
ABSTRACT
The scaffold protein AMBRA1 regulates the early steps of autophagosome formation and cell growth, and its deficiency is associated with neurodevelopmental defects and cancer. In a recent study, we show that AMBRA1 is a key factor in the upstream branch of the MYCN-MYC and CDK4-CDK6-dependent regulation of G1/S phase transition. Indeed, in the developing neuroepithelium, in neural stem cells, and in cancer cells, we demonstrate that AMBRA1 regulates the expression of D-type cyclins by controlling both their proteasomal degradation and their MYCN-MYC-mediated transcription. Also, we show that this regulation axis maintains genome integrity during DNA replication, and we identify a possible line of treatment for tumors downregulating AMBRA1 and/or overexpressing CCND1 (cyclin D1), by demonstrating that AMBRA1-depleted cells carry an AMBRA1-loss-specific lethal sensitivity to CHEK1 inhibition. Interestingly, we show that this aspect is specific for AMBRA1 loss, because ATG7 knockdown does not display the same response to CHEK1 inhibitors. Hence, our findings underscore that the AMBRA1-CCND1 pathway represents a novel crucial mechanism of cell cycle regulation, deeply interconnected with genomic stability in development and cancer.
Macroautophagy/autophagy directly cross-talks with other intracellular processes, such as the cell cycle and the DNA damage response, and several autophagy-regulating proteins also have autophagy-independent roles in the modulation of these pathways. AMBRA1 (autophagy and beclin 1 regulator 1) is a scaffold factor, member of the autophagy core complex and characterized by long intrinsically disordered regions that promote interaction with many proteins involved in several cellular pathways. For instance, we have previously reported that AMBRA1 interacts with distinct members of the CUL (cullin) E3 ubiquitin ligases, regulating the stability of autophagy key proteins. Further, besides its role in activating autophagy, we have reported that AMBRA1 controls the rate of cellular proliferation by mediating PPP2/PP2A-dependent dephosphorylation and consequent degradation of the proto-oncogene MYC. Indeed, Ambra1-deficient mouse embryos display enhanced cell proliferation in the developing neuroepithelium, which is followed by massive apoptosis, whereas Ambra1 heterozygous mice develop tumors in the lungs, liver, and kidney, with mutations of this gene being found in human cancers.
To better define the AMBRA1 autophagy (in)dependent role in neurodevelopment and cancer, we generated a nervous system ambra1 conditional knockout (cKO) model [Citation1]. We found that ambra1 cKO mice are characterized by perinatal mortality and the presence of frontal bone prominence, as a consequence of increased cell proliferation in both the cortex and the lateral ventricles. By isolating neural stem cells from the medial ganglionic eminences/MGE of control and cKO embryos, we observed overexpression of CCND1 and CCND2 in Ambra1-deficient cells. We determined that increased levels of D-type cyclins result from both increased MYCN-dependent transcription and increased protein stability. Next, we identified DDB1, a member of the CUL4-DDB1 E3 ubiquitin ligase complex, as the E3 ligase that mediates CCND1 degradation through its binding with AMBRA1. As a consequence of CCNDs overexpression, we show that AMBRA1 knockdown (KD) results in a shorter G1 phase, with faster entrance and longer persistence in the S phase, and to increased cell death.
Primed by these results, we investigated whether the post-translational that AMBRA1 exerts on control of CCNDs is a specific feature of nervous system development or if it represented a more general function in cell cycle progression. Through quantitative imaging cytometry of human fibroblast and U2OS FUCCI cells, we found that AMBRA1 silencing causes an upregulation of cells in the S phase and an increase of CCND1 distributed over all cell cycle phases.
The apoptotic phenotype observed in neural cells ex vivo and in vitro, together with the deregulation of cell cycle checkpoints and accumulation of cells in S phase upon AMBRA1 KD, prompted us to investigate a possible effect of AMBRA1 deregulation on genome stability. Indeed, we found that AMBRA1 KD induces an increase of DNA damage to a greater extent with respect to ATG7 KD. Of note, it has been previously shown that autophagy influences DNA repair and that autophagy defects cause an impairment of the homologous recombination (HR) pathway. However, analyses of HR efficiency showed that, at variance with ATG7 KD, AMBRA1 deficiency does not cause an HR impairment. We thus reasoned that accumulation of DNA damage was mainly the consequence of an aberrant cell cycle progression caused by AMBRA1 depletion. Consistently, we found that CCND1 exogenous overexpression induces H2AX phosphorylation. Given that AMBRA1-silenced cells are accumulating DNA damage in S and G2 phases, we investigated if this could correlate with mitotic defects. Through time-lapse imaging and detailed microscopy analysis we concluded that AMBRA1 deficiency significantly results in longer mitosis, appearance of anaphase bridges and cell death after cytokinesis.
The occurrence of DNA damage in S phase, anaphase bridges, and mitotic abnormalities are all hallmarks of replication stress (RS). Indeed, we found that AMBRA1 deficiency induces an RS phenotype characterized by increased replication fork speed, RPA foci formation, and CHEK1 phosphorylation. Next, we showed by RNA-Seq and q-PCR analyses that AMBRA1 deficiency causes an upregulation of genes involved in cell cycle progression and DNA repair. Of note, we found that AMBRA1 downregulation induces a significant increase in CHEK1 mRNA and protein levels.
Due to the importance of AMBRA1 in the regulation of pathways frequently impaired in cancer, we investigated the frequency of its transcriptional downregulation in cancer datasets deposited in The Cancer Genome Atlas/TCGA database. Interestingly, we detected in several datasets the presence of an “AMBRA1-low” cancer subgroup, characterized by a remarkably lower expression of AMBRA1 with respect to healthy tissues. We further investigated the effect of ambra1 knockout in a lung cancer mouse model conditionally primed by KRASG12D mutation. Indeed, tumors depleted of AMBRA1 exhibit a more aggressive growth phenotype with larger lesions than the AMBRA1-proficient ones, together with increased levels of CCND1, p-Ser62 MYC and replication stress markers.
Because AMBRA1 deficiency causes high levels of replication stress and endogenous DNA damage, we speculated that targeting a key kinase activated in response to replication stress could selectively kill AMBRA1-deficient cells. Indeed, we found that AMBRA1-depleted cells are highly sensitive to treatments with CHEK1 inhibitors (AZD7762 and LY2603618). To evaluate the contribution of autophagy impairment in AZD7762 cell sensitivity, we next inhibited autophagy by ATG7 knockdown. Strikingly, both control and ATG7-depleted cells are not affected by AZD7762 treatment. In turn, we found that CCND1-overexpressing cells are sensitive to CHEK1 inhibitors.
In sum, our findings reveal that AMBRA1 acts as a gatekeeper for the G1/S transition by regulating the stability of both CCND1 and MYC. This newly discovered regulatory axis has an important role in neurodevelopment and in neoplastic transformation (). Moreover, the finding that AMBRA1 expression influences the extent of RS supports a role for AMBRA1 as a promising prognostic biomarker for sensitivity to CHEK1 inhibition and strengthens the evidence pointing at AMBRA1 as an important tumor suppressor.
Figure 1. AMBRA1 regulates autophagy and cell cycle through the CUL4-DDB1 complex. (a) in physiological conditions, AMBRA1 acts as a DCAF substrate receptor protein for recognition of CCNDs and ubiquitination by the DDB1-CUL4 complex. Once ubiquitinated, both AMBRA1 and CCNDs are primed for proteasomal degradation, with this influencing both cell cycle orchestration and autophagy. (b) The absence of AMBRA1 impairs CCND degradation, thereby provoking premature S-phase entry and replication stress that leads to genomic instability, ultimately affecting both neurodevelopment and cancer onset and aggressiveness
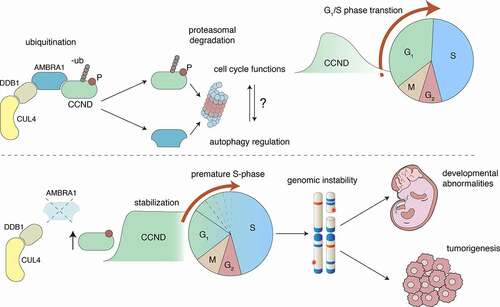
While we show that AMBRA1 regulates the S phase entry through D-type cyclins, a number of questions remain unanswered. How can different pools of AMBRA1 in parallel regulate different processes such as cell proliferation and autophagy? Both autophagy and G1/S transition deeply rely on nutrients and growth factor sensing. Despite being downstream of both pathways, can AMBRA1 represent a conjunction point between these two key cellular processes? What is the autophagy status of AMBRA1-low cancers? Is it possible to conceive a novel therapy based on autophagy targeting in these tumors? Further studies are needed to address these important questions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Maiani E, Milletti G, Nazio F, et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature. 2021;592(7856):799–803.