
ABSTRACT
EIF4A3 (eukaryotic translation initiation factor 4A3) is an RNA helicase and core component of the exon junction complex. While this RNA-binding protein (RBP) is well-characterized for its crucial roles in splicing, RNA trafficking and nonsense-mediated decay, its role in the regulation of metabolic signaling pathways remains elusive. In a recent study, we describe a new role for EIF4A3 as a negative regulator of macroautophagy/autophagy. Mechanistically, we report that EIF4A3, through its ability to safeguard splicing, can maintain low basal levels of autophagy through the cytosolic retention of the key autophagy transcription factor TFEB. Upon EIF4A3 depletion, the shuttling of TFEB to the nucleus results in an integrated transcriptional response, which induces both early and late steps of the autophagy pathway and enhances autophagic flux. We further report the upregulation of EIF4A3 across multiple cancer types and highlight the relevance of this newly identified EIF4A3-TFEB signaling axis in human tumors.
In recent years, the number of RBPs has increased substantially owing to the development of new technologies that enable global and precise mapping of protein-RNA interactions. RBPs are now estimated to represent up to 10% of the human proteome, presenting with diverse functions spanning transcription, translation, RNA processing, localization and decay. They have profound impacts on a majority of cellular processes include emerging roles in metabolic signaling pathways, such as the conserved degradation process of autophagy. In our previous work, we screened the RNA-binding proteome seeking to reveal new mechanistic understanding of autophagy. Among 1530 RBPs, we disclosed several interesting autophagy regulators, including the DEAD box-family RNA helicase EIF4A3, which is well-known for its broad impact on various aspects of RNA metabolism.
We recently reported that EIF4A3, through its ability to safeguard splicing, acts as a negative regulator of autophagy [Citation1] (). Our initial observations confirmed that depletion of EIF4A3 leads to an increased production of autophagosomes and lysosomes, which results in an enhanced autophagic flux. Investigating this further, we did not find any major changes in global translation; however, our transcriptome profiling analysis revealed a clear overrepresentation of autophagy- and lysosome-associated genes among upregulated transcripts after EIF4A3 depletion. These included multiple known targets of the autophagy transcription factor TFEB. Under fully fed conditions, TFEB is predominantly found in the cytosol in its phosphorylated form. Upon induction of autophagy, for instance by nutrient deprivation, TFEB is rapidly dephosphorylated and translocates to the nucleus, where it induces the transcription of a broad group of autophagy and lysosomal genes. We found that depletion of EIF4A3 can initiate this transcriptional response by inducing the dephosphorylation of TFEB, leading to its subsequent relocalization to the nucleus. Accordingly, we confirmed that the autophagy induction caused by loss of EIF4A3 is dependent on TFEB activity. Importantly, re-expression of EIF4A3 in an siRNA-depleted background can revert TFEB nuclear localization and rescue all autophagy-related phenotypes.
Figure 1. Schematic representation of the EIF4A3-GSK3B-TFEB signaling axis regulating the autophagy response. Upon loss of EIF4A3, an exon skipping event in the transcript encoding GSK3B leads to its reduced kinase activity, contributing to TFEB dephosphorylation and nuclear translocation. In the nucleus, TFEB binds to the promoters of several autophagy and lysosomal target genes, leading to an enhanced autophagic flux
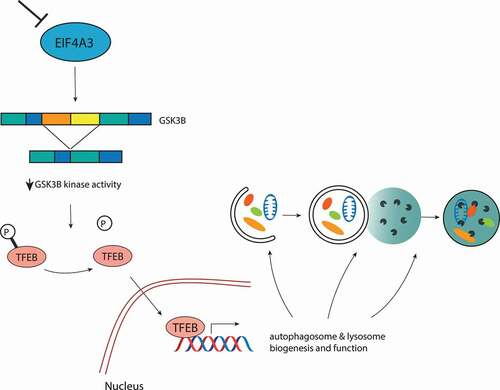
In accordance with previous studies, our transcriptome-wide analysis of alternative splicing events suggested that EIF4A3 regulates multiple types of splice events, including the predominant event of exon skipping. Among several kinase-encoding transcripts subjected to exon-skipping, we identified the transcript encoding GSK3B (glycogen synthase kinase 3 beta), a direct TFEB kinase. We confirmed that loss of EIF4A3 leads to the skipping of exons 6 and 7 in this transcript. A closer look at these splicing events suggests that they result in the introduction of a premature stop codon, likely leading to a truncated product which is degraded. In support of this, we found that knockdown of EIF4A3 led to a reduction in the full-length GSK3B RNA and protein levels as well as its reduced kinase activity toward known targets NFKBIA/IkBα and CTNNB1/β-catenin. We additionally confirmed that the skipping of these exons is followed by a reduced phosphorylation on the established GSK3B-targetted phospho-site of TFEB, Ser138. Phosphorylation at this site has been previously associated with TFEB’s cytoplasmic retention, and inhibition of GSK3B is known to enhance the nuclear translocation of TFEB. Through expression of correctly spliced GSK3B, the nuclear localization of TFEB caused by EIF4A3 depletion was effectively blocked, supporting the functional importance of this splicing event.
TFEB is phosphorylated on multiple serine residues by a number of independent upstream kinases besides GSK3B, including MTOR, MAPK/ERK, PRKC/PKC and AKT and hence a complicated interplay exists between these sites to ultimately control TFEB subcellular localization. It has been suggested that GSK3B mediates an initial phosphorylation event, allowing for subsequent phosphorylation by additional kinases, which would explain why a splicing defect which presents in a subset of GSK3B transcripts, could lead to an amplified effect. However, the precise relationship between these phospho-sites remains unclear. Of note, our RNA sequencing revealed a complex picture, in which is it likely that additional events, independent of the EIF4A3-GSK3B-TFEB signaling axis, may further influence the cellular autophagy response.
In this study, we report a significant upregulation of EIF4A3 expression across a panel of different tumor types, suggestive of its oncogenic potential. In line with this, its deficiency has previously been demonstrated to inhibit cell migration and decrease cell viability, whereas small molecule-based targeting of EIF4A3 has been shown to exhibit anti-tumor effects. By further scrutinizing the expression patterns in these tumors, we revealed a negative expression correlation between EIF4A3 and a cluster of established TFEB targets. Considering the broad inter- and intra-tumor heterogenicity among investigated tumors, as well as the previously reported complexity of the TFEB response, the consistency of this expression pattern suggests a potential role for the EIF4A3-TFEB signaling axis in cancer. However, whether this pathway represents a driving force in tumorigenesis remains elusive.
The recent development of selective EIF4A3 inhibitors opens the potential for directly targeting EIF4A3 as a means to alter the autophagy response in vivo. Due to an emerging clinical interest in boosting TFEB activity to enhance aggregate clearance in the context of neurodegenerative and lysosomal storage disorders, EIF4A3 may represent an attractive target of broader clinical relevance. Hence, the potential therapeutic benefits of targeting this newly identified signaling axis warrant further investigation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Sakellariou D, Tiberti M, Kleiber TH, et al. eIF4A3 regulates the TFEB-mediated transcriptional response via GSK3B to control autophagy. Cell Death Differ. 2021. DOI:https://doi.org/10.1038/s41418-021-00822-y.