase leads to mitochondrial damage, as evidenced by the impaired oxidative phosphorylation, decreased ATP production and increased ROS generation. Furthermore, PRKN, PINK1, MAP1LC3, polyubiquitin and SQSTM1/p62 are localized to these damaged mitochondria to initiate mitophagy [Citation295]. Xiong et al. reported a strong association between aberrant choline-PC metabolism and mitophagy in diffuse large B-cell lymphoma [Citation296]. Declined choline uptake and disordered PC bio-synthesis lead to the remodeling of mitochondrial lipidomic signature, decrease in ATP production and AMPK activation, which are critical for mitophagy initiation [Citation297]. Collectively, these studies thus shed light on a pathomechanistic link between choline-PC metabolism and mitophagy induction. Additionally, other studies reported that coenzyme Q [Citation298,Citation299] and bile acid [Citation300] could also impinge on mitochondrial autophagy in the corresponding models.
cAMP and mitophagy
cAMP (cyclic adenosine 3′,5′-monophosphate) is the first identified second messenger and plays fundamental roles in cellular signaling transduction, mainly by supporting the PRKA-CREB1 (cAMP responsive element binding protein 1) axis. ADCYs (adenylate cyclases), including transmembrane ADCYs (tmADCYs) and soluble ADCYs (sADCYs), are responsible for converting ATP into cAMP, while PDE (phosphodiesterase) catalyzes the hydrolysis of intracellular cAMP. It was reported that sADCYs reside at multiple subcellular organelles, including mitochondria [Citation301]. sADCYs-mediated cAMP synthesis inside mitochondria sustains the mitochondrial bioenergetics through supporting PRKA-dependent phosphorylation of respiratory chain complexes [Citation302]. Conversely, mitochondrial ATP, Ca2+ and bicarbonate could finely tune the enzymatic activity of sADCYs and facilitate the cAMP synthesis. Therefore, the mitochondrial health determines mitochondrial cAMP production and vice versa.
More than 100 mitochondrial proteins have been identified as potential PRKA substrates [Citation303]. Specifically, PRKA could phosphorylate DNM1L and restrain its translocation onto the mitochondrial surface [Citation304,Citation305]. During starvation, cellular cAMP increases and supports DNM1L phosphorylation by PRKA, leading to mitochondrial elongation and protecting mitochondria from autophagic degradation [Citation66,Citation306,Citation307]. The cAMP-PRKA also phosphorylates IMMT and CHCHD3/MIC19 (coiled-coil-helix-coiled-coil-helix domain containing 3), destabilizes PINK1 on the depolarized mitochondria, leading to the defect of the PINK1-PRKN-mediated mitophagy [Citation36]. Furthermore, PRKN could be directly phosphorylated by PRKA. Similarly, this phosphorylation prevents the recruitment of PRKN onto mitochondria, resulting in defective mitophagy [Citation308]. BNIP3L was recently reported as another PRKA substrate. PRKA phosphorylating BNIP3L abrogates BNIP3L-mediated mitochondrial fission and mitophagy [Citation115]. Additionally, cAMP-PRKA axis also suppresses general autophagy by phosphorylating MAP1LC3 at its N-terminal region and abrogating MAP1LC3 recruitment to autophagosomes [Citation309]. Collectively, these studies suggest a multifaceted modulation of mitophagy by the cAMP-PRKA axis ().
Figure 6. cAMP-PRKA signaling antagonizes mitophagy. Mitochondrial sADCYs are responsible for the biosynthesis of cAMP, which supports the PRKA activity. cAMP-PRKA phosphorylates CREB1 and activates the transcription of major mitochondrial biogenesis-associated factors. Mitochondrial fission protein DNM1L could be phosphorylated by PRKA, and this phosphorylation prevents its translocation onto the mitochondrial surface for mitochondrial fission. Furthermore, PRKA also phosphorylates IMMT, CHCHD3, PRKN and BNIP3L, abrogating PRKN- and BNIP3L-mediated mitophagy. MAP1LC3 phosphorylation by PRKA represses its translocation onto phagophores, thus inhibits general autophagy.
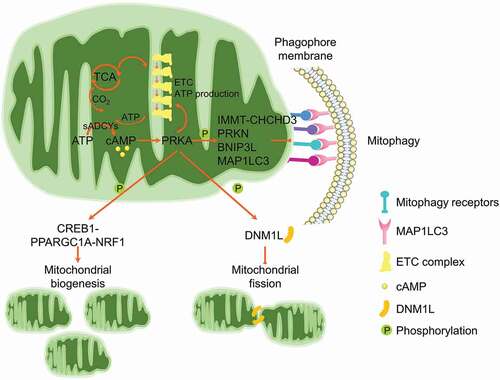
Concluding remarks and perspective
Mitochondria are highly dynamic organelles with constant fusion, fission, mobility, biogenesis and removal. These mitochondrial behaviors are coordinated to maintain the proper mitochondrial quality and quantity [Citation8]. As mentioned above, distinct pathways are sensing different mitochondrial stresses to coordinate mitophagy for mitochondrial quality control, suggesting a strict necessity to maintain a proper mitophagy activity for the cellular homeostasis [Citation15].
Programmed mitophagy refers to the physiological routine to remove partial or all mitochondria in certain developmental, metabolic and physiological circumstances. Specifically, BNIP3L-mediated mitophagy is physiologically important for the maturation of red blood cells [Citation111], while elimination of paternal mitochondria after fertilization is critical for the zygote development post fertilization and supports the maternal inheritance of the mitochondrial genome [Citation310]. Besides, basal mitophagy, referred to the continuous removal of damaged mitochondria under steady-state level, was revealed by two transgenic mouse models to detect mitophagy in vivo, while its activity is highly tissue- and cell-dependent [Citation311,Citation312]. The induced mitophagy has attracted tremendous concerns. Although numerous studies utilized distinct mitochondrial toxins to trigger mitophagy, it is worth noting that the metabolic dynamics could impinge on mitophagy program due to the altered metabolites as mentioned above. Conversely, the induced mitophagy could finely tune the mitochondrial integrity including quality and quantity, and subsequently remodel the cellular metabolism. However, a group of previous studies aimed to explore the physiological relevance of endogenous metabolites in mitophagy mainly rely on the exogenous administration of these metabolites or the metabolic precursors. These supplementations largely exceed the endogenous levels in some cases. Therefore, the exact physiological role of the endogenous metabolites in mitophagy regulation needs further investigations. The patients-derived cells with altered metabolic programs and metabolites are ideal models for these studies.
The defective mitophagy due to genetic susceptibility or environmental insults is manifested in several disease pathogenesis. Targeted normalization of proper mitophagy is believed to be a potent therapeutic design for these diseases. Exercise training is an effective nonpharmacological approach to resist pathogenesis. It has been reported that exercise could activate mitophagy to remove the damaged mitochondria and stimulate mitochondrial biogenesis to improve the mitochondrial functionalities in multiple tissues [Citation313,Citation314]. Furthermore, exercise-induced mitophagy is essential to reverse the high-fat diet-induced mitochondrial dysfunction [Citation315]. The ATP-AMPK-ULK1 and NAD+-SIRTs axes are involved in this mitophagy, although the exact mechanism is complicated and exercise type-dependent [Citation316]. Nevertheless, the optimal manipulation of endogenous metabolites as mentioned above could drive a proper level of mitophagy, which provides ideal nutritional designs for intervention of related diseases.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Tan JX, Finkel T. Mitochondria as intracellular signaling platforms in health and disease. J Cell Biol. 2020 May 4;219(5):e202002179. DOI:https://doi.org/10.1083/jcb.202002179.
- Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014 May 27;12:34. DOI:https://doi.org/10.1186/1741-7007-12-34.
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012 Oct 26;48(2):158–167.
- Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997 Nov 14;91(4):479–489.
- Du CY, Fang M, Li YC, et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000 Jul 7;102(1):33–42.
- Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018 Jan;25(1):27–36.
- Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Bio. 2020 Feb;21(2):85–100.
- Schapira AH. Mitochondrial diseases. Lancet. 2012 May 12;379(9828):1825–1834.
- Deshwal S, Fiedler KU, Langer T. Mitochondrial proteases: multifaceted regulators of mitochondrial plasticity. Annu Rev Biochem. 2020 Jun 20;89:501–528.
- Voos W. Chaperone-protease networks in mitochondrial protein homeostasis. BBA-Mol Cell Res. 2013 Feb;1833(2):388–399.
- Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Bio. 2018 Feb;19(2):109–120.
- Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol -Mech. 2020;15:235–259.
- Soubannier V, McLelland GL, Zunino R, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012 Jan 24;22(2):135–141.
- Sugiura A, McLelland GL, Fon EA, et al. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. Embo J. 2014 Oct 1;33(19):2142–2156.
- Wu H, Wei HF, Sehgal SA, et al. Mitophagy receptors sense stress signals and couple mitochondrial dynamic machinery for mitochondrial quality control. Free Radical Bio Med. 2016 Nov;100:199–209.
- Liu L, Sakakibara K, Chen Q, et al. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014 Jul;24(7):787–795.
- Hruban Z, Spargo B, Swift H, et al. Focal cytoplasmic degradation. Am J Pathol. 1963;42:657–683.
- Greene WB, Balentine JD, Hennigar GR. Selective mitochondrial degeneration in renal tubules following hyperbaric oxygen exposure. Am J Pathol. 1979 Sep;96(3):737–752.
- Oczypok EA, Oury TD, Chu CT. It’s a cell-eat-cell world: autophagy and phagocytosis. Am J Pathol. 2013 Mar;182(3):612–622.
- Tolkovsky AM, Xue LZ, Fletcher GC, et al. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002 Feb-Mar;84(2–3):233–240.
- Lemasters JJ. Perspective - Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuv Res. 2005 Spr;8(1):3–5.
- Killackey SA, Philpott DJ, Girardin SE. Mitophagy pathways in health and disease. J Cell Biol. 2020 Nov 2;219(11):e202004029. DOI:https://doi.org/10.1083/jcb.202004029.
- Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85:685–713.
- Clark IE, Dodson MW, Jiang CG, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006 Jun 28;441(7097):1162–1166.
- Greene JC, Whitworth AJ, Kuo I, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003 Apr 1;100(7):4078–4083.
- Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006 Jun 28;441(7097):1157–1161.
- Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008 Dec 1;183(5):795–803.
- Jin SM, Lazarou M, Wang CX, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010 Nov 29;191(5):933–942.
- Deas E, Plun-Favreau H, Gandhi S, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011 Mar 1;20(5):867–879.
- Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013 Nov 1;9(11):1758–1769.
- Lazarou M, Jin SM, Kane LA, et al. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 Ligase Parkin. Dev Cell. 2012 Feb 14;22(2):320–333.
- Okatsu K, Uno M, Koyano F, et al. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem. 2013 Dec 20;288(51):36372–36384.
- Okatsu K, Kimura M, Oka T, et al. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J Cell Sci. 2015 Mar 1;128(5):964–978.
- Sekine S, Wang C, Sideris DP, et al. Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol Cell. 2019 Mar 7;73(5):1028–1043 e5.
- Hasson SA, Kane LA, Yamano K, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013 Dec 12;504(7479):291–295.
- Akabane S, Uno M, Tani N, et al. PKA regulates PINK1 stability and Parkin recruitment to damaged mitochondria through phosphorylation of MIC60. Mol Cell. 2016 May 5;62(3):371–384.
- Sekine S. PINK1 import regulation at a crossroad of mitochondrial fate: the molecular mechanisms of PINK1 import. J Biochem. 2020 Mar;167(3):217–224.
- Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014 Apr 28;205(2):143–153.
- Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014 Jun 5;510(7503):162–166.
- Trempe JF, Sauve V, Grenier K, et al. Structure of Parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013 Jun 21;340(6139):1451–1455.
- Ordureau A, Sarraf SA, Duda DM, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014 Nov 6;56(3):360–375.
- Riley BE, Lougheed JC, Callaway K, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013 Jun;4:1982. DOI:https://doi.org/10.1038/ncomms2982.
- Sauve V, Sung G, Soya N, et al. Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol. 2018 Jul;25(7):623–630.
- Kumar A, Aguirre JD, Condos TEC, et al. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. Embo J. 2015 Oct 14;34(20):2506–2521.
- Gladkova C, Maslen SL, Skehel JM, et al. Mechanism of parkin activation by PINK1. Nature. 2018 Jul 19;559(7714):411–414.
- Okatsu K, Koyano F, Kimura M, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015 Apr 13;209(1):111–128.
- Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013 Apr 18;496(7445):372–376.
- Chen Y, Dorn GW. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013 Apr 26;340(6131):471–475.
- Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010 Feb;12(2):119–131.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015 Aug 20;524(7565):309–314.
- Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015 Oct 1;60(1):7–20.
- Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015 Jan 21;85(2):257–273.
- Lee JJ, Sanchez-Martinez A, Martinez Zarate A, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018 May 7;217(5):1613–1622.
- McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018 Feb 6;27(2):439–449 e5.
- Wang XN, Winter D, Ashrafi G, et al. PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011 Nov 11;147(4):893–906.
- Shlevkov E, Kramer T, Schapansky J, et al. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci USA. 2016 Oct 11;113(41):E6097–E6106.
- Shin JH, Ko HS, Kang H, et al. PARIS (ZNF746) repression of PGC-1 alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011 Mar 4;144(5):689–702.
- Stevens DA, Lee Y, Kang HC, et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci USA. 2015 Sep 15;112(37):11696–11701.
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA. 2008 Aug 12;105(32):11364–11369.
- Morais VA, Haddad D, Craessaerts K, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014 Apr 11;344(6180):203–207.
- Deng H, Dodson MW, Huang H, et al. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008 Sep 23;105(38):14503–14508.
- Poole AC, Thomas RE, Andrews LA, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008 Feb 5;105(5):1638–1643.
- Yang YF, Ouyang YS, Yang LH, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008 May 13;105(19):7070–7075.
- Han HL, Tan JDO, Wang RX, et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. Embo Rep. 2020 Aug 5;21(8):e48686. DOI:https://doi.org/10.15252/embr.201948686.
- Dagda RK, Pien I, Wang R, et al. Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem. 2014 Mar;128(6):864–877.
- Dagda RK, Gusdon AM, Pien I, et al. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011 Dec;18(12):1914–1923.
- Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009 Mar 13;33(5):627–638.
- Huang E, Qu D, Huang T, et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun. 2017 Nov 9;8(1):1399.
- Pridgeon JW, Olzmann JA, Chin LS, et al. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007 Jul;5(7):e172.
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014 Jun;16(6):495–501.
- Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013 Aug 1;126(15):3237–3247.
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009 Jul 21;17(1):87–97.
- Kanki T, Wang K, Cao Y, et al. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009 Jul 21;17(1):98–109.
- Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012 Feb;14(2):177–185.
- Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014 May 8;54(3):362–377.
- Kuang Y, Ma KL, Zhou CQ, et al. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy. 2016;12(12):2363–2373.
- Wu H, Xue DF, Chen G, et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy. 2014 Oct;10(10):1712–1725.
- Ma KL, Zhang Z, Chang R, et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 2020 Mar;27(3):1036–1051.
- Wu WX, Tian WL, Hu Z, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. Embo Rep. 2014 May;15(5):566–575.
- Chen ZH, Liu L, Cheng Q, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. Embo Rep. 2017 Mar;18(3):495–509.
- Lim Y, Berry B, Viteri S, et al. FNDC-1-mediated mitophagy and ATFS-1 coordinate to protect against hypoxia-reoxygenation. Autophagy. 2021 Jan 19:1–13. DOI:https://doi.org/10.1080/15548627.2021.1872885.
- Lim Y, Rubio-Pena K, Sobraske PJ, et al. Fndc-1 contributes to paternal mitochondria elimination in C. elegans. Dev Biol. 2019 Oct 1;454(1):15–20.
- Xu GY, Shen H, Nibona E, et al. Fundc1 is necessary for proper body axis formation during embryogenesis in zebrafish. Sci Rep-UK. 2019 Dec 11;9(1):18910.
- Liu L, Li YJ, Wang JN, et al. Mitophagy receptor FUNDC1 is regulated by PGC-1 alpha/NRF1 to fine tune mitochondrial homeostasis. Embo Rep. 2021 Feb 8;22(3):e50629. DOI:https://doi.org/10.15252/embr.202050629.
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C-elegans. Nature. 2015 May 28;521(7553):525–528.
- Li W, Zhang XL, Zhuang HX, et al. MicroRNA-137 is a novel hypoxia-responsive MicroRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol Chem. 2014 Apr 11;289(15):10691–10701.
- Chen M, Chen ZH, Wang YY, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12(4):689–702.
- Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Bio. 2012 Oct;13(10):607–615.
- Friedman JR, Lackner LL, West M, et al. ER tubules mark sites of mitochondrial division. Science. 2011 Oct 21;334(6054):358–362.
- Murley A, Lackner LL, Osman C, et al. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife. 2013 May 14;2:e00422. DOI:https://doi.org/10.7554/eLife.00422.
- Bockler S, Westermann B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell. 2014 Feb 24;28(4):450–458.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013 Mar 21;495(7441):389–393.
- Wu SN, Lu QL, Wang QL, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017 Dec 5;136(23):2248–2266.
- Wu WX, Lin CX, Wu K, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. Embo J. 2016 Jul 1;35(13):1368–1384.
- Wu H, Wang Y, Li W, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019 Nov;15(11):1882–1898.
- Fu TT, Xu ZS, Liu L, et al. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 2018 May 1;23(5):1357–1372.
- Cai Y, Yang EY, Yao XH, et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021 Jan;38:101792.
- Zhou H, Zhu PJ, Guo J, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507.
- Ren J, Sun MM, Zhou H, et al. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv. 2020 Sep;6(38):eabc8561.
- Zhou H, Zhu PJ, Wang J, et al. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2 alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018 Jun;25(6):1080–1093.
- Zhou H, Zhu PJ, Wang J, et al. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduction Tar. 2019 Dec 6;4:56.
- Wang J, Zhu PJ, Li RB, et al. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020 Feb;30:101415.
- Li SQ, Zhou Y, Gu XC, et al. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Proliferat. 2021 Jan 11;54(3):e12986.
- Li WH, Li YJ, Siraj S, et al. FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology. 2019 Feb;69(2):604–621.
- Boyd JM, Malstrom S, Subramanian T, et al. Adenovirus-E1b 19-Kda and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994 Oct 21;79(2):341–351.
- Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012 Jun 1;287(23):19094–19104.
- Quinsay MN, Thomas RL, Lee Y, et al. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2010 Oct 1;6(7):855–862.
- Tracy K, Dibling BC, Spike BT, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007 Sep;27(17):6229–6242.
- Zhu Y, Massen S, Terenzio M, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013 Jan 11;288(2):1099–1113.
- Diwan A, Koesters AG, Odley AM, et al. Unrestrained erythroblast development in Nix(-/-) mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA. 2007 Apr 17;104(16):6794–6799.
- Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007 Dec 4;104(49):19500–19505.
- Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008 Jul 10;454(7201):232–235.
- Schwarten M, Mohrluder J, Ma PX, et al. Nix directly binds to GABARAP A possible crosstalk between apoptosis and autophagy. Autophagy. 2009 Jul 1;5(5):690–698.
- Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. Embo Rep. 2010 Jan;11(1):45–51.
- Rosa SCD, Martens MD, Field JT, et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy. 2020 Oct 12;1–16.
- Rogov VV, Suzuki H, Marinkovic M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep-UK. 2017 Apr 25;7(1):1131.
- Yuan Y, Zheng YR, Zhang XN, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13(10):1754–1766.
- Marinkovic M, Sprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy. 2020 Apr 25;17(5):1232–1243.
- Sowter HM, Ratcliffe PJ, Watson P, et al. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001 Sep 15;61(18):6669–6673.
- Zhang HF, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008 Apr 18;283(16):10892–10903.
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009 Jul;16(7):939–946.
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. 2000 Aug 1;97(16):9082–9087.
- Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007 Dec;6(6):458–471.
- Li EM, Li XY, Huang J, et al. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell. 2020 Sep;11(9):661–679.
- Chourasia AH, Macleod KF. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy. 2015;11(10):1937–1938.
- Murai M, Toyota M, Suzuki H, et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin Cancer Res. 2005 Feb 1;11(3):1021–1027.
- Okami J, Simeone DM, Logsdon CD. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 2004 Aug 1;64(15):5338–5346.
- Wu XL, Zheng YR, Liu MR, et al. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy. 2020 Aug 13;17(8):1934–1946.
- Fu ZJ, Wang ZY, Xu L, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020 Sep;36:101671.
- Tang CY, Han HL, Liu ZW, et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019 Sep 12;10(9):677. DOI:https://doi.org/10.1038/s41419-019-1899-0.
- Jin QH, Li RB, Hu N, et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018 Apr;14:576–587.
- Chourasia AH, Tracy K, Frankenberger C, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. Embo Rep. 2015 Sep;16(9):1145–1163.
- Bacon AL, Fox S, Turley H, et al. Selective silencing of the hypoxia-inducible factor 1 target gene BNIP3 by histone deacetylation and methylation in colorectal cancer. Oncogene. 2007 Jan 4;26(1):132–141.
- Humpton TJ, Alagesan B, DeNicola GM, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019 Sep;9(9):1268–1287.
- Naik PP, Birbrair A, Bhutia SK. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell Mol Life Sci. 2019 Jan;76(1):27–43.
- Kataoka T, Holler N, Micheau O, et al. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J Biol Chem. 2001 Jun 1;276(22):19548–19554.
- Kim JY, So KJ, Lee S, et al. Bcl-rambo induces apoptosis via interaction with the adenine nucleotide translocator. Febs Lett. 2012 Sep 21;586(19):3142–3149.
- Matsubara H, Tanaka R, Tateishi T, et al. The human Bcl-2 family member Bcl-rambo and voltage-dependent anion channels manifest a genetic interaction in Drosophila and cooperatively promote the activation of effector caspases in human cultured cells. Exp Cell Res. 2019 Aug 15;381(2):223–234.
- Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015 Jul;6:7527. DOI:https://doi.org/10.1038/ncomms8527.
- Li MQ, Jia J, Zhang XC, et al. Selective binding of mitophagy receptor protein Bcl-rambo to LC3/GABARAP family proteins. Biochem Biophys Res Commun. 2020 Sep 10;530(1):292–300.
- Murakawa T, Okamoto K, Omiya S, et al. A mammalian mitophagy receptor, Bcl2-L-13, recruits the ULK1 complex to induce mitophagy. Cell Rep. 2019 Jan 8;26(2):338–345.
- Meng F, Sun N, Liu D, et al. BCL2L13: physiological and pathological meanings. Cell Mol Life Sci. 2021 Mar;78(6):2419–2428.
- Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol. 2003 Jan;5(1):28–37.
- Shirane-Kitsuji M, Nakayama KI. Mitochondria: FKBP38 and mitochondrial degradation. Int J Biochem Cell B. 2014 Jun;51:19–22.
- Bhujabal Z, Birgisdottir AB, Sjottem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. Embo Rep. 2017 Jun;18(6):947–961.
- Yoo SM, Yamashita S, Kim H, et al. FKBP8 LIRL-dependent mitochondrial fragmentation facilitates mitophagy under stress conditions. Faseb J. 2020 Feb;34(2):2944–2957.
- Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nat Commun. 2013 Jan;4:1410. DOI:https://doi.org/10.1038/ncomms2400.
- Bavelloni A, Piazzi M, Raffini M, et al. Prohibitin 2: at a communications crossroads. Iubmb Life. 2015 Apr;67(4):239–254.
- Wei YJ, Chiang WC, Sumpter R, et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017 Jan 12;168(1–2):224–238.
- Yan CJ, Gong LL, Chen L, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy. 2020 Mar 3;16(3):419–434.
- Paradies G, Paradies V, Ruggiero FM, et al. Cardiolipin and mitochondrial function in health and disease. Antioxid Redox Sign. 2014 Apr 20;20(12):1925–1953.
- Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013 Oct;15(10):1197–1205.
- Anton Z, Landajuela A, Hervas JH, et al. Human Atg8-cardiolipin interactions in mitophagy: specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy. 2016;12(12):2386–2403.
- Kagan VE, Jiang J, Huang Z, et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016 Jul;23(7):1140–1151.
- Dagvadorj J, Mikulska-Ruminska K, Tumurkhuu G, et al. Recruitment of pro-IL-1alpha to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc Natl Acad Sci USA. 2021 Jan 5;118(1):e2015632118. DOI:https://doi.org/10.1073/pnas.2015632118.
- Sentelle RD, Senkal CE, Jiang WH, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012 Oct;8(10):831–838.
- Dany M, Gencer S, Nganga R, et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood. 2016 Oct 13;128(15):1944–1958.
- Wang ZT, Lu MH, Zhang Y, et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell. 2019 Feb;18(1):e12860.
- Cen XF, Chen YY, Xu XY, et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat Commun. 2020 Nov 12;11(1):5731. DOI:https://doi.org/10.1038/s41467-020-19547-6.
- Strappazzon F, Nazio F, Corrado M, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1 (vol 22, pg 419, 2015). Cell Death Differ. 2015 Mar;22(3):517–517. DOI:https://doi.org/10.1038/cdd.2014.190.
- Shu L, Hu C, Xu M, et al. ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. Embo J. 2021 Mar 5;40(8):e106283.
- Schofield JH, Schafer ZT. Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid Redox Signal. 2021 Mar 1;34(7):517–530.
- Dagda RK, Cherra SJ 3rd, Kulich SM, et al. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009 May 15;284(20):13843–13855.
- Wang Y, Nartiss Y, Steipe B, et al. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012 Oct;8(10):1462–1476.
- Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018 Feb;19(2):121–135.
- Richardson DR, Lane DJR, Becker EM, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci USA. 2010 Jun 15;107(24):10775–10782.
- Ward DM, Cloonan SM. Mitochondrial iron in human health and disease. Annu Rev Physiol. 2019;81:453–482.
- Allen GFG, Toth R, James J, et al. Loss of iron triggers PINK1/Parkin-independent mitophagy. Embo Rep. 2013 Dec;14(12):1127–1135.
- Mancias JD, Wang XX, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014 May 1;509(7498):105–109.
- Hara Y, Yanatori I, Tanaka A, et al. Iron loss triggers mitophagy through induction of mitochondrial ferritin. Embo Rep. 2020 Sep 25;21(11):e50202. DOI:https://doi.org/10.15252/embr.202050202.
- Nagi M, Tanabe K, Nakayama H, et al. Iron-depletion promotes mitophagy to maintain mitochondrial integrity in pathogenic yeast Candida glabrata. Autophagy. 2016;12(8):1259–1271.
- Schiavi A, Maglioni S, Palikaras K, et al. Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr Biol. 2015 Jul 20;25(14):1810–1822.
- Kirienko NV, Ausubel FM, Ruvkun G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2015 Feb 10;112(6):1821–1826.
- Wu H, Wei H, Zhang D, et al. Defective mitochondrial ISCs biogenesis switches on IRP1 to fine tune selective mitophagy. Redox Biol. 2020 Sep;36:101661.
- Fader CM, Salassa BN, Grosso RA, et al. Hemin induces mitophagy in a leukemic erythroblast cell line. Biol Cell. 2016 Apr;108(4):77–95.
- Hu ST, Li XY, Chen H, et al. [Knockdown of ALAS2 affects erythroid differentiation by down-regulating mitophagy receptor BNIP3L]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2020 Oct;28(5):1710–1717.
- Suliman HB, Keenan JE, Piantadosi CA. Mitochondrial quality-control dysregulation in conditional HO-1(-/-) mice. JCI Insight. 2017 Feb 9;2(3):e89676.
- Hull TD, Boddu R, Guo L, et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight. 2016;1(2):e85817.
- Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006 Apr;86(2):583–650.
- Kim HJ, Joe Y, Rah SY, et al. Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis. 2018 Oct 17;9(11):1060. DOI:https://doi.org/10.1038/s41419-018-1112-x.
- Bogdan AR, Miyazawa M, Hashimoto K, et al. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016 Mar;41(3):274–286.
- Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium (vol 19, pg 713, 2018). Nat Rev Mol Cell Bio. 2018 Nov;19(11):746–746.
- Rizzuto R, De Stefani D, Raffaello A, et al. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Bio. 2012 Sep;13(9):566–578.
- Bootman MD, Chehab T, Bultynck G, et al. The regulation of autophagy by calcium signals: do we have a consensus? Cell Calcium. 2018 Mar;70:32–46.
- Xia HG, Zhang LH, Chen G, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010 Jan 1;6(1):61–66.
- Cardenas C, Miller RA, Smith I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010 Jul 23;142(2):270–283.
- Mallilankaraman K, Cardenas C, Doonan PJ, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012 Dec;14(12):1336–1343.
- Wang JQ, Zhu S, Wang YH, et al. Miro2 supplies a platform for Parkin translocation to damaged mitochondria. Sci Bull. 2019 Jun 15;64(11):730–747.
- Safiulina D, Kuum M, Choubey V, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. Embo J. 2019 Jan 15;38(2):e99384. DOI:https://doi.org/10.15252/embj.201899384.
- Wang XN, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009 Jan 9;136(1):163–174.
- Bonadonna RC, Defronzo RA. Glucose-metabolism in obesity and type-2 diabetes. Diabetes Metab. 1991;17(1bis):112–135.
- Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. 1929;23(3):536–545.
- Lee SH, Du J, Stitham J, et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. Embo Mol Med. 2016 Jul;8(7):779–795.
- Onphachanh X, Lee HJ, Lim JR, et al. Enhancement of high glucose-induced PINK1 expression by melatonin stimulates neuronal cell survival: involvement of MT2/Akt/NF-kappa B pathway. J Pineal Res. 2017 Sep;63(2):e12427.
- Yu TZ, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Sign. 2011 Feb;14(3):425–437.
- Marfella R, D’Amico M, Di Filippo C, et al. Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia. 2002 Aug;45(8):1172–1181.
- Catrina SB, Okamoto K, Pereira T, et al. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes. 2004 Dec;53(12):3226–3232.
- Tang Y, Liu JK, Long JG. Phosphatase and tensin homolog-induced putative kinase 1 and Parkin in diabetic heart: role of mitophagy. J Diabetes Invest. 2015 May;6(3):250–255.
- Zhou WZ, Yang J, Zhang D, et al. Role of Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 in myocardial cells in diabetes. Exp Ther Med. 2015 Jul;10(1):67–73.
- Rabinowitz JD, Enerback S. Lactate: the ugly duckling of energy metabolism. Nat Metab. 2020 Jul;2(7):566–571.
- Hashimoto T, Hussien R, Oommen S, et al. Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. Faseb J. 2007 Aug;21(10):2602–2612.
- Zhu Y, Ji JJ, Yang R, et al. Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal. 2019 Jun;58:53–64.
- Zhu Y, Han XQ, Sun XJ, et al. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. 2020 Jun;25(5–6):321–340.
- Vanderperre B, Bender T, Kunji ERS, et al. Mitochondrial pyruvate import and its effects on homeostasis. Curr Opin Cell Biol. 2015 Apr;33:35–41.
- Koh E, Kim YK, Shin D, et al. MPC1 is essential for PGC-1alpha-induced mitochondrial respiration and biogenesis. Biochem J. 2018 May 18;475(10):1687–1699.
- Wilson L, Yang Q, Szustakowski JD, et al. Pyruvate induces mitochondrial biogenesis by a PGC-1 alpha-independent mechanism. Am J Physiol Cell Physiol. 2007 May;292(5):C1599–605.
- Park S, Choi SG, Yoo SM, et al. Pyruvate stimulates mitophagy via PINK1 stabilization. Cell Signal. 2015 Sep;27(9):1824–1830.
- Kim JY, Lee SH, Bae IH, et al. Pyruvate protects against cellular senescence through the control of mitochondrial and lysosomal function in dermal fibroblasts. J Invest Dermatol. 2018 Dec;138(12):2522–2530.
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, et al. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015 Jun 2;21(6):805–821.
- Marino G, Pietrocola F, Eisenberg T, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014 Mar 6;53(5):710–725.
- He A, Chen X, Tan M, et al. Acetyl-CoA derived from hepatic peroxisomal beta-oxidation inhibits autophagy and promotes steatosis via mTORC1 activation. Mol Cell. 2020 Jul 2;79(1):30–42 e4.
- Son SM, Park SJ, Lee H, et al. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 2019 Jan 8;29(1):192–201 e7.
- Webster BR, Scott I, Han K, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci. 2013 Nov 1;126(21):4843–4849.
- Bosch-Presegue L, Raurell-Vila H, Marazuela-Duque A, et al. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol Cell. 2011 Apr 22;42(2):210–223.
- Zhang YR, Zhang MD, Zhu W, et al. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox Biol. 2020 Jan;28:101365.
- Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013 Apr 11;496(7444):238–242.
- Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014 Nov 20;515(7527):431–435.
- Zhang ZH, Tan MJ, Xie ZY, et al. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011 Jan;7(1):58–63.
- Xie N, Zhang L, Gao W, et al. NAD(+)metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduction Tar. 2020 Oct 7;5(1):227.
- Covarrubias AJ, Perrone R, Grozio A, et al. NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Bio. 2021 Feb;22(2):119–141.
- Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015 Dec 4;350(6265):1208–1213.
- Luongo TS, Eller JM, Lu MJ, et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature. 2020 Dec 3;588(7836):174–179.
- Zhang HB, Ryu D, Wu YB, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016 Jun 17;352(6292):1436–1443.
- Canto C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012 Jun 6;15(6):838–847.
- Pirinen E, Canto C, Jo YS, et al. Pharmacological inhibition of Poly(ADP-Ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014 Jun 3;19(6):1034–1041.
- Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/Sirtuin Pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013 Jul 18;154(2):430–441.
- Gomes AP, Price NL, Ling AJY, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013 Dec 19;155(7):1624–1638.
- Jang SY, Kang HT, Hwang ES. Nicotinamide-induced mitophagy event mediated by high NAD(+)/NADH ratio and SIRT1 protein activation. J Biol Chem. 2012 Jun 1;287(23):19304–19314.
- Kang HT, Hwang ES. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell. 2009 Aug;8(4):426–438.
- Fang EF, Scheibye-Knudsen M, Brace LE, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014 May 8;157(4):882–896.
- Fang EF, Kassahun H, Croteau DL, et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016 Oct 11;24(4):566–581.
- Fang EF, Hou YJ, Lautrup S, et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat Commun. 2019 Nov 21;10(1):5284. DOI:https://doi.org/10.1038/s41467-019-13172-8.
- Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019 Mar;22(3):401–412.
- Toyama EQ, Herzig S, Courchet J, et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016 Jan 15;351(6270):275–281.
- Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011 Jan 28;331(6016):456–461.
- Fang EF, Scheibye-Knudsen M, Chua KF, et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Bio. 2016 May;17(5):308–321.
- Price NL, Gomes AP, Ling AJY, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012 May 2;15(5):675–690.
- Huang R, Xu YF, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015 Feb 5;57(3):456–466.
- Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008 Mar 4;105(9):3374–3379.
- Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013 Mar;14(3):133–139.
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008 Jan;7(1):11–20.
- Eng CH, Yu K, Lucas J, et al. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010 Apr 27;3(119):ra31–ra31.
- Polletta L, Vernucci E, Carnevale I, et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. 2015;11(2):253–270.
- Kappler M, Pabst U, Rot S, et al. Normoxic accumulation of HIF1alpha is associated with glutaminolysis. Clin Oral Investig. 2017 Jan;21(1):211–224.
- Van Laar VS, Roy N, Liu A, et al. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol Dis. 2015 Feb;74:180–193.
- Wong CG, Bottiglieri T, Snead OC 3rd. GABA, gamma-hydroxybutyric acid, and neurological disease. Ann Neurol. 2003;54(Suppl 6):S3–12.
- Lakhani R, Vogel KR, Till A, et al. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med. 2014 Apr;6(4):551–566.
- Salimian Rizi B, Achreja A, Nagrath D. Nitric oxide: the forgotten child of tumor metabolism. Trends Cancer. 2017 Sep;3(9):659–672.
- Ghasemi M, Mayasi Y, Hannoun A, et al. Nitric oxide and mitochondrial function in neurological diseases. Neuroscience. 2018 Apr 15;376:48–71.
- Han JY, Kang MJ, Kim KH, et al. Nitric oxide induction of Parkin translocation in PTEN-induced putative kinase 1 (PINK1) deficiency: functional role of neuronal nitric oxide synthase during mitophagy. J Biol Chem. 2015 Apr 17;290(16):10325–10335.
- Ozawa K, Komatsubara AT, Nishimura Y, et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep-Uk. 2013 Jul 16;3:2202.
- Oh CK, Sultan A, Platzer J, et al. S-Nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep. 2017 Nov 21;21(8):2171–2182.
- Rizza S, Cardaci S, Montagna C, et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc Natl Acad Sci U S A. 2018 Apr 10;115(15):E3388–E3397.
- Sarkar S, Korolchuk VI, Renna M, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011 Jul 8;43(1):19–32.
- Madeo F, Eisenberg T, Pietrocola F, et al. Spermidine in health and disease. Science. 2018 Jan 26;359(6374):eaan2788.
- Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009 Nov;11(11):1305–1314.
- Yang X, Zhang M, Dai Y, et al. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging (Albany NY). 2020 Sep 9;12(17):16852–16866.
- Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016 Dec;22(12):1428–1438.
- Tyrrell DJ, Blin MG, Song J, et al. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ Res. 2020 Jan 31;126(3):298–314.
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016 Jan 7;529(7584):37–42.
- Qi Y, Qiu Q, Gu X, et al. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci Rep. 2016 Apr 19;6:24700.
- Varghese N, Werner S, Grimm A, et al. Dietary mitophagy enhancer: a strategy for healthy brain aging? Antioxidants (Basel). 2020 Sep 29;9(10):932.
- Zhu HL, Shi XT, Xu XF, et al. Melatonin protects against environmental stress-induced fetal growth restriction via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. Redox Biol. 2021 Apr;40:101854.
- Zhou H, Li D, Zhu P, et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res. 2017 Nov;63(4):e12438.
- Zhou H, Zhang Y, Hu S, et al. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017 Aug;63(1):e12413.
- Wang SY, Wang L, Qin X, et al. ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal Transduction Tar. 2020 Jul 24;5(1):119.
- Zhou H, Du W, Li Y, et al. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018 Jan;64(1):e12450.
- Platten M, Nollen EAA, Rohrig UF, et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019 May;18(5):379–401.
- Castro-Portuguez R, Sutphin GL. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: targeting tryptophan metabolism to promote longevity and healthspan. Exp Gerontol. 2020 Apr;132:110841.
- Katsyuba E, Mottis A, Zietak M, et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature. 2018 Nov 15;563(7731):354–359.
- Shi HJ, Enriquez A, Rapadas M, et al. NAD deficiency, congenital malformations, and niacin supplementation. New Engl J Med. 2017 Aug 10;377(6):544–552.
- Ivatt RM, Sanchez-Martinez A, Godena VK, et al. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc Natl Acad Sci USA. 2014 Jun 10;111(23):8494–8499.
- Maddison DC, Alfonso-Nunez M, Swaih AM, et al. A novel role for kynurenine 3-monooxygenase in mitochondrial dynamics. Plos Genet. 2020 Nov;16(11):e1009129.
- Townsend DM, Tew KD, Tapiero H. Sulfur containing amino acids and human disease. Biomed Pharmacother. 2004 Jan;58(1):47–55.
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012 Apr;92(2):791–896.
- Paul BD, Snyder SH, Kashfi K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021 Jan;38:101772.
- Liu N, Wu J, Zhang L, et al. Hydrogen Sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J Cell Mol Med. 2017 Dec;21(12):3190–3203.
- Sun Y, Lu F, Yu X, et al. Exogenous H2S promoted USP8 sulfhydration to regulate mitophagy in the hearts of db/db mice. Aging Dis. 2020 Apr;11(2):269–285.
- Vandiver MS, Paul BD, Xu R, et al. Sulfhydration mediates neuroprotective actions of parkin. Nat Commun. 2013;4:1626.
- Sen U, Sathnur PB, Kundu S, et al. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol. 2012 Jul 1;303(1):C41–51.
- Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009 Feb-Apr;30(1–2):42–59.
- Calabrese G, Morgan B, Riemer J. Mitochondrial glutathione: regulation and functions. Antioxid Redox Signal. 2017 Nov 20;27(15):1162–1177.
- Deffieu M, Bhatia-Kissova I, Salin B, et al. Glutathione participates in the regulation of mitophagy in yeast. J Biol Chem. 2009 May 29;284(22):14828–14837.
- Sakakibara K, Eiyama A, Suzuki SW, et al. Phospholipid methylation controls Atg32-mediated mitophagy and Atg8 recycling. Embo J. 2015 Nov 3;34(21):2703–2719.
- Levine RL, Mosoni L, Berlett BS, et al. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci USA. 1996 Dec 24;93(26):15036–15040.
- Plummer JD, Johnson JE. Extension of cellular lifespan by methionine restriction involves alterations in central carbon metabolism and is mitophagy-dependent. Front Cell Dev Biol. 2019;7:301.
- Seite S, Mourier A, Camougrand N, et al. Dietary methionine deficiency affects oxidative status, mitochondrial integrity and mitophagy in the liver of rainbow trout (Oncorhynchus mykiss). Sci Rep. 2018 Jul 5;8(1):10151.
- Sutter BM, Wu X, Laxman S, et al. Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell. 2013 Jul 18;154(2):403–415.
- Li R, Xin T, Li D, et al. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018 Sep;18:229–243.
- Zhou T, Chang L, Luo Y, et al. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019 Feb;21:101120.
- Baulies A, Ribas V, Nunez S, et al. Lysosomal cholesterol accumulation sensitizes to acetaminophen hepatotoxicity by impairing mitophagy. Sci Rep. 2015 Dec 11;5:18017.
- Ordonez MP, Roberts EA, Kidwell CU, et al. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum Mol Genet. 2012 Jun 15;21(12):2651–2662.
- Davis OB, Shin HR, Lim CY, et al. NPC1-mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in Niemann-Pick Type C. Dev Cell. 2021 Feb 8;56(3):260–276 e7.
- Roca-Agujetas V, Barbero-Camps E, de Dios C, et al. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol Neurodegener. 2021 Mar 8;16(1):15.
- Mitsuhashi S, Hatakeyama H, Karahashi M, et al. Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum Mol Genet. 2011 Oct 1;20(19):3841–3851.
- Xiong J, Wang L, Fei XC, et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017 Jul 28;7:e582–e582.
- Sanchez-Lopez E, Zhong Z, Stubelius A, et al. Choline uptake and metabolism modulate macrophage IL-1beta and IL-18 production. Cell Metab. 2019 Jun 4;29(6):1350–1362 e7.
- Rodriguez-Hernandez A, Cordero MD, Salviati L, et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009 Jan;5(1):19–32.
- Cotan D, Cordero MD, Garrido-Maraver J, et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. Faseb J. 2011 Aug;25(8):2669–2687.
- Xiao YT, Zhou Y, Lu Y, et al. PHB2 interacts with LC3 and SQSTM1 is required for bile acids-induced mitophagy in cholestatic liver. Cell Death Dis. 2018 Feb 7;9(2):160. DOI:https://doi.org/10.1038/s41419-017-0228-8.
- Zippin JH, Chen YQ, Nahirney P, et al. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. Faseb J. 2002 Nov;16(13):82–84.
- Valsecchi F, Ramos-Espiritu LS, Buck J, et al. cAMP and mitochondria. Physiology. 2013 May;28(3):199–209.
- Grimsrud PA, Carson JJ, Hebert AS, et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012 Nov 7;16(5):672–683.
- Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. Embo Rep. 2007 Oct;8(10):939–944.
- Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007 Jul 27;282(30):21583–21587.
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011 May;13(5):589–598.
- Rambold AS, Kostelecky B, Elia N, et al. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011 Jun 21;108(25):10190–10195.
- Lu XD, Altshuler-Keylin S, Wang Q, et al. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent and UCP1-independent mechanism. Sci Signal. 2018 Apr 24;11(527): eaap8526.
- Cherra SJ, Kulich SM, Uechi G, et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010 Aug 23;190(4):533–539.
- Song WH, Yi YJ, Sutovsky M, et al. The ART and science of sperm mitophagy. Autophagy. 2016 Dec;12(12):2510–2511.
- Sun N, Yun J, Liu J, et al. Measuring in vivo mitophagy. Mol Cell. 2015 Nov 19;60(4):685–696.
- McWilliams TG, Prescott AR, Allen GF, et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol. 2016 Aug 1;214(3):333–345.
- Memme JM, Erlich AT, Hood DA, et al. Exercise and mitochondrial health. J Physiol-London. 2021 Feb;599(3):803–817.
- Moreira OC, Estebanez B, Martinez-Florez S, et al. Mitochondrial function and mitophagy in the elderly: effects of exercise. Oxid Med Cell Longev. 2017;2017:1–13.
- Laker RC, Xu P, Ryall KA, et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 2014 Apr 25;289(17):12005–12015.
- Laker RC, Drake JC, Wilson RJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017 Sep 15;8(1):548. DOI:https://doi.org/10.1038/s41467-017-00520-9.