
ABSTRACT
Macroautophagy/autophagy is an evolutionarily well-conserved recycling process in response to stress conditions, including a burst of reactive oxygen species (ROS) production. High level of ROS attack key cellular macromolecules. Protein cysteinyl thiols or non-protein thiols as the major redox-sensitive targets thus constitute the first-line defense. Autophagy is unique, because it removes not only oxidized/damaged proteins but also bulky ROS-generating organelles (such as mitochondria and peroxisome) to restrict further ROS production. The oxidative regulations of autophagy occur in all processes of autophagy, from induction, phagophore nucleation, phagophore expansion, autophagosome maturation, cargo delivery to the lysosome, and finally to degradation of the cargo and recycling of the products, as well as autophagy gene transcription. Mechanically, these regulations are achieved through direct or indirect manners. Direct thiol oxidation of key proteins such as ATG4, ATM and TFEB are responsible for specific regulations in phagophore expansion, cargo recognition and autophagy gene transcription, respectively. Meanwhile, oxidation of certain redox-sensitive chaperone-like proteins (e.g. PRDX family members and PARK7) may impair a nonspecifically local reducing environment in the phagophore membrane, and influence BECN1-involved phagophore nucleation and mitophagy recognition. However, ROS do exhibit some inhibitory effects on autophagy through direct oxidation of key autophagy regulators such as ATG3, ATG7 and SENP3 proteins. SQSTM1 provides an alternative antioxidant mechanism when autophagy is unavailable or impaired. However, it is yet to be unraveled how cells evolve to equip proteins with different redox susceptibility and in their correct subcellular positions, and how cells fine-tune autophagy machinery in response to different levels of ROS.
Abbreviations: AKT1/PKB: AKT serine/threonine kinase 1; AMPK: AMP-activated protein kinase; ATG: autophagy related; ATM: ATM serine/threonine kinase; BAX: BCL2 associated X, apoptosis regulator; BECN1: beclin 1; BH3: BCL2-homology-3; CAV1: caveolin 1; CCCP: carbonyl cyanide m-chlorophenylhydrazone; CTSB: cathepsin B; CTSL: cathepsin L; DAPK: death associated protein kinase; ER: endoplasmic reticulum; ETC: electron transport chain; GSH: glutathione; GSTP1: glutathione S-transferase pi 1; H2O2: hydrogen peroxide; HK2: hexokinase 2; KEAP1: kelch like ECH associated protein 1; MAMs: mitochondria-associated ER membranes; MAP1LC3B/LC3: microtubule associated protein 1 light chain 3 beta; MAPK8/JNK1: mitogen-activated protein kinase 8; MAP3K5/ASK1: mitogen-activated protein kinase kinase kinase 5; MCOLN1: mucolipin 1; MMP: mitochondrial membrane potential; MTOR: mechanistic target of rapamycin kinase; NFE2L2/NRF2: nuclear factor, erythroid 2 like 2; NFKB1: nuclear factor kappa B subunit 1; NOX: NADPH oxidase; O2-: superoxide radical anion; p-Ub: phosphorylated Ub; PARK7/DJ-1: Parkinsonism associated deglycase; PE: phosphatidylethanolamine; PEX5: peroxisomal biogenesis factor 5; PINK1: PTEN induced kinase 1; PPP3CA/calcineurin: protein phosphatase 3 catalytic subunit beta; PRDX: peroxiredoxin; PRKAA1: protein kinase AMP-activated catalytic subunit alpha 1; PRKD/PKD: protein kinase D; PRKN/parkin: parkin RBR E3 ubiquitin protein ligase; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol-3-phosphate; PTEN: phosphatase and tensin homolog; ROS: reactive oxygen species; SENP3: SUMO specific peptidase 3; SIRT1: sirtuin 1; SOD1: superoxide dismutase 1; SQSTM1/p62: sequestosome 1; SUMO: small ubiquitin like modifier; TFEB: transcription factor EB; TRAF6: TNF receptor associated factor 6; TSC2: TSC complex subunit 2; TXN: thioredoxin; TXNRD1: thioredoxin reductase 1; TXNIP: thioredoxin interacting protein; Ub: ubiquitin; ULK1: unc-51 like autophagy activating kinase 1.
Autophagy and ROS: a brief introduction
Autophagy
Autophagy is a cellular recycling process that is evolutionarily well-conserved from yeast to human. The entire autophagy processes from phagophore nucleation, phagophore expansion, autophagosome maturation, cargo delivery to the lysosome, then cargo degradation and recycling have been well established [Citation1]. Autophagy machinery enables a cell to eat part of itself in response to different types of stress such as nutrient starvation, growth factor deprivation, infection etc [Citation2]. Although autophagy had long been considered as a nonselective process, an accumulating number of findings revealed that autophagy has selective “appetites” to eat protein aggregates, damaged organelles, invading pathogens [Citation2], and unnecessary mitochondria particularly during egg fertilization [Citation3] or during red blood cell production [Citation4].
The process of autophagy has been well reviewed elsewhere [Citation1,Citation2,Citation5,Citation6]. In brief, autophagy machinery is sparked by stress or physiological signals. The autophagy-initiating signals commonly converge on activation of class III phosphatidylinositol 3-kinase (PtdIns3K) complex and downstream ATG (autophagy related) proteins [Citation7]. Beside the catalytic kinase of the PtdIns3K complex, PIK3C3, other important components include BECN1 (beclin 1), ATG14, AMBRA1, PIK3R4/VPS15 and NRBF2. The catalytic activity of PtdIns3K and the inter-components interactions of PtdIns3K complex members are essential for autophagy induction and phagophore nucleation. MTOR (mechanistic target of rapamycin kinase) complex 1 and its substrate, the ULK1 (unc-51 like autophagy activating kinase 1) complex, reside upstream of PtdIns3K and phosphorylate key components of the PtdIns3K complex. The activation of the PtdIns3K lipid kinase results in sequential steps including local phosphatidylinositol-3-phosphate (PtdIns3P) production, recruitment of ATG proteins to a specific phagophore assembly site (in yeast) or omegasome (in mammalian cells) at the endoplasmic reticulum (ER), and membrane nucleation to form a phagophore (a cup-shaped double-membrane structure). Next, the recruited ATG7 and ATG3 promote conjugations of Atg8-family proteins such as MAP1LC3B/LC3B-Ι (microtubule associated protein 1 light chain 3 beta; hereafter referred to as LC3-I) with membrane-residing phosphatidylethanolamine (PE) to generate membrane-bound lipidated LC3 (LC3-I, a characteristic autophagic signature). LC3 and other Atg8-family proteins are essential for gradual phagophore expansion into a structure that extends around a portion of cytoplasm or specific cargos. After nonspecific/specific cargos are recognized and enclosed within a sealed phagophore membrane, a nascent double-membrane vacuole called the autophagosome is generated. Then, the outer membrane of the autophagosome fuses with an endosome (to form an amphisome) and/or a lysosome to form a degradative autolysosome. Finally, the delivered contents in the autophagosome are digested by lysosomal acidic hydrolytic enzymes. Selective autophagy is termed according to the specific cargos engulfed within the sealed autophagosome. For example, autophagy specific for mitochondria is called mitophagy, whereas that for peroxisomes is termed pexophagy, sequestration of invading pathogens is denoted as xenophagy, that of lipid droplets as lipophagy, and so on [Citation2,Citation5,Citation6].
ROS
Reactive oxygen species (ROS) comprise nonradical hydrogen peroxide (H2O2), hydroxyl radical (·OH) and superoxide radical anion (O2−). They can be generated in physiological conditions as by-products of cellular metabolism and in different stressed conditions. It is generally accepted that mitochondrial respiratory chain is the predominant cellular source of ROS, where leaky electrons are readily conjugated with molecular oxygen to form O2− radicals (approximately 1–2% of mitochondrial oxygen consumed) [Citation8]. SOD1 (superoxide dismutase 1) and SOD2, which reside in the mitochondrial intermembrane space and mitochondrial matrix respectively, convert O2− to H2O2. These redox nonradical/radicals react with key cellular macromolecules such as protein, DNA, and lipid by oxidatively modifying their sensitive residues [Citation9]. Mild levels of ROS may act as second messengers in cellular physiological signaling. If subjected to intensive oxidative stress, however, key macromolecules are impaired by irreversible oxidative modifications such as carbonylation (protein), 8-hydroxy-2-deoxyguanosine (DNA) and isoprostane (lipid) [Citation9]. To counteract the harmful effects of ROS, cells are equipped with several antioxidant defense systems.
Some key questions are yet unresolved and warrant future mechanical studies. For example, it is yet unclear which species of ROS is the major messenger to spark autophagy. Chen et al. suggested that O2− is more important because overexpression of SOD2 reduced O2−, increased H2O2 levels and more importantly reduced starvation-induced autophagy [Citation10]. However, most studies presumed H2O2 as the major messenger to deliver oxidative signals [Citation11]. It is mainly because nonradical H2O2, compared to radical O2−, is more stable and long-lived, so that it can affect protein targets far from the intrinsic sources where these redox radicals/non-radicals are generated. But this long-distance signal transduction seems to be nonspecific and susceptible to antioxidant buffering. In this case, a gradient decrease of oxidation or reversely a gradient increase of reduction might be observed surrounding mitochondria and/or peroxisome, the two major bulky ROS-generating organelles. Different species of ROS are not mutually exclusive. They may affect proteins at different accessible and susceptible levels. It may be inferred that if O2− is more important for autophagy the key protein receiver should reside on or relocate to mitochondrial outer membrane. Otherwise, these radicals can be readily neutralized by reducing system. If specific oxidative modifications of certain autophagic proteins are more important, these protein targets relatively away from mitochondrion may have comparatively higher sensitivities to H2O2. Future studies investigating how the susceptibilities of different proteins to oxidative modification regulate autophagy signaling and how cells spatially regulate the autophagic proteins may shed more light on this question.
Since the thought-provoking review by Scherz-Shouval and Elazar one decade ago [Citation11], accumulating results have unraveled the mechanical interplays between autophagy and ROS. In this review, we aim to summarize and interpret recent advances in how ROS mechanically ignite autophagy, and how autophagy dampens ROS-induced damages.
Two major antioxidant defenses against ROS
Proteins and non-protein thiols: the first-line antioxidant defense
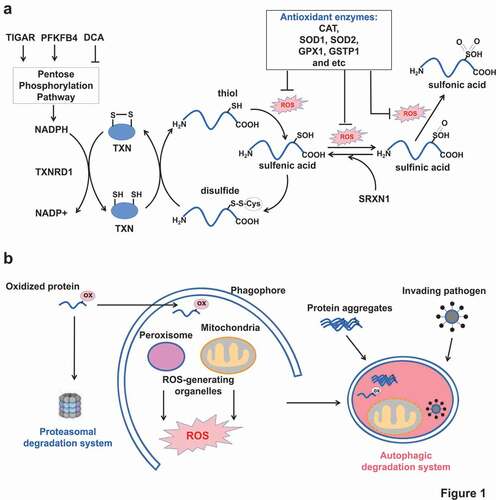
Importantly, it has been estimated that proteins are the major targets (accounting for 50–75%) of reactive oxidants and other radicals in vivo [Citation12]. Protein oxidation commonly occurs at thiol-containing cysteine residues. The protein cysteinyl thiolate anions (P-SH) can be readily oxidized to sulfenic acid (P-SOH) which is very unstable. If not rapidly converted to a disulfide bridge with another cysteine, sulfenic acid can be further oxidized to sulfinic (P-SO2H) and sulfonic acid (P-SO3H). It had been believed that sulfinic acid and sulfonic acid could not be recovered until the discovery of SRXN1 which is able to restore PRDX (peroxiredoxin) from sulfinic acid oxidation [Citation13]. Protein oxidation, if unrepaired, will affect protein conformational structure, protein-protein interaction, and post-translational modifications of nearby residues, all of which leading to functional abnormalities [Citation9].
The reduction of disulfide bridges is generally achieved by small-molecule antioxidant glutathione (GSH) or antioxidant proteins such as TXN (thioredoxin) and GLRX (glutaredoxin). However, their own regenerations are dependent on NADPH availability. Besides, cells are also equipped with antioxidant enzymes including SOD1, SOD2, catalase, glutathione peroxidase, GSTP1 (glutathione S-transferase pi 1) to detoxify ROS [Citation9]. Altogether, these ROS-neutralizing protein thiols, non-protein thiols and detoxifying enzymes constitute the first-line defense against ROS, as summarized in . This function seems to be nonspecific. But some evidence showed that certain antioxidant chaperon proteins could specifically recognize and bind with key autophagy proteins to affect autophagy. More importantly, some key autophagic proteins themselves also contain sensitive cysteinyl thiols [Citation11]. Oxidations on these thiols affect the protein activity and subsequent autophagy from induction to maturation.
Figure 1. Two cellular antioxidant defenses. (A) The upper panel showing protein and non-protein thiols as the first-line antioxidant defense. Increasing concentrations of ROS cause protein cysteinyl oxidation from thiol to sulfenic acid, sulfinic acid and sulfonic acid. If unrepaired, the structure and activity of oxidized protein are affected. However, cells have evolved reducing systems (TXN, TXNRD1 and SRXN1) to recover from sulfenic acid and sulfinic acid. Any impairments of the reducing enzymes or cofactor NADPH production decrease the cellular antioxidant buffering capacity. (B) The below panel showing proteasome and autophagy as the second-line antioxidant defense. Proteasome removes oxidized proteins. Autophagy is responsible for removal of large junks such as protein aggregates, damaged organelles, and invading pathogens, beside oxidized proteins. ox in pink cycle: oxidative modification.
Autophagy and proteasome: the second-line antioxidant defense
The first-line antioxidant defense by protein and nonprotein thiols could neutralize small ROS burst, but it is helpless to handle irreversible oxidations in protein and other macromolecules, nonetheless damaged organelles. Cells have evolved to develop ubiquitin (Ub)-proteasome and autophagy systems as the second-line antioxidant defense, as depicted in . Proteasomes are multi-subunit enzyme complexes that degrade ubiquitin-conjugated proteins [Citation14]. In response to metabolic or environmental ROS, cells increase 26S and 20S proteasome activity [Citation15]. Persistent ROS further facilitate dissociation of 26S proteasome into two subunits: 20S core and 19S regulatory protein, which enables 20S proteasome to degrade oxidized proteins in a Ub- and ATP-independent manner [Citation16]. But extensive oxidative modifications (carbonylation or 4-hydroxy-2-nonenal modification) of the 20S proteasome also cause impaired proteolytic activities [Citation17].
Due to small capacity and limited function, proteasomes, however, are incapable to recycle large protein aggregates, damaged organelles, invading pathogen, and lipid droplets [Citation14]. These large junks must resort to the autophagy-lysosome system (). Autophagy is such a unique system by not only removing oxidized macromolecules but also controlling ROS-generating organelles such as mitochondria. On the one hand, autophagy may behave like an electric resistor by removing existing oxidized proteins or damaged organelles through specific cargo recognition and respective autophagic degradation. On the other hand, upon persistent oxidative stress, cells induce autophagy to restrict ROS production by active removal of the two major ROS-generating organelles mitochondria and peroxisomes.
The finding that mitochondria-associated ER membranes (MAMs) or the ER-mitochondria contact site play a central role in phagosome formation [Citation18,Citation19] strengthened the association of autophagy with ROS-generating organelles. Phagophores are found to be initially formed at the phagophore assembly site (or omegasome) predominantly on the ER and other organelles such as mitochondria, the Golgi complex and the plasma membrane. Mitochondria connect with ER though MAMs to facilitate cross-organelle communications that are necessary for calcium transport, lipid synthesis/transfer, macroautophagy and mitophagy. Upon autophagy induction by amino acid starvation, both ATG14 (a key component of the PtdIns3K complex and a pre-autophagosome marker) and ATG5 (a hallmark protein of autophagy, which binds to the phagophore membrane but then detaches from the membrane after autophagosome formation is complete) translocate to MAMs [Citation18,Citation19]. Notably, disruption of MAMs by knocking down of PACS2 (a cytosolic sorting protein that is crucial for MAMs formation) interrupts ATG14 recruitment and subsequent autophagosome formation. This finding may discover a direct link between ROS-generating mitochondria with autophagy induction. However, whether ROS are involved in this very early stage of autophagy initiation is yet unknown. Exogenous oxidized low-density lipoproteins have been given to vascular smooth muscle cells in vitro to study the involvement of MAMs in mitophagy [Citation20]. This treatment caused increased MAMs abundance and recruitment of BECN1 at MAMs. PACS2 knockdown again impaired mitophagosome formation. However, this study did not address the direct effect of oxidation on MAMs and phagophore formation.
Rheostat effects of protein thiol oxidation to regulate autophagy
It has been observed for decades that many cellular stresses such as nutrient starvation, hypoxia, mitochondrial toxins, and therapeutic agents could induce simultaneously both ROS production and autophagy [Citation21]. Defects in antioxidant proteins contribute to uncontrolled ROS burst and autophagy induction. But replenishment of cellular GSH pool by N-acetyl L-cysteine are sufficient to restore redox balance and inhibit autophagy [Citation21].
As an example of antioxidant proteins, TXN is important for the reduction of protein disulfide oxidation. Thioredoxin and its upstream enzymes () influence protein thiols and autophagy in different levels. First, yeasts with mutated TXN are more sensitive to autophagy induction by MTOR complex 1 inhibitor rapamycin, compared to those wild-type yeasts [Citation22]. TXN mechanically reduces disulfide bonds between Cys338 and Cys394 in yeast ATG4 and impairs autophagosome biogenesis. Second, oxidized TXN could be recovered by TXNRD1 (thioredoxin reductase 1) with cofactor NADPH. Heart specific deletion of mitochondrial Txnrd1 causes mitochondrial degeneration and accumulation of autophagic bodies [Citation23]. Another study showed that deletion of Txnrd1 repressed lysosomal activity in nutritionally starved SH-SY5Y cells, which in turn impaired autophagy maturation and induced apoptosis [Citation24]. Third, deletion of Txnrd1 (thioredoxin interacting protein; an endogenous inhibitor of TXN) prevents high glucose induced mitophagy [Citation25], strengthening the importance of TXN in the regulation of autophagy. Lastly, manipulation of pentose phosphate pathway affects the production of NADPH (the essential cofactor for TXN reduction) and subsequent autophagy induction. Increased NADPH production can be achieved by genetic approaches e.g. upregulation of the upstream promoters TIGAR [Citation26] and PFKFB4 [Citation27]. In contrast, pharmacological agent dichloroacetate can decrease NADPH production, block TXN reduction, and consequently promote autophagy induction [Citation28].
Moreover, many redox-sensitive chaperone-like proteins (such as PRDX1, PRDX2, PRDX6 and GSTP1) were found to reside on the phagophore membrane [Citation29]. PRDX1, PRDX2 and PRDX6 belong to a family of ubiquitous antioxidant proteins. They scavenge H2O2 by sacrificing their own sensitive cysteinyl thiols. The presence of PRDXs thus help maintain a local reducing environment in autophagosome in basal conditions. But when ROS reach to a higher level beyond PRDXs’ maximal buffering capacity, other proteins residing on or translocated to autophagosome membrane become accessible to ROS. Furthermore, it has been also suggested that oxidized PRDX in the form of sulfinic or sulfonic acid may have altered protein structure to facilitate autophagy induction [Citation30].
Taken together, the first-line defense protein thiols behave like rheostats mediated by their own oxidations that regulate the switch between anti-autophagy and pro-autophagy effects. The distinct susceptibilities of ATG3/7 vs. ATG4 to thiol oxidation, as discussed below, may further support this notion. The affected proteins and their relevant functions according to different processes of autophagy will be discussed in later sessions and listed in .
Table 1. Essential autophagy proteins that are susceptible to oxidative stress directly or indirectly
Oxidative regulations of autophagy initiation and phagophore nucleation
Oxidative regulations of the TSC2-MTOR pathway
The oxidative regulation of autophagy was firstly envisaged as an indirect mode by PTEN (phosphatase and tensin homolog) [Citation31]. PTEN, as well as other protein tyrosine phosphatases, contain a catalytic cysteine in a signature motif of HCXXXXXR, which are highly susceptible to ROS [Citation32]. Exogenous (0.1–1 mM H2O2) and endogenous ROS can oxidize PTEN by forming an internal disulfide bond between active-site Cys124 and nearby Cys71 [Citation33,Citation34]. PTEN oxidation thus impairs the lipid phosphatase activity and allows activation of AKT1/PKB (AKT serine/threonine kinase 1) and the downstream signaling pathways including MTOR and PtdIns3K.
Walker’s laboratory [Citation35] reported a direct ROS-dependent pathway to initiate autophagy. They found that cytoplasmic ATM (ATM serine/threonine kinase) may act as a redox-sensitive protein linking ROS with increased PtdIns3K complex activity. This finding is intriguing because ATM is a well-studied cellular sensor of DNA damage upon oxidative stress. Low doses of H2O2 (0.1–0.4 mM) induced sequential reactions including ATM phosphorylation, TSC2 (TSC complex subunit 2) phosphorylation, MTOR repression and consequently autophagy. In the same year, Paull’s laboratory discovered that ATM oxidation at Cys2991 directly induces ATM activation by forming a disulfide cross-linked ATM dimer [Citation36]. This action is independent of DNA double-strand breaks. Moreover, Walker’s laboratory later demonstrated that PEX5 (peroxisomal biogenesis factor 5) recruits both ATM and TSC2 at the peroxisome surface [Citation37]. This proximal protein interaction may facilitate TSC2 phosphorylation by ATM and subsequent autophagy induction. In contrast, commonly-observed cancerous TSC2 mutations could decrease TSC2-PEX5 interaction and disrupt ROS-induced TSC2 phosphorylation. Collectively, these results consistently suggested that ROS could directly activate ATM and its downstream TSC2-MTOR signaling pathway to initiate autophagy.
Some other upstream regulators of MTOR activity have been reported to be redox sensitive and responsible for autophagy induction. For example, SIRT1 (sirtuin 1; an NAD-dependent deacetylase) is redox sensitive [Citation38]. Oxidative stress causes SIRT1 cysteinyl carbonylation (an irreversible form of protein oxidation) on Cys482, which decreases SIRT1 enzymatic activity and protein stability. Importantly, SIRT1-deficient cells are more sensitive to exogenous H2O2 treatment to induce autophagy, compared to the parent cells with wild-type SIRT1 [Citation39]. It was found SIRT1 could increase PtdIns3K and MTOR activity, although the exact mechanisms are unclear.
Converged oxidative regulations of BECN1
Mounting evidence suggested that among all PtdIns3K complex components BECN1 (mammalian homolog of Vps30/Atg6) may exist in a nexus position connecting ROS and autophagy induction. BECN1 is a coiled-coil protein containing a BCL2-homology-3 (BH3) domain, so that this protein was originally discovered as an interacting partner of BCL2. The antiapoptotic protein BCL2 also functions as an autophagy inhibitor through repressive binding with BECN1.
Levine’s laboratory [Citation40] showed that starvation-induced BCL2 phosphorylation (Thr69, Ser70 and Ser87) could release BECN1 from the inhibitory BCL2-BECN1 complex and consequently stimulate autophagy. The authors further demonstrated that the essential kinase to phosphorylate specifically BCL2 was MAPK8/JNK1 (mitogen-activated protein kinase 8; a redox-sensitive stress-activated kinase). It is well reviewed that both ROS and reactive nitrogen species can activate MAPK8 and the upstream MAP3K5/ASK1 (mitogen-activated protein kinase kinase kinase 5) indirectly by oxidations of their inhibitory binding partners GSTP1 and TXN, respectively [Citation41]. Beside direct phosphorylation of MAPK8, MAP3K5 can also activate MAPK8 indirectly through ATF-mediated transcription of DAPK (death associated protein kinase) and consequent phosphorylation of PRKD/PKD (protein kinase D) [Citation42,Citation43]. Because BCL2 binds with either BECN1 or BAX (BCL2 associated X, apoptosis regulator) to repress autophagy and apoptosis, it was thus proposed by Levine’s laboratory that the different levels of MAPK8-mediatd BCL2 phosphorylation may contribute to the dual effects of ROS in autophagy and apoptosis [Citation44]. Transient MAPK8 activation due to short-term and mild stress causes moderate level of BCL2 phosphorylation, which dissociates BCL2-BECN1 complex and activates autophagy. The BCL2-BAX complex is undisrupted, possibly because of their stronger binding affinities. But long-term stress and extensive MAPK8 activation results in complete BCL2 phosphorylation, which impairs the BCL2-BAX complex and finally initiates apoptosis.
The oxidative regulation of BECN1 can also be achieved by interaction with CAV1 (caveolin 1, an integral-membrane scaffold protein in caveolae membranes). In response to a high dose of H2O2 treatment (3 mM for 30 min), CAV1 gets phosphorylational modification at Tyr14, which can recruit BECN1 through its scaffolding domain at the surface of mitochondria and further promote autophagosome formation [Citation45]. Consistently, ectopic expression of PTPN1 (a phosphatase acting on CAV1) reduced CAV1 phosphorylation and repressed autophagy. cav1 knockout mice also have impaired autophagy and aggregated cerebral infarct damage. However, an earlier study by our laboratory showed opposite results. By using mice embryonic fibroblasts with/without CAV1, we demonstrated that CAV1 deficiency promotes both basal and inducible (amino acid deprivation) autophagy through enhanced autophagosome-lysosome fusion and lysosomal function [Citation46]. Disruption of lipid rafts due to loss of CAV1 mediated the observed phenomenon. More importantly, CAV1 downregulation was associated with enhanced autophagy in human breast cancer cells and tissues. It is worthy-noting that these two seemingly-inconsistent studies might reflect dual functions of CAV1 in autophagy: the promoting effect of CAV1 phosphorylation in the early stage of autophagosome formation and the inhibitory effect through lipid rafts in the late stage of autophagosome-lysosome fusion. Whether the inconsistent results are possibly caused by different types and levels of stress (H2O2 vs. amino acid deprivation) are yet unclear.
It is noteworthy that post-translational modifications of BECN1 (phosphorylation, ubiquitination and SUMOylation) directly affect PtdIns3K phagophore-nucleating activity. First, activational phosphorylation of BECN1 is essential for the formation of PtdIns3K complex. Energy starvation induces autophagy through activation of AMPK (AMP-activated protein kinase) and deactivation of its downstream serine/threonine kinase, MTOR. AMPK can also bypass MTOR and directly phosphorylate ULK1 and BECN1 [Citation47]. It has been reported that ROS (H2O2) could oxidize PRKAA1 (protein kinase AMP-activated catalytic subunit alpha 1) on Cys299 and Cys304 residues and thus increase its kinase activity [Citation48]. PRKAA1C299A or PRKAA1C304A mutation reduced H2O2-induced PRKAA1 activation and autophagy. However, this conclusion was challenged by another report revealing that PRKAA1 activation does not resulted from direct oxidation but from impaired ATP production [Citation49].
Second, Lys117 residue of BECN1, which locates in the BH3 domain, is a critical ubiquitination site [Citation50]. Lys117 ubiquitination can disrupt BCL2-BECN1 inhibitory complex and facilitate BECN1-involved autophagy. In the process of inflammation associated autophagy, TRAF6 (TNF receptor associated factor 6, an E3 ubiquitin ligase) binds to BECN1 through two TRAF6-binding motifs in BECN1 and then executes Lys117 ubiquitination. The molecular mechanism of how oxidative stress affects TRAF6-mediated BECN1 ubiquitination was recently revealed [Citation51]. In unstressed condition, redox-sensitive PRDX1 interacts with TRAF6 through a ring finger domain. This inhibitory binding abolishes the E3 ubiquitin ligase activity of TRAF6. ROS could abolish TRAF6-PRDX1 complex and then release TRAF6 to promote BECN1 ubiquitination. The resultant Lys117 ubiquitination on BECN1 in turn releases BECN1 from the inhibitory BCL2-BECN1 complex and consequently promotes autophagy. Consistently, in cells silenced with PRDX1, there were increased levels of ROS, BECN1 ubiquitination and autophagy.
Lastly, SUMOylation (a type of post-translational modification like ubiquitination) is achieved by protein conjugation of a SUMO (small ubiquitin like modifier) on lysine residues. This process may affect several cellular functions. A recent paper by Liu and colleagues showed that starvation increased BECN1 SUMOylation at Lys380, which in turn promoted the formation of PtdIns3K complex and its catalytic activity [Citation52]. The authors demonstrated that the mutant BECN1K380R had weakened interactions with UVRAG and other PtdIns3K complex components. Furthermore, Lys380 SUMOylation on BECN1 was found to be removed by SENP3 (SUMO specific peptidase 3). SENP3 was reported as a redox-sensitive protein in an earlier study by the same research group [Citation53]. Upon starvation or mild oxidative stress (H2O2 from 0.05 to 0.2 mM), SENP3 was oxidized at two cysteine residues (Cys243 and Cys274). These oxidative modifications enable SENP3 to bind with HSP90AA1 and co-chaperone/ubiquitin ligase STUB1/CHIP, which may increase SENP3 protein stability. Therefore, mild ROS may facilitate BECN1 deSUMOylation and consequently impair BECN1-involved autophagy.
Oxidative regulation of PtdIns3K
Like BECN1, another PtdIns3K complex component PtdIns3K gets activational phosphorylation through PRKD/PKD [Citation54]. It was reported that upon exposure to H2O2 treatment, DAPK, a calcium/calmodulin-regulated serine/threonine kinase, initiated PRKD/PKD phosphorylation and subsequent PtdIns3K phosphorylation, PtdIns3P formation and autophagy initiation [Citation54].
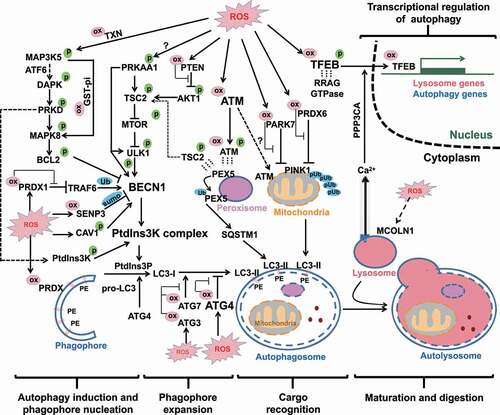
Taken together, ROS deliver autophagy-initiating signals that are converged on the PtdIns3K complex particularly BECN1, through different modes including protein-protein interactions (with BCL2 and CAV1), phosphorylation (by upstream TSC2-MTOR and possibly PRKAA1), ubiquitination (by TRAF6), and deSUMOylation (by SENP3). The relevant oxidative regulations of autophagy initiation and phagophore nucleation are summarized in the left panel of .
Figure 2. Full-coverage oxidative regulations of autophagy. All the four processes of autophagy and its transcriptional regulation are regulated by ROS. In the first process of autophagy induction and phagophore nucleation, the oxidative regulations converge on BECN1 and PtdIns3K activities through post-translational modifications and protein-protein interactions. In the second process of phagophore expansion, mild level of ROS induces ATG3 and ATG7 oxidation (but spares ATG4), contributing to inhibition of LC3 lipidation. In contrast, higher levels of ROS result in ATG4 oxidation and inhibition of its LC3-deconjugation function, which consequently allows phagophore expansion. In the third process of cargo recognition, ATM oxidation is responsible for mitophagy through unclear mechanism. Chaperone-like proteins PRDX6 and PARK7 take part in the repression of mitophagy, so that inhibitory oxidations of these two proteins are necessary for the process of mitophagy. In pexophagy, ATM-mediated PEX5 phosphorylation facilitates PEX5 ubiquitination and consequent cargo recognition by SQSTM1. In the last process of autophagy maturation, only limited evidence suggested that lysosomal CTSL is sensitive to ROS. Moreover, the TFEB-mediated transcriptions of a battery of lysosomal and autophagic genes are also regulated by ROS. Direct cysteinyl oxidation on TFEB can facilitate its nuclear translocation and transcriptional activity. Another indirect modulation pathway by ROS is achieved sequentially by MCOLN1-mediated lysosomal Ca2+ release, PPP3-mediated TFEB dephosphorylation and nuclear translocation. ox in pink cycle: oxidative modification; p in green cycle: phosphorylation modification; pUb in blue cycle: phosphorylated ubiquitin chain; sumo in blue cycle: SUMOylation; Ub in blue cycle: ubiquitin modification; dashed short lines represent protein-protein interaction.
Phagophore expansion: differential oxidative regulations of ATG4, ATG3 and ATG7
After autophagy initiation, the nucleated phagophore expands and then engulfs cargos within an enclosed double-membrane structure. The process of phagophore expansion is also regulated by ROS. Among all ATG family members, ATG4 was the first identified protein susceptible to oxidation [Citation55]. There are four members in the ATG4 family: ATG4A, ATG4B, ATG4C and ATG4D. ATG4 proteins have two major functions [Citation2]. First, the cysteine protease activity of ATG4 enables it to process nascent pro-Atg8-family proteins at the C terminus and expose a conserved glycine residue that is necessary for conjugation of Atg8-family proteins with membrane-residing PE. Second, ATG4 limits phagophore expansion by deconjugating Atg8-family proteins from membranal PE to recycle this protein. ATG4B recognizes all Atg8-family proteins, including MAP1LC3/LC3, GABARAP and GABARAPL2/GATE-16. ATG4A is more specific to recognize GABARAP.
Oxidative regulation of ATG4
Elazar’s laboratory [Citation55] firstly found that starvation-induced ROS (particularly H2O2) or exogenous H2O2 treatment (1 mM) decreased ATG4 deconjugating activity, but not its protease activity. This ROS-dependent reduction in ATG4 deconjugating activity promoted autophagosome formation in CHO cells and HeLa cells. Mutations of the conserved redox-sensitive Cys residues (Cys81 of ATG4A and Cys78 of ATG4B) to Ser significantly reduced LC3 lipidation and autophagosome formation. The authors thus concluded that the inhibitory oxidation of ATG4 promoted autophagy. A recent study by Münz’s group [Citation56] confirmed that ATG4B-dependet delipidation was repressed by Cys78 oxidation during the processes of LC3-acssociated phagocytosis in human macrophage. They further demonstrated NOX2 (which resides in the phagosome membrane) was responsible for ROS production, ATG4B oxidation and consequent ATG4B aggregation. This mechanism may prolong MHC class II restricted antigen presentation.
Moreover, a recent study by Li group concurred the stimulatory effects of ATG4B oxidation on autophagy [Citation57]. However, this study demonstrated that two other cysteine residues Cys292 and Cys361, rather than the previously reported Cys78 [Citation55,Citation56], were essential for the oxidative regulation of ATG4B in HEK293 and HeLa cells. Upon H2O2 oxidation (1 mM), ATG4B becomes inactive oligomers by forming intermolecular disulfide bonds between Cys292 and Cys361. Double mutations of ATG4BC292S,C361S inhibited ATG4B deconjugating activity and promoted autophagic flux. But ATG4BC78S mutation failed to affect in the same condition. Similarly, another study conducted in yeast found that ATG4 was oxidized by forming a disulfide bond between Cys338 and Cys394 [Citation22]. The presence of TXN efficiently protected ATG4 from oxidation by reducing the disulfide bond. Consistently, TXNIP, an inhibitor of TXN antioxidative activity, was found to play an opposite function by promoting LC3 lipidation and autophagy [Citation58]. Stress conditions could up-regulate TXNIP and another MTOR inhibitor REDD1, which in turn suppress ATG4-mediated deconjugating activity and induce LC3 lipidation and consequently autophagy.
Oxidative regulations of ATG3 and ATG7
Surprisingly, the stimulatory role of ROS on autophagy through ATG4 oxidation was challenged by the findings of Burgoyne’s laboratory [Citation59]. The authors reported that ROS impaired autophagy through oxidations of ATG7 (Cys572) and ATG3 (Cys264), but not ATG4 in HEK293 and SMC cells. At basal conditions, ATG7 and ATG3 are covalently conjugated with their substrate ATG8 [Citation2]. Upon autophagy initiation, the ATG4-processed ATG8 is further activated by E1-like enzyme ATG7, and then conjugated with membrane-resident PE through E2-like enzyme ATG3. The unbound ATG7 and ATG3 are susceptible to inhibitory oxidation on Cys572 and Cys264 respectively, which prevent ATG8 lipidation and autophagosome maturation [Citation59]. Impairment of the reducing activity of TXN by overexpressing its endogenous inhibitor TXNIP could inhibit LC3 lipidation. The results that increased ATG7 oxidation was accompanied with decreased LC3 lipidation in aged mouse aorta further substantiated the inhibitory roles of ATG7/ATG3 oxidation on autophagy.
The distinctive effects of ATG4 oxidation vs. ATG3/7 oxidation
It is noteworthy that the disparity of the above two conclusions might be resulted from two major differences in the systems applied to induce autophagy and in the levels of ROS. The two studies conducted on ATG4 [Citation55,Citation57] used higher doses of H2O2 (0.5 and 1 mM) to induce autophagy in HEK293 cells. In contrast, the study on ATG3/ATG7 [Citation59] used EBSS to starve the same cell line HEK293 to induce autophagy. The role of oxidation on ATG activity and autophagy was explored by addition of low dose of H2O2 (0.01 ~ 0.2 mM). Burgoyne’s group [Citation59] further determined cysteinyl oxidations of ATG3, ATG4B and ATG7 through a mobility shift coupled with a PEG-switch assay. In the untreated or EBSS-starved conditions, there were low levels of ATG3 and ATG7 oxidation (around 20% and 10% respectively) and undetectable ATG4 oxidation. Addition of 0.1 mM H2O2 in the presence of EBSS treatment significantly increased ATG3 and ATG7 oxidation (as high as 70% and 30% respectively). Higher doses of H2O2 (0.2 and 0.5 mM) cannot further increase ATG3 and ATG7 oxidation, suggesting that 0.1 mM H2O2 in the presence of EBSS already reached the plateau level of ATG3/7 oxidation. In contrast, the same treatments of H2O2 (0.2 and 0.5 mM) dose-dependently increased ATG4B oxidation. Thus, it is conceivable that ATG3/7 and ATG4B may have different levels of susceptibility to oxidation. ATG3 and ATG7 may be more redox sensitive than ATG4. The higher sensitivity of ATG3/ATG7 to oxidation allows cells to repress autophagy induction and then maintain low levels of ROS as signaling molecules. But when ROS reach to a higher level to oxidize ATG4 which is comparatively less redox-sensitive, ATG4 oxidation becomes dominant and in turn promotes LC3 lipidation and phagophore expansion. This switch () may promote autophagy induction to restrict the harmful level of ROS. However, this speculation needs to be tested in future studies.
Oxidative regulations of selective autophagic cargo recognition
Because mitochondrion and peroxisome are the two major ROS-generating organelles, cells may resort to an additional control measure to restrict upstream ROS sources through autophagy-dependent recycling of these two organelles, besides continuous removal of downstream oxidized/damaged proteins and organelles. Although there are increasing understandings of how ROS promote phagophore nucleation and expansion, the mechanisms of how ROS label damaged or superfluous mitochondrion/peroxisome and then how autophagy machinery recognizes these oxidized cargos are yet less studied. NOX (NADPH oxidase), as another cellular ROS source in generating superoxide across the membranes of neutrophils and phagosomes, is more important for the activation of xenophagy [Citation60].
Oxidative regulation of mitophagy
By taking up glucose and oxygen, mitochondrion is the powerhouse of energy production in all eukaryotic cells. This activity is achieved by electron transfer consecutively through five electron transport chain (ETC) complexes of oxidative phosphorylation. Maintenance of proton gradients across mitochondrial membranes is thus important for energy production. However, accidental escapes of electrons from ETC complex Ι or lead to ROS formation. Upon metabolic stress or treatment of mitochondrial poison, mitochondria experience loss of mitochondrial membrane potential (MMP) and a burst of ROS production [Citation61,Citation62]. As a result, mitochondria become the major source of cellular ROS production. In order to maintain mitochondria quality and quantity, cells have evolved a quality control system called mitophagy [Citation63]. Upon impairment of MMP, mitochondrial kinase PINK1 (PTEN induced kinase 1) gets auto-phosphorylation and protein stabilization [Citation64,Citation65]. It then moves to the outer mitochondrial membrane surface and recruits a cellular E3 ubiquitin ligase PRKN/parkin (parkin RBR E3 ubiquitin protein ligase). These two proteins cooperate to form a feedforward loop and generate phosphorylated Ub (pUb) chains on several proteins at the outer mitochondrial membrane surface. The pUB chains function as “eat-me” signals to recruit cargo receptors such as SQSTM1, OPTN and CALCOCO2/NDP52 etc. Besides the well-studied PINK1-PRKN pathway, mitophagy also occurs in PRKN-independent or Ub-independent signaling manners through direct interactions of phagophore-residing LC3 with several mitochondrial proteins, such as BNIP3, BNIP3L/NIX and FUNDC1 [Citation2,Citation66].
Like the aforementioned upstream autophagic steps, cargo recognition of mitophagy is also affected by ROS (). Park’s group reported that ROS play a critical role in PINK1-dependent PRKN translocation in mouse cortical neurons and in embryonic fibroblasts [Citation67]. Expression of wild-type PARK7/DJ-1 (Parkinsonism associated deglycase, a redox-sensitive chaperone), but not the redox-resistant mutant PARK7C106A, reduced PRKN recruitment. PRDX6 is another redox-sensitive protein found to be recruited to the depolarized mitochondria in a ROS-dependent manner [Citation68]. This translocation can decrease PINK1 stability and LC3 lipidation. However, how mitophagy specifically recruits these two proteins to the damaged mitochondria is not known. More importantly, it is yet to be resolved whether these two mitochondria-recruited proteins could neutralize unspecifically mitochondrial ROS or inhibit specifically certain mitophagy proteins.
Furthermore, Kastan’s laboratory found that ATM was responsible for mitophagy and the maintenance of mitochondrial homeostasis [Citation69]. They draw this conclusion based on two observations. First, a fraction of ATM resides on mitochondria and gets activated upon mitochondrial depolarization after carbonyl cyanide m-chlorophenylhydrazone (CCCP) treatment. Second, ATM-deficient thymocytes have impaired mitophagy and increased oxidative stress, but they still maintain efficient macroautophagy. Although ATM had been reported as a redox sensitive protein [Citation36], the authors did not explore the connections between ROS and activation of mitochondrial-residing ATM and subsequent mitophagy. Moreover, it is yet unknow whether ATM oxidation (e.g. Cys2991) affects mitophagy and whether there is any mitochondrial protein phosphorylated by ATM to act as “eat-me” signal in mitophagy.
Oxidative regulation of pexophagy
Peroxisome owes the name because of its activities in generation and decomposition of hydrogen peroxide. It plays key roles in catabolism of very-long-chain fatty acids via beta-oxidation, and phytanic acid via alpha-oxidation. These metabolic processes generate large amount of H2O2 as toxic by-products. But peroxisome also contains H2O2-decomposing enzymes such as catalase that converts H2O2 to water and oxygen. Walker’s laboratory found that ATM played an important role in cargo recognition in pexophagy (peroxisome autophagy) in response to ROS [Citation70]. They further identified PEX5 (a peroxisomal import receptor) as the specific peroxisomal “eat-me” signal. After oxidants (0.4 mM H2O2) treatment, PEX5 recruits ATM to the peroxisome surface where ATM gets activated by peroxisomal ROS. The activated ATM have two roles at peroxisomal surface. First, it sparks autophagy machinery by phosphorylating TSC2 and repressing MTOR [Citation35]. Second, it phosphorylates PEX5 at Ser141 which in turn facilitates mono-ubiquitination of PEX5 at Lys209 [Citation70]. The “eat-me” signal of ubiquitinated PEX5 is then recognized by autophagy receptor protein SQSTM1, so that the bulky peroxisome is specifically engulfed in LC3-enriched autophagosome.
Oxidative regulation of lipophagy
Lipophagy (a specific form of autophagy to digest lipid droplets) might be also regulated by ROS. It was found that PRDX1 deficiency caused oxidative stress and impairment of lipophagy-mediated cholesterol hydrolysis in macrophage [Citation71]. Treatment of 2-Cys PRDX-mimics ebselen and gliotoxin could restore lipophagy and cholesterol efflux. It is yet unresolved whether PRDX1 plays a specific role in lipophagy or just neutralizes unspecifically ROS and then affects different forms of autophagy.
Feedback loops between mitochondria/peroxisomes and autophagy
From birth to death, life is characterized by constant exchange of materials between life body and environment, and by maintaining anabolic and catabolic metabolism. Among intracellular organelles, mitochondrion and peroxisome are actively involved in glucose, amino acid, purine, and fatty acid metabolism [Citation62]. As a result, ROS are endogenously generated by many oxidases as metabolic by-products within these two organelles. Moreover, the discovery of mitochondria-dependent apoptosis resulted from mitochondrial depolarization and release of cytochrome C (an ETC complex component) demonstrates that ROS-involved metabolism can crosstalk with signaling pathways [Citation72].
ROS induce mitophagy and pexophagy. In turn, autophagy digests ROS-generating mitochondria/peroxisomes, forming a negative feedback loop. On the one hand, in the nutrition-rich condition, both glucose and amino acid metabolism activate MTOR and suppress autophagy, as well-reviewed elsewhere [Citation73]. However, starved cells experience sudden energetic stress and mitochondrial overburden, resulting in depolarization-mediated electron leakage and ROS overproduction [Citation74]. The oxidative regulations of autophagy initiation and phagophore expansion have been described above, but how ROS label damaged mitochondrial/peroxisome is comparatively less studied. ATM, which resides on mitochondrial membrane [Citation69] or translocates to peroxisomal membrane [Citation70], participates in ROS-involved autophagic recognition. Upon mitochondrial depolarization, PINK1 gets stabilized and relocates to mitochondrial outer membrane, which facilitates mitophagy recognition [Citation64,Citation65]. Whether ROS play a role in or just accompany PINK1 stabilization/relocation after mitochondrial depolarization is yet unclear. Another candidate protein to connect mitochondria with autophagy could be HK2 (hexokinase 2; a mitochondrial enzyme responsible for the first step of glycolysis). Miyamoto’s laboratory found that glucose starvation caused inhibitory binding of HK2 with MTOR and subsequent autophagy induction [Citation75,Citation76]. This interaction was mediated through an MTOR signaling (TOS) motif which is critical for MTOR binding to raptor and subsequent substrate phosphorylation. Glucose starvation, particularly the decrease of HK2-mediated glucose-6-phosphate, further disrupt ROS homeostasis by blocking NADPH production in the pentose phosphate pathway [Citation77].
On the other hand, autophagy may support essential mitochondrial function and digest damaged mitochondrion/peroxisome. First, autophagy is a recycling process by breaking down nonessential protein, lipid, and organelle. During metabolic deprivation, this action may donor mitochondria with catabolized amino acids and fatty acids to prevent energy crisis and meet survival needs [Citation1,Citation2]. Second, autophagy eliminates ROS overproduction by removing damaged mitochondria and peroxisomes. Consistently, cells with impaired macroautophagy or selective autophagy (e.g., Atg5 or Pink1 deletion) accumulate dysfunctional mitochondria and ROS levels that would be normally removed and recycled [Citation70,Citation78,Citation79]. Thus, cells have evolved to develop a fine mechanism of feedback loop regulation by which mitochondria and autophagy together maintain a balanced energetic/metabolic supply and ROS homeostasis.
Oxidative regulations of autophagosome maturation and autophagy gene transcription
Oxidative regulations of lysosomal cathepsins
After successful cargo engulfment and phagophore sealing, the autophagy machinery reaches to its final step autophagy maturation. At this step, the outer membrane of autophagosome fuses with lysosomal membrane to form digestive autolysosome. There are only limited evidence suggesting that this process might be regulated by ROS. It has been reported that lysosomal protease cathepsins had different susceptibilities to ROS [Citation80]. CTSL (cathepsin L) that is involved in autophagic digestion decreased its hydrolytic function in response to auranofin-induced ROS or exogenous H2O2 treatment (0.5 mM); while CTSB (cathepsin B) that is involved in apoptosis was not affected. Overall, auranofin-induced ROS inhibited autophagy but increased lysosomal leakage of CTSB into cytoplasm and consequently induced apoptotic cell death. Whether ROS-involved CTSL deactivation affect autophagic digestion in different stress conditions or in different cell systems are yet to be confirmed.
Autophagy gene transcription: oxidative regulations of TFEB
TFEB (transcription factor EB) is a master transcription factor of a gene network involved in autophagy and lysosome biogenesis [Citation2]. In nutrient-rich conditions, RRAG GTPase recruits both TFEB and the kinase MTOR to lysosomal surface [Citation81]. It is where TFEB is phosphorylated (mainly at Ser211) by MTOR. The phosphorylated TFEB is in turn sequestered in cytosol in an inhibitory binding complex with chaperone YWHA/14-3-3. Upon MTOR gets inactivated in nutrient-poor starvation, TFEB dissociates from the TFEB-YWHA complex, translocates to nuclei and then activates autophagy and lysosome biogenesis.
Wang and colleagues recently reported a direct oxidative regulatory mechanism of TFEB [Citation82]. TFEB and family members TFE3, MITF are redox-sensitive proteins. Within a few minutes’ exposure to H2O2 (1 mM), TFEB is oxidized at a conserved Cys212 residue which can abolish its interaction with RRAG GTPase. This oxidation enables TFEB to translocate to nuclei for active transcription of a battery of autophagic (e.g. ATG9B, ATG16L1 and UVRAG) and lysosomal genes (e.g., ATP6V0D2, ATP6V0E1, ATP6V1H, CTSA, CTSF, etc). Consistently, single mutation of TFEBC212A abolishes TFEB oxidation, and in turn maintains its interaction with RRAG GTPase and cytosol localization. TFE3 and MITF with redox-sensitive cysteines (Cys322 and Cys281, respectively) have similar results. Note that TFEB Cys212 oxidation does not affect the neighboring Ser211 phosphorylation by MTOR complex 1. The authors proposed that Cys212 oxidation and Ser211 phosphorylation may cooperate to regulate TFEB. Triple knockdown of TFEB, TFE3 and MITF in HEK293 cells further confirmed that they are essential for ROS-induced autophagy/lysosome gene transcriptions. This study well established a direct transcriptional link between ROS and autophagy.
An earlier study proposed another regulatory model of ROS to indirectly activate TFEB [Citation7]. Treatments with mitochondrial poison CCCP or exogenous oxidants increased mitochondrial ROS and induced lysosomal Ca2+ release through activation of MCOLN1 (mucolipin 1; a lysosomal ion channel protein that belongs to a transient receptor potential channel superfamily). After binding with Ca2+, PPP3CA/calcineurin (protein phosphatase 3 catalytic subunit alpha) gets activated and then dephosphorylates TFEB at Ser211. The dephosphorylated TFEB, in turn, translocated to nuclei and initiated lysosomal biogenesis. Either genetic deletion of Mcoln1 or treatment with pharmacological inhibitors (ML-SI3 for MCOLN1 or BAPTA-AM for calcium chelation) can block ROS-induced autophagy, suggesting the essential roles of MCOLN1 and Ca2+ release. However, how MCOLN1 is activated by ROS is not yet unraveled. Wang et al tested the involvement of MCOLN1 [Citation82], but they found that the rapid H2O2-induced TFEB activation was independent on MCOLN1 and PPP3CA. It is worthy-noting that the earlier study [Citation7] demonstrated that MCOLN1 was activated in a whole endolysosome electrophysiological experiment by higher concentrations of H2O2 (1–10 mM). Future studies are thus needed to solve this inconsistency.
Taken together, ROS can activate TFEB directly through Cys212 oxidation or indirectly through MCOLN1 -mediated lysosomal Ca2+ release and PPP3CA-mediated TFEB dephosphorylation (). Except TFEB and family members, there are yet limited mechanical evidence to connect ROS with other known lysosome/autophagy transcription factors including FOXO1, EGR1, FXR.
SQSTM1: standing at the crossroad of autophagy and antioxidant gene transcription
SQSTM1/p62 (sequestosome 1) is a Ub-binding protein. It is a multifunctional protein that plays different roles in autophagy and antioxidant gene transcription. In autophagy, SQSTM1 acts as an autophagy receptor protein by interacting with phagophore-residing LC3 to enable autophagic cargo recognition [Citation2]. Since SQSTM1 itself is degraded by lysosomal hydrolytic enzymes, its degradation is commonly used as a signature marker to study autophagic flux. The antioxidant effect of SQSTM1 is mainly achieved by its activational effects on NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) and NFKB1 (nuclear factor kappa B subunit 1) [Citation83].
First, NFE2L2, as a basic region leucine-zipper transcription factor, is a master regulator of antioxidant gene transcription. In nuclei, it binds to antioxidant response elements (ARE) and in turn transcribe a battery of genes involved in antioxidant and anti-inflammatory defenses [Citation84]. But at unstressed conditions, NRF2’s transcriptional activity is constitutively repressed by inhibitory interaction with KEAP1 (kelch like ECH associated protein 1). KEAP1 further recruits CUL3 (cullin 3) E3 ubiquitin ligase to form a complex to ubiquitinate NFE2L2 and enables NFE2L2 degradation. In response to oxidative or electrophilic stress, KEAP1 is oxidized at its cysteine residues (Cys273 and Cys288). These oxidations however repress KEAP1 E3 ligase activity that consequently contribute to NFE2L2 stabilization and transcriptional activation of antioxidant genes. Besides the classical ROS-dependent KEAP1 inactivation pathway, Yamamoto’s group found that SQSTM1 can release NFE2L2 by competitive interaction with KEAP1 at the NFE2L2-binding site [Citation85]. Both enforced SQSTM1 over-production or impaired SQSTM1 autophagic degradation contribute to its cellular accumulation, which in turn hyperactivate NFE2L2-mediated transcriptions of antioxidant genes. Moreover, Yamamoto and colleagues later in another study reported that MTOR-involved SQSTM1 phosphorylation (Ser351) could further enhance SQSTM1 binding affinity with KEAP1 [Citation86].
Second, Duran and coauthors reported another SQSTM1-NFKB1 antioxidant gene transcription pathway [Citation87]. They demonstrated that oncogene Ras could induce SQSTM1 in human cancers. Importantly, SQSTM1-mediated NFKB1 activation neutralizes Ras-induced ROS production and support tumor cell survival. Mechanically, SQSTM1 triggers IκB kinase through TRAF6 polyubiquitination and activation. In contrast, transgenic mice with SQSTM1 deficiency are protected from Ras-induced lung adenocarcinoma because tumor cells are particularly susceptible to oxidative stress induced cell death.
Taken together, SQSTM1 takes part in antioxidant defenses in either autophagy -dependent or -independent manners, a kind of switch effect (). In cells with functional autophagy, autophagy machinery utilizes SQSTM1 to recognize phagophore-residing LC3 to recycle damaged (oxidized) proteins and organelles. SQSTM1 itself is degraded with the delivered cargos to decrease its cellular abundancy. If both protein thiols and autophagy antioxidant defenses failed to restrict ROS, SQSTM1 provides an additional mechanism to tame ROS by activating NFE2L2-dependent or NFKB1-dependent antioxidant gene transcription. Unfortunately, the latter pathway is hijacked by Ras-induced oncogenesis to sustain tumor cell survival from ROS-induced cell death and consequently promote unrestricted cell proliferation.
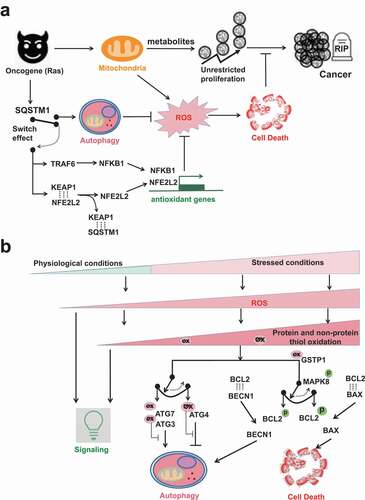
Figure 3. The ROS involved switch between autophagy and cell death. (A) SQSTM1 provides an alternative antioxidant pathway by functioning like an electric switch to connect either autophagy or NFE2L2-dependent antioxidant gene transcription. When autophagy is functional, SQSTM1 is a bona fide autophagic receptor protein. SQSTM1 itself is also recycled by autophagy. But when autophagy is impaired, SQSTM1 aggregates in cells and then switches to turn on NFE2L2- or NFKB1-dependent antioxidant gene transcriptions. Ras-driven oncogenesis over-expresses SQSTM1. The cancer cells in turn hijack SQSTM1 to activate either autophagy or antioxidant gene transcriptions to tame ROS, so that their unrestricted cancer cell proliferation will not be interfered by ROS-induced cell death. (B) ROS exist at different levels from physiological to pathological/stressed conditions. At low levels, ROS act as signaling messengers. Higher levels of ROS are restricted by the first-line protein thiols system and the second-line autophagy to maintain redox homeostasis. The rheostat effects of protein thiols in the regulation of autophagy inhibition/induction and in the regulation of autophagy/apoptosis are shown by two examples. The first switch is mediated by different susceptibilities of ATG3/7 vs. ATG4 to oxidation. ATG3/7 are more sensitive than ATG4 to oxidation, so that low levels of ROS oxidize ATG3/7 but spare ATG4. The second switch is mediated by different BCL2 complexes formation. MAPK8 phosphorylates BCL2 and thus interfere the association of BCL2 with binding partners. Notably, MAPK8 activation is affected by inhibitory binding with antioxidant GSTP1. Comparatively, the association of BCL2-BECN1 complex is weaker than that of BCL2-BAX complex. Thus, low levels of stress and GSTP1 oxidation cause transient MAPK8 activation and dissociation of BCL2-BECN1 complex but not BCL2-BAX complex. Dissociated BECN1 then turn on autophagy. But sustained MAPK8 activation frees BAX from the inhibitory complex BCL2-BAX and switches on apoptosis.
The interplays among ROS, protein thiol oxidation, autophagy and cell death
With increasing concentrations of ROS in stressed conditions, there are tilted balance toward oxidative stress and eventually cell death (). Protein and non-protein thiols are the first-line antioxidant defense to neutralize low level of ROS. Thiol oxidations of the very susceptible autophagic proteins like ATG3, ATG7 and SENP3 may keep autophagy inactivated and allow ROS to execute their signaling tasks. But under stress by moderate levels of ROS, less susceptible autophagy-involved proteins like ATG4, ATM and TFEB are affected, so that autophagy is activated to remove oxidized proteins/organelles and restrict further ROS generation from mitochondria and peroxisome sources. The rheostatic effects of protein thiols could be shown by two examples. First, differential susceptibilities to protein thiol oxidations may mediate the switch between the anti-phagophore expansion effect of ATG3 and ATG7 oxidation [Citation59] and the pro-phagophore expansion effect of ATG4 oxidation [Citation55,Citation57] (). ATG3 and ATG7 have higher susceptibility to thiol oxidation so that the autophagy-inhibiting effects of their oxidation allow ROS to further deliver messenger signals. But increased levels of ROS oxidize the relatively resistant ATG4 and flip the switch toward autophagy induction. In turn, autophagy machinery clears oxidized proteins and bulky ROS-generating mitochondria and peroxisome. Second, as suggested by Levine’s group [Citation44], different BCL2 complexes formation may mediate the switch between autophagy and apoptosis (). This action is indirectly affected by GSTP1 thiol oxidation and then MAPK8 phosphorylational activation. Low levels of stress oxidize GSTP1 and cause transient MAPK8 activation. The latter phosphorylates BCL2, which may dissociate BECN1 from the inhibitory complex BCL2-BECN1 and consequently allow PtdIns3K activation and autophagy induction. Comparatively, the binding of BCL2-BAX complex is much tighter. However, due to long-term and higher levels of stress and GSTP1 oxidation, sustained MAPK8 activation further phosphorylate BCL2. In this case, BAX dissociates from the inhibitory complex BCL2-BAX and then switches on apoptosis.
The interplay between autophagy and apoptosis has been well-reviewed elsewhere [Citation88,Citation89]. Autophagosome is often observed in dying cells. In most circumstances, autophagy precedes cell death, playing a rheostatic effect to limit stress and recycle damaged cellular components. It is yet a debate whether autophagy results in or just accompany cell death [Citation89]. The accumulation of autophagic vacuolization in cell death may be resulted from defects in autophagosome maturation [Citation90]. When cells succumb to apoptotic cell death, activated caspases can even cleave essential autophagic proteins (e.g. BECN1, ATG4D and ATG16L1) and consequently inactivate autophagy [Citation91]. BCL2L11/BIM (a proapoptotic BH3-only protein) also binds to BECN1 and sequesters it on microtubule, resulting in autophagy inhibition [Citation92]. However, consumption of indispensable cellular components by autophagy facilitates apoptotic or necrotic cell death [Citation89,Citation93]. Experimentally, excessive autophagy induced by genetic models did increase apoptotic cell death, which could be prevented by knocking-down of ATG1 or ATG5 [Citation94–96]. This is regarded as autophagic (or autophagy-mediated) cell death, a separate form of programmed cell death. Levine’s group further found a type of autophagy-dependent cell death (autosis) that was dependent on Na+, K+-ATPase activity [Citation97]. The Nomenclature Committee on Cell Death (NCDD) recently suggested that autophagic cell death, distinct from apoptosis and necrosis, could be mechanically repressed through genetic and pharmacological inhibitions of the autophagy signaling pathway, rather than inhibitions of other types of programmed cell death [Citation98]. Besides, autophagic digestion of FTL (ferritin light chain) and/or FTH may release free iron, which facilitates lipid peroxidation and consequent ferroptotic cell death [Citation99–101]. This specific autophagic process is called ferritinophagy, further supporting the causative role of autophagy in cell death at least in certain circumstances.
ROS overproduction have been linked to immunity, neurodegenerative diseases, metabolic diseases, cancer, premature aging, and many other types of pathologies, besides cell death. The rheostatic effects of protein thiols also exist in these pathological systems to establish redox/reduction homeostasis. Beyond the buffering capacities of protein thiols and autophagy, excessive ROS cause reversible or irreversible oxidative damages and consequent pathological changes.
Concluding remarks and perspectives
ROS exist ubiquitously and incessantly in cells. At physiological conditions, low levels of ROS act as signaling messengers. As illustrated in , almost all processes of autophagy from autophagy induction till autophagy maturation are affected by ROS. On the one hand, these actions can be executed through direct cysteinyl thiol oxidations of key autophagic proteins including ATG4 and ATM for phagophore expansion, ATM for cargo recognition, and TFEB for autophagic and lysosomal gene transcription. On the other hand, some redox-sensitive chaperone-like proteins (e.g. PARK7, GSTP1, PRDX1, PRDX6) may indirectly affect activities of key autophagic proteins by altering their post-translational modifications and/or protein-protein interactions. In general, a majority of results suggest that ROS promote autophagy execution. But some did support the opposite direction, e.g. oxidations of ATG3, ATG7 and SENP3 exhibiting inhibitory functions on autophagy. It is yet to be confirmed whether these two autophagy-promoting and autophagy-inhibiting effects by ROS are resulted from different doses of ROS or from different genetic backgrounds of redox homeostasis systems.
Some other open questions await future investigations. First, as aforementioned it is yet unclear which species of ROS (nonradical H2O2 or radical O2−) is more important to ignite autophagy? Future studies using specific oxidation indicators may help trace the source and movement of different oxidation species. Spatial investigations of protein oxidation and their basal susceptibilities will help generate cellular geographic information system to define the critical autophagic proteins to spark and boost autophagy machinery. Second, whether ROS-induced autophagy is initiated from the ER-mitochondria or ER-peroxisome contact site? First come, first served, simply because mitochondria and peroxisome are the major cellular ROS sources. Laser directed mitochondria damages with the aid of phototoxic fluorescent protein (e.g., KillerRed conjugated with mitochondrial protein [Citation102]) can cause local mitochondrial ROS production. This technique may shed some light on the spatial mechanisms involved. Third, the molecular mechanisms of ROS-initiated mitophagy and pexophagy are yet unclear. Although previous studies have identified antioxidant proteins like PRDXs which reside on or relocate to phagophore acting as rheostats (or gatekeepers) to prevent ROS-induced autophagy, can we find any mitochondrial/peroxisomal protein to behave as an ignitor for autophagy induction upon mitochondrial/peroxisomal ROS overproduction? Furthermore, what is the specific “eat-me” signal in ROS-induced mitophagy? Last but not the least, whether macroautophagy and mitophagy is induced simultaneously by the same level of ROS or separately by different levels of ROS?
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;17:1–382.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Rawi SA, Louvet-Vallee S, Djeddi A, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147.
- Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235.
- Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014;12:839–852.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774.
- Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109.
- Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605.
- Harwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007;1147–1150.
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–1052.
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38.
- Davies MJ, Fu S, Wang H, et al. Stable markers of oxidant damage to proteins and their application in the study of human disease. Free Radic Biol Med. 2000;27:1151–1163.
- Biteau B, Labarre J, Toledano MB, et al. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984.
- Aiken CT, Kaake RM, Wang X, et al. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics. 2011;10:1–11.
- Pickering AM, Koop AL, Teoh CY, et al. The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J. 2010;432:585–594.
- Wang X, Yen J, Kaiser P, et al. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 2010;3:ra88.
- Bulteau AL, Lundberg KC, Humphries KM, et al. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063.
- Hailey DW, Rambold AS, Satpute-krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667.
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393.
- Moulis M, Grousset E, Faccini J, et al. The multifunctional sorting protein PACS-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells. 2019;8:638.
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–790.
- Perez-Perez ME, Zaffagnini M, Marchand CH, et al. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy. 2014;10:1953–1964.
- Kiermayer C, Northrup E, Schrewe A, et al. Heart-specific knockout of the mitochondrial thioredoxin reductase (Txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. J Am Heart Assoc Cardiovasc Cerebrovascular Dis. 2015;4:e002153.
- Nagakannan P, Iqbal MA, Yeung A, et al. Perturbation of redox balance after thioredoxin reductase deficiency interrupts autophagy-lysosomal degradation pathway and enhances cell death in nutritionally stressed SH-SY5Y cells. Free Radic Biol Med. 2016;101:53–70.
- Devi TS, Somayajulu M, Kowluru RA, et al. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis. 2017;8:e2777.
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–3026.
- Strohecker AM, Joshi S, Possemato R, et al. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene. 2015;34:5662–5676.
- Lin G, Hill DK, Andrejeva G. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br J Cancer. 2014;111:375–385.
- Overbye A, Brinchmann MF, Seglen PO. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy. 2007;3:300–322.
- Filomeni G, Desideri E, Cardaci S, et al. Under the ROS … thiol network is the principal suspect for autophagy commitment. Autophagy. 2014;6:999–1005.
- Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7:560–577.
- Knebel A, Rahmsdorf HJ, Ullrich A, et al. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996;15:5314–5325.
- Leslie NR. Redox regulation of PI 3‐kinase signalling via inactivation of PTEN. EMBO J. 2014;22:5501–5510.
- Lee SR, Yang KS, Kwon J, et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342.
- Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–4158.
- Guo Z, Kozlov S, Lavin MF, et al. ATM activation by oxidative stress. Science. 2010;330:517–521.
- Zhang J, Kim J, Alexander A, et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol. 2013;15:1186–1196.
- Caito S, Rajendrasozhan S, Cook S, et al. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010;24:3145–3159.
- Ou X, Lee MR, Huang X, et al. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells. 2014;32:1183–1194.
- Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688.
- Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939.
- Gade P, Manjegowda SB, Nallar SC, et al. Regulation of the death-associated protein Kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating Kinase 1. Mol Cell Biol. 2014;34:4033–4048.
- Eisenberg-Lerner A, Kimchi A. DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ. 2007;14:1908–1915.
- Wei Y, Sinha SC, Levine B. Dual Role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951.
- Nah J, Yoo SM, Jung S, et al. Phosphorylated CAV1 activates autophagy through an interaction with BECN1 under oxidative stress. Cell Death Dis. 2017;8:e2822.
- Shi Y, Tan SH, Ng S, et al. Critical role of CAV1/caveolin-1 in cell stress responses in human breast cancer cells via modulation of lysosomal function and autophagy. Autophagy. 2015;11:769–784.
- Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303.
- Zmijewski JW, Banerjee S, Bae H, et al. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285:33154–33164.
- Hinchy EC, Gruszczyk AV, Willows R, et al. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J Biol Chem. 2018;293:17208–17217.
- Shi CS, Kehrl JH. TRAF6 and A20 regulate Lysine 63–linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42.
- Min Y, Mi-Jeong K, Sena L, et al. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy. 2018;14:1347–1358.
- Kejia L, Chu G, Yimin L, et al. A fine-tuning mechanism underlying self-control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy. 2020;16:975–990.
- Yan S, Sun X, Xiang B, et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J. 2014;29:3773–3786.
- Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012;19:788–797.
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760.
- Ligeon LA, Pena-Francesch M, Vanoaica LD, et al. Oxidation inhibits autophagy protein deconjugation from phagosomes to sustain MHC class II restricted antigen presentation. Nat Commun. 2021;12:1–13.
- Zheng X, Yang Z, Gu Q, et al. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy. 2019;16:1838–1850.
- Qiao S, Dennis M, Song X, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014.
- Frudd K, Burgoyne T, Burgoyne JR. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun. 2018;9:1–15.
- Ju H, Canadien V, Lam GY, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–6231.
- Bock FJ, Tait S. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100.
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167.
- Youle RJ. Mitochondria — striking a balance between host and endosymbiont. Science. 2019;365:eaaw9855.
- Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R185.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314.
- Villa E, Marchetti S, Ricci JE. No Parkin zone: mitophagy without Parkin. Trends Cell Biol. 2018;28:882–895.
- Joselin AP, Hewitt SJ, Callaghan SM, et al. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet. 2012;21:4888–4903.
- Ma S, Zhang X, Zheng L, et al. Peroxiredoxin 6 is a crucial factor in the initial step of mitochondrial clearance and is upstream of the PINK1-Parkin pathway. Antioxid Redox Signal. 2016;24:486–501.
- Valentin-Vega YA, MacLean KH, Tait-Mulder J. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119:1490–1500.
- Zhang J, Tripathi DN, Jing J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269.
- Jeong SJ, Kim S, Park JG, et al. Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux. Autophagy. 2017;14:120–133.
- Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377-88.
- Roberts DJ, Tan-Sah VP, Ding EY, et al. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 2014;53:521–533.
- Sun L, Shukair S, Naik TJ, et al. Glucose phosphorylation and mitochondrial binding are required for the protective effects of Hexokinases I and II. Mol Cell Biol. 2008;28:1007–1017.
- Wu R, Wyatt E, Chawla K, et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012;4:633–646.
- Pua HH, Guo J, Komatsu M, et al. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055.
- Billia F, Hauck L, Konecny F, et al. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572–9577.
- Nagakannan P, Eftekharpour E. Differential redox sensitivity of cathepsin B and L holds the key to autophagy-apoptosis interplay after Thioredoxin reductase inhibition in nutritionally stressed SH-SY5Y cells. Free Radic Biol Med. 2017;108:819–831.
- Martina JA, Puertollano R. Rag GTPases mediate amino acid–dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200:475–491.
- Wang H, Wang N, Xu D, et al. Oxidation of multiple MiT/TFE transcription factors links oxidative stress to transcriptional control of autophagy and lysosome biogenesis. Autophagy. 2020;16:1683–1696.
- Diaz-Meco MMT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004.
- Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123-40.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223.
- Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol Cell. 2013;51:618–631.
- Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354.
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010.
- Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
- Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–462.
- Luo S, Garcia-Arencibia M, Zhao R, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–370.
- Galluzzi L, Aaronson SA, Abrams J, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107.
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in drosophila. Cell. 2007;131:1137–1148.
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939.
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95.
- Liu Y, Shoji-Kawata S, Sumpter RM, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110:20364–20371.
- Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
- Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428.
- Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–282.
- Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109.
- Bulina ME, Chudakov DM, Britanova OV, et al. A genetically encoded photosensitizer. Nat Biotechnol. 2006;24:95–99.