
ABSTRACT
Free spread is a classical mode for mammalian virus transmission. However, the efficiency of this transmission approach is generally low as there are structural barriers or immunological surveillances in the extracellular environment under physiological conditions. In this study, we systematically analyzed the spreading of classical swine fever virus (CSFV) using multiple viral replication analysis in combination with antibody neutralization, transwell assay, and electron microscopy, and identified an extracellular vesicle (EV)-mediated spreading of CSFV in cell cultures. In this approach, intact CSFV virions are enclosed within EVs and transferred into uninfected cells with the movement of EVs, leading to an antibody-resistant infection of the virus. Using fractionation assays, immunostaining, and electron microscopy, we characterized the CSFV-containing EVs and demonstrated that the EVs originated from macroautophagy/autophagy. Taken together, our results showed a new spreading mechanism for CSFV and demonstrated that the EVs in CSFV spreading are closely related to autophagy. These findings shed light on the immune evasion mechanisms of CSFV transmission, as well as new functions of cellular vesicles in virus lifecycles.
Abbreviations: 3-MA: 3-methyladenine; CCK-8: Cell Counting Kit-8; CSF: classical swine fever; CQ: chloroquine; CSFV: classical swine fever virus; DAPI, 4-,6-diamidino-2-phenylindole; EVs: extracellular vesicles; hpi: h post infection; IEM: immunoelectron microscopy; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MOI: multiplicity of infection; MVs: microvesicles; ND50: half neutralizing dose; PCR: polymerase chain reaction; PBS: phosphate-buffered saline; SEC: size-exclusion chromatography; siRNA: small interfering RNA; TEM: transmission electron microscopy.
Introduction
Classical swine fever (CSF) is an acute contagious disease caused by classical swine fever virus (CSFV), which seriously restrains the development of pig industry [Citation1]. The control strategy for CSF is generally a combination of compulsory vaccine immunization and elimination of diseased pigs [Citation2], but under the high pressure of compulsory vaccination, CSF is still repeatedly emerged. CSFV, an enveloped, positive-sense, single-stranded RNA virus, belongs to the Pestivirus genus of the Flaviviridae family [Citation3]. The genome of CSFV contains a 5′-noncoding region, a large open reading frame, and a 3′-noncoding region. The open reading frame is translated into a precursor polyprotein which is cleaved into four structural proteins (C, Erns, E1, and E2) and eight non-structural proteins (Npro, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) [Citation4]. In addition to acute infection, CSFV usually induces chronic persistent infection. Pigs with chronic infection shed the virus, and specific antibodies will be detected but without elimination of the virus [Citation5], suggesting that CSFV can counteract the host antibody response through a specific mechanism. However, the mechanism is not clear at present.
The initial step for mammalian viruses to transmit and establish an infection is to get into the target cells. In a classical transmission mode, virus particles in the extracellular environment diffuse freely and bind directly onto the surface of target cells, then the virions are internalized. This mode of virus transmission is called cell-free virus spread [Citation6]. However, it has been reported that some viruses spreading between susceptible cells do not need to release and spread free viral particles. Instead, communication between cells is required to transfer complete virions or infectious viral components from infected to uninfected susceptible cells [Citation7,Citation8]. This mode of transmission is called viral cell-to-cell spread mode. Several viruses spread in a cell-to-cell way with different mechanisms, for example, retroviruses spread through pores formed by the fusion of adjacent cells [Citation9], paramyxoviruses spread through synapses [Citation10], and herpes viruses utilize the tight junction structure of epithelial cells to spread [Citation11]. One important type of cell-to-cell transmission pattern depends on extracellular vesicles (EVs) and is therefore called viral EV-dependent spread. In this way, viruses hijack EVs to carry virions or infectious components to neighboring cells resulting a local infection, and more importantly, the EVs can also enter adjoining bodily fluids and carry the viruses to distant sites to establish new infections [Citation12,Citation13]. For instance, porcine reproductive and respiratory syndrome virus-infected cells produce exosomes contain viral genomic RNA and partial viral proteins, and the exosomes establish productive infection in both porcine reproductive and respiratory syndrome virus-susceptible and -nonsusceptible cells [Citation14]. Besides, exosomes in the sera of hepatitis B virus patients spread viral nucleic acids and proteins to hepatocytes, and notably, the exosomes could be transferred into natural killer cells and impair natural killer-cell functions [Citation15]. Unlike in the cell-free spread mode, virions are only limitedly exposed to the outside of cells in cell-to-cell spread approaches, thus effectively avoid the neutralization of specific antibodies or activation of immune response by virions, facilitating virus transmission and persistent infection. While it is observed that CSFV frequently causes chronic long-term infections, the spreading patterns other than the cell-free mode of CSFV has not been studied so far.
To address this fundamental issue for infection, we systematically analyzed the spreading of CSFV in a cell culture model. Using multiple viral replication analysis in combination with antibody neutralization, transwell assay, and electron microscopy, we identified a new EV-dependent spreading approach for CSFV infection. We further characterized the features and origins of the CSFV-containing EVs by fractionation assays, immunostaining, and electron microscopy, which supports the idea that CSFV hijacks the EVs originating from autophagy to transfer virions from infected to uninfected cells. These findings add new knowledges to CSFV spread pathway as well as functions of cellular vesicles in virus infection, revealing a potential immune evasion mechanism of CSFV to resist neutralizing antibodies by EV-dependent spread.
Results
Identification of alternative spread modes for CSFV transmission
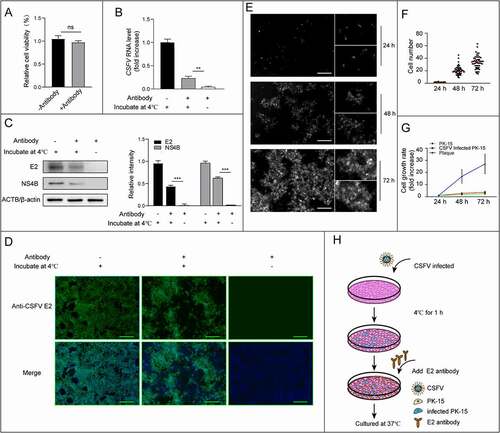
Neutralizing antibodies can make virions lose the infectivity, thus preventing the infection of cells by free virions. The envelope glycoprotein E2 of CSFV is the main protective antigen to induce neutralizing antibodies. To find out whether CSFV could use other transmission approaches rather than the free virus spread, we performed an antibody neutralization experiment. Firstly, we tested the concentration of neutralizing antibodies for fully blocking of CSFV particle infection (Figure S1). The E2 antibody with dilutions of 10−2 to 10−9 were used to block infection of PK-15 cells with CSFV at 1 multiplicity of infection (MOI), and virus replication was measured by real-time PCR, western blot, as well as infected plaques detected by immunofluorescence. Quantitative data showed that dilution of the E2 antibody up to 1: 100 (10× ND50) is still sufficient to neutralizing CSFV infection at 1 MOI (Figure S1). To ensure the free CSFV virions are fully blocked by neutralizing antibodies, excessive amount of antibody (10× ND50) was used to block the free spread of CSFV at 0.1 MOI. Then we incubated the cells with 0.1 MOI of CSFV at 4°C for 1 h, which allowed the virus to attach and establish a minimal infection, followed by excessive amount of neutralizing antibody treatment to block subsequent infection caused by newly formed and released free virions (). Virus infection and replication were identified by real-time PCR, western blot so as infected cells by immunofluorescence. As expected, antibody treatment significantly reduced the number of infected cells in comparison to the untreated group, confirming the transmission of CSFV by the free spreading mode, but the infection was not fully blocked, virus replication and newly infected cells were both identified (–D). When we look at the distribution of infected cell plaques, it is quite clear that these plaques are clustered in the presence of neutralizing antibodies, unlike the scattering distribution in absence of antibodies (). As those clustered plaques could also be formed due to division of infected cells, we then take a close look at the cell proliferation rate and new infection rate. The new infection rate was measured by counting cells in each plaque at indicated times (24, 48 and 72 h post infection (hpi)) (–F); the division rates of CSFV-infected and uninfected cells were also counted under the same conditions. As shown in , although the division rate of PK-15 cells was slightly increased with CSFV infection, it was much lower than the new infection rate. In other words, viral transmission is faster than cell proliferation. These results were also confirmed by analysis of CSFV new infections in co-culturing of infected and uninfected cells. Similarly, we observed a higher infection rate than cell proliferation rate even in the presence of CSFV neutralizing antibodies (Figure S2). From those experiments, we concluded that, besides the cell free mode, there are cell-to-cell ways for CSFV transmission, which are responsible for the antibody-resistant spreading of the virus.
Figure 1. Identification of an antibody-resistant spreading of CSFV. PK-15 cells were infected with CSFV (MOI = 0.1) at 4°C for 1 h and then cultured in a medium containing E2 antibody for 72 h. The replication and infected plaques of CSFV was analyzed. (A) Detection of the cell viability on the treatment of antibody (1:100 dilution) by CCK-8 test. (B) Detection of CSFV RNA levels in antibody treated and control cells by real-time PCR. (C) Detection and quantification of structural protein E2 and non-structural protein NS4B by western blot. ACTB/β-actin served as the internal reference, and the gray value was measured by ImageJ software. (D) Detection of plaques formed by infection of CSFV in antibody-treated and control cells by immunofluorescence at 72 hpi. Scale bar: 200 μm. (E) Representative images of infected plaques in antibody treated cells at 24, 48, and 72 hpi. Scale bar: 200 μm. (F) Quantification of the size of plaques. The number of cells in plaque was counted from at least 50 plaques at indicated time points, with the maximum and minimum values removed. The red horizontal line marks the median. (G) Comparison of plaque-size increase rate and cell growth rate. The curve that cell number in plaques with time and the single-cell growth curves of infected and uninfected PK-15 cells were made in a coordinate system. The increase rate of cell number in plaques was higher than the growth rate of infected and uninfected cell. (H) Schematic representation of the experimental schedules. Results are shown as the mean ± SD (n = 3). **, P < 0.01; ***, P < 0.001; ns, not significant.
Cell-contact is not a prerequisite for CSFV transmission
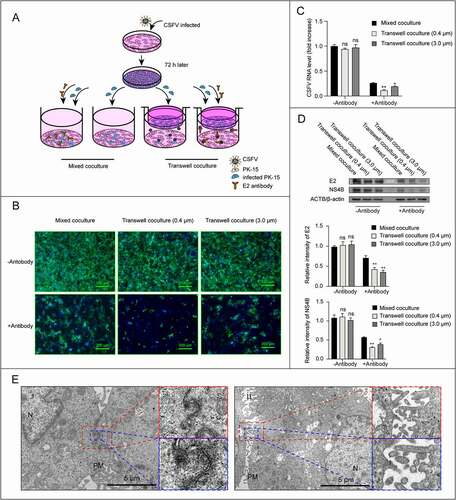
There are different mechanisms for cell-to-cell transmission approaches, such as spreading mediated by cell junctions, virological synapses, membrane pores or filopodia, which all depend on direct cell contacts. But one of cell-to-cell transmission ways does not require cell contact, that is EV-mediated virus transmission. To verify whether CSFV virions tend to spread through direct cell-to-cell contact, we used a transwell coculture system to physically separate infected cells from uninfected target cells. CSFV-infected cells were plated on a cell culture insert, and uninfected cells were plated on the bottom well; then the cells were cocultured for 72 h in media with or without neutralizing antibodies (). Virus spread from infected to uninfected target cells was analyzed by real-time PCR, western blot, and immunofluorescence. The results showed that, in the presence of antibodies, the total viral RNA and protein levels in transwell-coculture system were lower than that in direct mixed-coculture setup control, but the infected target cells were observed clearly in transwell-coculture system, both in 0.4 μm and 3.0 μm pore size (–D), indicating that direct cell-to-cell contact is not a prerequisite for CSFV antibody-resistant spreading. To further determine whether the direct cell-to-cell contact contribute to CSFV transmission, infected cells were collected for transmission electron microscopy (TEM). Indeed, no virion was observed at the contact site (). Instead, CSFV virions seem to prefer to gather on the side that has no contact with other cells (data not shown), indicating cell contact is not required for the cell-to-cell transmission of CSFV.
Figure 2. Cell-cell contact is not a prerequisite for CSFV transmission. (A) Schematic representation of the transwell assay. CSFV infected PK-15 cells in transwell and uninfected cells in bottom were cocultured in the absence or in the presence of neutralizing antibodies for 72 h, then virus replication and infected cells were analyzed. Cocultures of the mixed infected/uninfected PK-15 cells were used as controls for virus transmission. (B) Representative image of infected plaques in bottom wells detected by immunofluorescence. Scale bar: 200 μm. (C) Detection of CSFV RNA levels in both transwells and bottom cells by real-time PCR. (D) Detection and quantification of structural protein E2 and non-structural protein NS4B levels by western blot. ACTB/β-actin served as the internal reference, and the gray value was measured by ImageJ software. (E) TEM images of PK-15 cells infected with CSFV (MOI = 1). Cells were collected at 12 hpi and imaged using TEM (FEI, 80kV). N, nucleus, PM, cell plasma membrane. Results are shown as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01.
EVs secreted from CSFV-infected cells carry viral particles
At this step, we proposed that CSFV was transmitted via extracellular vesicles. If this hypothesis is true, the supernatant from infected cells should be infectious and contains vesicles loaded with viral infectious components or virions. So, we collected the fresh culture supernatant and measured the virus titer, which is about 105 half tissue culture infection dose/mL. Then PK-15 cells were co-cultured with the supernatant in the presence of antibodies, and virus replication and new infection were detected by real-time PCR, western blot, and immunofluorescence. The results showed that the fresh supernatant from CSFV infected cells could lead to new infections in an antibody-resistant way, and a simple freeze-thawing process which destroyed the membrane structure of potential EVs but not the virions abolished the antibody-resistant infectivity of the supernatant, indicating an infection mediated by membraned vesicles (–D). Next, we collected the infected cells and observed the possible vesicles secreted from infected PK-15 cells by TEM at 12 hpi. As expected, vesicles containing virions were observed outside the cell (), with some containing only one or a small number of virions, and some others containing multiple virions. Therefore, CSFV could release newly formed virions in the EVs. The TEM data clearly showed that CSFV could utilize the EVs to exit cells. The similar phenomenon was also observed in infected-ST cells (Figure S3), indicating that EV-mediated release of virions is a general mode of CSFV transmission.
Figure 3. CSFV virions were released in EVs. The freeze-thawed culture supernatant of CSFV infected cells was inoculated into the uninfected cells which cultured in the presence of antibodies, the replication of CSFV was detected at 72 hpi. Unfreeze-thawed fresh culture supernatant was used as control. (A-C) The fresh culture supernatant of infected cells has an antibody-resistant infectivity. (A) Detection of CSFV genome replication by real-time PCR. (B) Detection and quantification of structural protein E2 and non-structural protein NS4B levels by western blot. ACTB/β-actin served as the internal reference, and the gray value was measured by ImageJ software. (C) Detection of infected plaques by immunofluorescence. Scale bar: 200 μμm. (D) Schematic representation of the experimental schedules. (E) TEM images of the EVs secreted from infected PK-15 cells (12 hpi). The dashed box shows the vesicles containing virions. N, nucleus, PM, cell plasma membrane. Results are shown as the mean ± SD (n = 3). ***, P < 0.001.
EVs mediate the antibody-resistant spreading of CSFV
In order to determine whether the EVs in the infected culture supernatant are responsible for this antibody-resistant virus transmission, we isolated the EVs using differential centrifugation in the cell culture supernatants from both infected and uninfected cells. Using TEM, we did observe the intact vesicles isolated from the supernatants of both cell cultures (). To check the infectivity of these EVs, the isolated EVs were inoculated to PK-15 cells and cultured with or without antibodies. As expected, viral RNA, protein and infected plaques were all detected in both antibody-treated or not group (–D), indicating that the isolated EVs had an antibody-resistant infectivity.
Figure 4. Identification of the EVs isolated from CSFV-infected cells. (A) Negatively stained TEM images of the isolated EVs. The isolated EVs was inoculated into PK-15 cell monolayer and cultured with or without E2 antibody for 72 h. (B-D) The infectivity of the isolated EVs was analyzed by detection of the CSFV replication. (B) Detection of CSFV genome replication by real-time PCR. *, P < 0.05; ns, not significant. (C) Detection and quantification of structural protein E2 and non-structural protein NS4B levels by western blot. ACTB/β-actin served as the internal reference. (D) CSFV infected plaques formed by isolated EVs were detected by immunofluorescence. Scale bar: 100 μm. (E-G) TEM images of the isolated EVs attached on the surface of recipient cells. The dashed box shows the vesicles containing virions. The blue arrows point to the vesicles and the red arrows point to the virions. N, nucleus, PM, cell plasma membrane.
Next, we tried to observe infection of cells by these isolated EVs directly using TEM. The isolated EVs were layered onto PK-15 cells at 4°C for 1 h and incubated at 37°C for 15 min, followed by observation through TEM. It was found that virion-containing EVs attached to the cell surface, with some containing only one virion () and others containing multiple virions (). shows that the virions enclosed within EVs enter the target cells through the fusion of EVs and plasm membrane of target cells. These results demonstrated that CSFV virions indeed spread through EVs in cell culture.
Autophagy markers are present in EVs containing virions
Previous study reported that autophagy is important for the release of CSFV [Citation16], so we wonder whether these CSFV-containing vesicles are closely related to autophagy. To verify this conjecture, we need to detect autophagy markers in the CSFV-containing EVs. Given that the EVs isolated by differential centrifugation is heterogeneous, we further purified the EVs using a size-based separation method – size-exclusion chromatography (SEC). The EVs isolated by differential centrifugation were divided into 30 sequential fractions by SEC (). As shown in , the start material was highly dense and varied in size, and the size of the vesicles became homogeneous and smaller with the grading, even invisible. Then, the fractions were detected by real-time PCR, western blot, and immunofluorescence. Results of immunofluorescence showed that the infected plaques were detected in fraction 5–9, of which fraction 5 and 6 had the highest content (); the distribution of CSFV RNA was similar to the plaques results (). Western blot showed that the viral protein E2 was detected in fraction 5 and 6 (). Further, E2 was also detected in vesicles of fraction 5–6 by immunoelectron microscopy (IEM) (). That is, the fraction 5 and 6, which positive for both viral RNA and protein, as well as the infectivity of CSFV, contained the EVs containing CSFV virions. The size of vesicles in fraction 5–6 is about 400 to 800 nm (). Then we detected autophagic proteins in the fractions, western blot results showed that autophagy marker microtubule associated protein 1 light chain 3B (MAP1LC3B/LC3B) and BECN1/beclin 1 also presented in fraction 5 and 6 (), implying the CSFV-containing vesicles may be autophagic. Furthermore, through laser scanning confocal microscopy, we found many more LC3B-positive vesicles outside the CSFV-infected cells (). These results suggested that the infection of CSFV promotes the secretion of autophagic vesicles, which may contain CSFV virions.
Figure 5. Identification of the EVs purified by SEC. (A) Schematic diagram of SEC. Start material, the EVs isolated by differential centrifugation, is divided into 30 sequential fractions. (B) Negatively stained TEM images of the fractions. Vesicles, which was heterogeneous in the start, became homogeneous and smaller with fraction. Scale bar: 500 nm. (C) Detection of the infectivity of CSFV in each fraction by immunofluorescence. The fraction 5–9 were detected positive for plaques. Scale bar: 200 μm. (D) Detection of the proteins in each fraction by western blot. Autophagy marker proteins, including LC3B and BECN1, as well as CSFV envelope protein E2, were all clearly identified in fraction 5 and 6, indicating CSFV-containing EVs are closely related to autophagy. The exosome marker CD81 was present in fraction 1–24, CD63 was not detected in the start and fractions (not shown). (E) Detection of CSFV genome copies in each fraction by real-time PCR. (F) Detection of CSFV in vesicles of fraction 5–6 by IEM. The red arrow points to gold nanoparticle (10 nm)-labeled CSFV envelope protein E2. Scale bar: 100 nm. (G) Detection the particle size of start and fraction 5–6 using laser particle diameter analyzer (MALVERN).
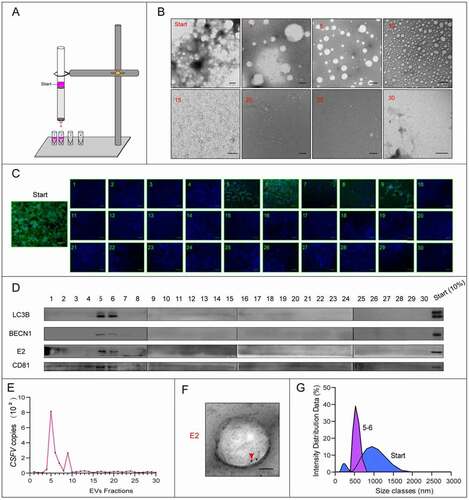
Figure 6. The infection of CSFV promotes the secretion of LC3B-positive vesicles. Immunofluorescence detection of autophagy proteins in EVs by confocal microscopy. PK-15 cells were infected with CSFV for 24 h, and LC3B protein which is a marker for autophagy were detected by a specific antibody (anti-LC3B, in red). Extracellular vesicles with LC3B staining signal were detected after CSFV infection. Cell nuclei were stained with DAPI (in blue).
EVs positive for LC3B contain CSFV virions
Given that the virion-containing EVs might be autophagic, we then wonder if the autophagic EVs contain virions. To check that, we tried to isolate the autophagic EVs from fraction 5 and 6 by using immunoprecipitation. Immunoprecipitation was performed using the anti-LC3B antibody, and confirmed using the anti-BECN1 antibody. Then we checked the LC3B-positive vesicles for CSFV virions using real-time PCR, western blot, and immunofluorescence. Results showed that the LC3B-positive vesicles contained CSFV RNA and protein (–B), and were infectious (). Using IEM, we also detected E2 in LC3B-positive vesicles (). Further, PK-15 cells were incubated with the LC3B-positive vesicles at 4°C for 1 h, followed by immunostaining with anti-LC3B antibody and anti-E2 antibody. Through laser scanning confocal microscopy, CSFV was found to be enclosed within LC3B-positive vesicles (). These results identified the presence of intact infectious virions in the autophagic EVs, proving that autophagy-derived vesicles could transmit CSFV virions to uninfected cells.
Figure 7. Autophagic EVs isolated by immunoprecipitation contain CSFV virions. The autophagic EVs were isolated from fraction 5–6 by immunoprecipitation, LC3B antibody was used to capture the target vesicles. (A) Identification and detection the autophagic proteins and viral protein from isolated EVs by western blot. CSFV envelope protein E2 was detected in the isolated EVs, which was positive for autophagic proteins LC3B and BECN1. (B) Detection of CSFV genome copies in isolated LC3B-positive EVs by real-time PCR. (C) The infectivity of CSFV in LC3B-positive EVs checked by detection of E2 protein with immunofluorescence. Scale bar: 100 μm. (D) Detection of CSFV in isolated LC3B-positive EVs by IEM. The red arrow points to gold nanoparticles (10 nm)-labeled CSFV envelope protein E2. The blue arrow points to gold nanoparticles (20 nm)-labeled LC3B. Scale bar: 150 nm. (E) Immunofluorescence detection of CSFV virions in LC3B-positive EVs by confocal microscopy. The isolated vesicles were attached to recipient cells and stained by immunofluorescence. Vesicles were detected by anti-LC3B antibody (in red), and CSFV virions were detected by anti-E2 antibody (in green). The dashed box shows the virions enclosed in LC3B-positive vesicles. Cell nuclei were stained with DAPI (in blue).
CSFV stays in autophagosomes before releasing
Given that the EVs for CSFV transmission are autophagic, CSFV virions should be closely related to autophagy pathway before leaving the host cells. To prove this hypothesis, we first examined the colocalization between CSFV and autophagic proteins at 9 hpi, a timepoint before assembled virions releasing [Citation17]. Immunofluorescence showed that CSFV was colocalized with autophagic protein LC3B, which correlates with the formation of autophagosomes [Citation18], ATG5, which associates with the precursor autophagosomes [Citation19], BECN1, which involves in the early step of autophagy pathway [Citation20], and SQSTM1, which is a marker of autophagy-mediated protein degradation pathway [Citation21] (). At the same time, through TEM, it was found that a large number of virions gathered in autophagosome-like vesicles (). These results indicated that CSFV stays in autophagosome before releasing.
Figure 8. CSFV virions aggregate on autophagosomes. (A) Immunofluorescence detection of the colocalization of CSFV and autophagosomes by confocal microscopy. CSFV was detected by anti-E2 antibody (in green), and autophagosomes were detected by anti-LC3B, anti-ATG5, anti-BECN1, and anti-SQSTM1 antibodies (in red). Cell nuclei were stained with DAPI (in blue). Yellow arrows point to the confocal fluorescence puncta. Scale bar: 10 μm. (B) Infected cells were collected at 9 hpi and imaged using TEM. Black arrows point to virions, which gathered in the autophagosome-like vesicles near the plasm membrane. Scale bar: 500 nm. PM, cell plasma membrane.
Autophagy involves in CSFV releasing
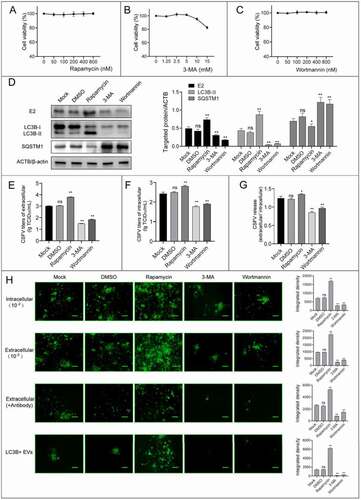
To determine the role of autophagy in CSFV release and transmission, we used the pharmacological methods to regulate autophagy pathway. We first performed CCK-8 assay to ensure that the concentrations (rapamycin, 400 nM; 3-methyladenine [3-MA], 5 mM; wortmannin, 400 nM) used in this study did not affect cell viability (–C). Then we examined viral protein E2 expression and viral progeny yields in these regulator-treated cells. The results showed that the induction of autophagy with rapamycin increased E2 expression and virus yields, while 3-MA and wortmannin-mediated autophagy suppression decreased E2 expression and virus production (–F). Because both the viral progeny yields in extracellular and intracellular were affected with these regulators, we used the ratio of virus titers in extracellular and intracellular to evaluate virus release. The results showed that viral release level was increased with rapamycin-treatment and decreased with 3-MA and wortmannin-treatment, indicating that autophagy is involved in CSFV releasing (). Meanwhile, we found that these drugs also affected the antibody-resistant infectivity of progeny viruses, as well as the infectivity of the autophagic EVs by performing immunofluorescence assay (), indicating that the EV-mediated transmission of CSFV was regulated by autophagy pathway.
Figure 9. The regulators of autophagy pathway affect CSFV release. (A-C) Detection of the cell viability with the treatment of rapamycin(A), 3-MA(B), and wortmannin(C) by CCK-8 test. (D-H) PK-15 cells were infected with CSFV at 1 MOI, and treated with rapamycin (400 nM), 3-MA (5 mM), and wortmannin (400 nM) at 20 hpi for 4 h. The cell samples were analyzed by western blot with anti-E2, anti-LC3B, anti-SQSTM1, and anti-ACTB/β-actin (loading control) antibodies (D). The virus titers of extracellular and intracellular were measured by endpoint dilution titrations by using immunofluorescence (E-F), and the ratios of extracellular and intracellular were calculated to evaluate viral release(G). (H) CSFV infected plaques formed by intracellular cell lysates, extracellular culture supernatants (with or without antibody), and isolated autophagic EVs were detected by immunofluorescence. Scale bar: 200 μm. Results are shown as the mean ± SD (n = 3). ns, not significant; *, P < 0.05; **, P < 0.01.
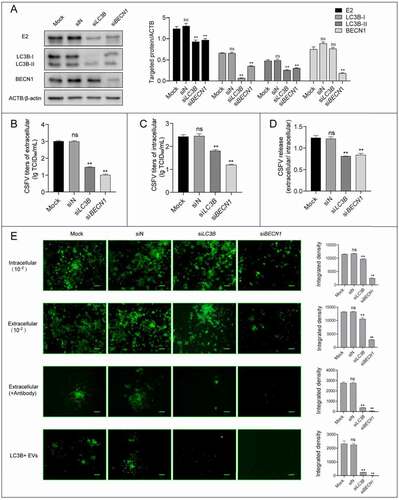
To further confirm the correlation between the autophagy and viral release and transmission, we performed small interfering RNA (siRNA)-mediated knockdown to decrease the activity of autophagy. As shown in , PK-15 cells treated with siRNAs presented significantly decreased levels of endogenous LC3B and BECN1 compared with cells transfected with siN. Notably, the level of LC3B-II was decreased with BECN1 knockdown because of the autophagy inhibition caused by siBECN1. We then combined multiple assays to detect CSFV level in knockdown cells, and found that CSFV-E2 expression (), progeny virus yields (–C), as well as viral release () were all strongly decreased compared with those in control cells. In addition, viral antibody-resistant transmission and the infectivity of autophagic EVs were significantly suppressed with LC3B and BECN1 knockdown (). These results fully demonstrated that autophagy pathway is involved in the release and antibody-resistant transmission of CSFV.
Figure 10. Inhibition of autophagy with specific siRNAs targeting LC3B and BECN1 reduces CSFV release. PK-15 cells were transfected with siRNAs targeting LC3B (siLC3B) and BECN1 (siBECN1) or control siRNA(siN), and infected with CSFV (1 MOI) at 24 h post transfection. The cell samples were analyzed by western blot with anti-E2, anti-LC3B, anti-BECN1, and anti-ACTB/β-actin (loading control) antibodies (A). The virus titers of extracellular and intracellular were measured by endpoint dilution titrations by using immunofluorescence (B-C). (D) The ratios of CSFV titers in extracellular and intracellular were calculated to evaluate viral release. (E) CSFV infected plaques formed by intracellular cell lysates, extracellular culture supernatants (with or without antibody), and isolated autophagic EVs were detected by immunofluorescence. Scale bar: 200 μm. Results are shown as the mean ± SD (n = 3). ns, not significant; **, P < 0.01.
CSFV releasing requires the formation of autolysosomes
To determine whether the release of CSFV depends on the fusion of autophagosomes and lysosomes, we first examined the effects of chloroquine (CQ), which represses the autophagic degradation through neutralizing lysosomal acidic pH [Citation22], on CSFV proliferation. The cytotoxicity of CQ was detected by performing CCK-8 test (). Cells infected with CSFV were treated with CQ (40 μM) for 4 h, followed by immunoblotting, real-time PCR, and immunofluorescence assays. The results showed that CQ significantly inhibited E2 expression (), progeny virus yields (–D), and the levels of viral releasing (). Notably, the antibody-resistant transmission and the infectivity of autophagic EVs were almost blocked with CQ treatment (), indicating that lysosomal acidic pH is a prerequisite for CSFV transmission and release.
Figure 11. CSFV release requires the formation of autolysosomes. (A) Detection of the cell viability with the treatment of CQ by CCK-8 test. (B-F) CSFV infected PK-15 cells were treated with CQ for 4 h at 20 hpi, then the cell samples were analyzed by western blot with anti-E2, anti-LC3B, anti-SQSTM1, and anti-ACTB/β-actin (loading control) antibodies(B), the virus titers of extracellular and intracellular were measured by endpoint dilution titrations by using immunofluorescence (C-D). (E) The ratios of CSFV titers in extracellular and intracellular were calculated to evaluate viral release. (F) CSFV infected plaques formed by intracellular cell lysates, extracellular culture supernatants (with or without antibody), and isolated autophagic EVs were detected by immunofluorescence. Scale bar: 200 μm. Results are shown as the mean ± SD (n = 3). ns, not significant; **, P < 0.01. (G) Detection of the autolysosomes in CSFV infected cells by using mCherry-GFP-LC3B. Transfected cells treated with DMSO, rapamycin, 3-MA, and CQ were used as control. Red arrows point to autolysosomes. (H) Detection of the autophagosome-lysosome fusion in CSFV-infected cells by confocal microscope. Autophagosomes were detected by anti-LC3B antibody (in green), lysosomes were detected by anti-LAMP2 antibody (in red). Cells treated with rapamycin and CQ used as control. (I) Detection of the CSFV in lysosomes. CSFV was detected by anti-E2 antibody (in green), lysosomes were detected by anti-LAMP2 antibody (in red). Cells treated with CQ used as control. Cell nuclei were stained with DAPI (in blue). Scale bar: 10 μm.
To confirm the formation of autolysosomes in CSFV-infected cells, we performed multiple fluorescence assays at 9 hpi. Firstly, the tandem-tagged reporter mCherry-GFP-LC3B, which appears red fluorescence in autolysosomes because of the loss of the GFP fluorescence in low-pH environment in lysosomes, was transfected into PK-15 cells and followed by DMSO, rapamycin, 3-MA, and CQ treatment or CSFV infection. The images of confocal showed several red fluorescence puncta in rapamycin-treated and CSFV-infected cells, but showed only yellow fluorescence in DMSO-, 3-MA-, and CQ-treated cells, indicating the formation of autolysosomes with CSFV infection (). To confirm this conclusion, we then examined the colocalization of autophagosome protein LC3B and lysosome protein LAMP2 to monitor the formation of autolysosomes. The results showed that the confocal puncta of LC3B and LAMP2, which appeared with rapamycin treatment and disappeared with CQ treatment, were observed in CSFV infected cells, indicating that the infection of CSFV indeed induces the formation of autolysosomes (). We finally detected the localization of CSFV in lysosomes, and found that CSFV was strongly localized in lysosomes. Interestingly, this localization disappeared with CQ treatment, indicating that the transportation of CSFV to lysosomes depends on the acidic pH of lysosomes (). These results demonstrated that the release and antibody-resistant transmission of CSFV requires the formation of autolysosomes. In conclusion, this study shows that the EVs originating from autophagy could mediate the antibody-resistant transmission of CSFV virions from infected to uninfected cells.
Discussion
The free spread is the basic transmission way for viral infections. However, this mode of transmission is limited by many barrier systems in the body, and therefore the efficiency is relatively low. Here, we described a new EV-dependent transmission of CSFV in cells in additional to the cell free spread mode. With the cell-to-cell contact or EV-dependent spreading mode, viruses can avoid the neutralization of antibodies and thus exerts an effective immune escape [Citation23,Citation24]. The antibody neutralizing experiments performed in this research confirmed this mechanism for virus immune evasion, in similar to previous studies in other virus species [Citation25–27]. As cell invaders, free virus particles are also relatively fragile to the extracellular environment, it could be affected not only by antibodies or the immune system, but also sensitive to chemicals or pH [Citation28]. With membrane structures, EVs could insulate the virions from the intercellular environment and provide protections for the virus during spread processes. From the point of infection, single free virus particle is also not an efficient way to establish new infections, the rate for successful infection by one single virus particle should be very low. But with EVs, multiple virions could be delivered to target cells, and therefore facilitate virus transmission and establishment of new infection. In this research, we observed EVs containing both single and multiple viruses, and captured the binding of multiple virions on the surface of target cells mediated by EVs, these findings support the idea of CSFV using the EVs system for successful transmission. Although we did not observe any CSFV particles at the cell contact areas with TEM, further studies are required to explore whether there are other cell-to-cell spreading mechanisms besides EVs for CSFV transmission.
EVs are membranous vesicles that can be secreted by all types of cells. They can transmit information and substances between cells, and even change the function of target cells [Citation29,Citation30]. According to the size of the vesicle, the EVs were divided into three subcategories: exosomes (30–150 nm), microvesicles (MVs; 100–1000 nm) and apoptotic body (>1000 nm) [Citation31]. From the results of electron microscope and vesicle size analysis, the diameter range of CSFV-encapsulated vesicles belongs to subcategories of MVs, so the differential centrifugation, a widely used protocol for MVs isolation [Citation32–34], was used to isolate the EVs containing virions. Further experiments to determine the origin and composition of CSFV-containing vesicles is necessary, but also difficult, because the size of vesicles is not clearly defined, and different types of EVs share labeling molecules. For example, the exosome marker CD81 was also detectable in CSFV-positive vesicles ().
To determine the origin of the virions-containing EVs, a more precise purification protocol is needed. Sucrose density-gradient ultracentrifugation and SEC are the most widely applied protocols for vesical purification [Citation35,Citation36]. Considering a loss of biological function of vesicles purified by sucrose density-gradient ultracentrifugation [Citation35,Citation37], we used the SEC for purifying specific vesicles in this study. The large particles that cannot enter the beads eluted early and the small delayed due to the increased length in pores of beads. In this study, the EVs are divided into 30 fractions of different size using SEC. Among them, the fraction 5 and 6 which contained CSFV virions were positive for autophagy marker LC3B and BECN1. Furthermore, CSFV virions were detectable in LC3B positive vesicles purified through immunoprecipitation, fully demonstrating that virions-containing EVs were originated from autophagy.
Autophagy is originally found to play major roles in keeping the cell balance by removing misfolded protein, clearing damaged cellular organelles, and also importantly, degradation of intracellular pathogens [Citation38]. While autophagy is a powerful tool to defend against viral infection in host cells, viruses have also developed a variety of strategies to fight against or utilize autophagy to promote their lifecycles [Citation39,Citation40]. Previous studies have shown that the macroautophagy pathway is activated by infection of CSFV, which facilitates viral intracellular release, replication and maturation in host cells [Citation16,Citation41,Citation42]. Besides traditional auto-digestive autophagies, in recent years it has been identified a non-degradative pathway, secretory autophagy, in which a single membrane vesicle is released to extracellular through the fusion of autophagosomes and plasma membrane [Citation43]. The secretory autophagy has been reported to possibly involve in extracellular release of several viruses, such as enteroviruses, coxsackievirus, and rhinovirus [Citation8,Citation44,Citation45]. However, these studies did not provide evidence of the association between such autophagy-related virus releasing and autolysosomes formation. In this study, we demonstrated that the autophagosome-lysosome fusion is a prerequisite for CSFV release in such autophagy-mediated way. That is, the autophagy-mediated CSFV release and transmission is independent of secretory type. Therefore, originations and biogenesis of the virion containing EVs should be explored to fully understand the EV-mediated virus spread mechanism.
Unlike the human immune deficiency virus type 1 virions preferentially at sites of cell contact [Citation46], CSFV seems to prefer to gather on the side away from contact with adjacent cells. As shown in immunohistochemistry, CSFV virions mainly distributed in the distal renal tubules, small intestinal gland cells, gastric gland cells, and other cells directly contacting the circulating fluid in the body, rather than stromal cells with high cell density and small gaps (Figure S4). This may be a strategy evolved to facilitate viral spreading. In this strategy, virions wrapped in vesicles can escape the neutralization of antibodies and thus spread quickly to all parts of the body with the circulation of bodily fluids.
In the context of infection, the transportation of viral components may make EVs a double-edged sword [Citation47]. On the one hand, EVs support the spread and pathogenicity of the virus; on the other hand, EVs can also transfer viral antigens to immune cells to induce an acquired immunity that can counteract the spread of the virus [Citation48,Citation49]. For examples, it has been reported that EVs released from hepatitis B virus-infected hepatocytes could transfer viral RNA [Citation50]. However, when the EVs is transferred to macrophages, NKG2D ligands would express, thus inducing the production of interferon-γ [Citation51,Citation52]. In addition, polyomavirus-induced vesicles can transfer micro-RNA from infected cells to uninfected cells, allowing the latter acquired immunity to polyomavirus [Citation53]. In CSFV-induced EVs, how the immune system participates in this battle is a subject worth studying.
In conclusion, this study proposes a new mechanism for CSFV spread, by which CSFV virions were shed in the autophagic EVs to escape the neutralization of antibodies ().
Figure 12. The schematic of EV-dependent transmission pattern in this study. In CSFV-infected cells, the newly formed virions accumulate in autophagosomes, and then enclosed into EVs through some unclear way. EVs carry the virions to uninfected cells. In this process of viral transmission, vesicles can protect virions from the neutralization of antibodies.
Materials and methods
Cells and viruses
Porcine kidney (PK-15) cells (ATCC, CCL-33) and swine testicle (ST) cells (National Infrastructure of Cell Line Resource of China, 1102SWI-NIFDC00090) were cultured in DMEM (Gibco, 11995040) containing 10% fetal bovine serum (Gibco, 10099141) at 37°C. The CSFV Shimen strain (GenBank: AF092448) was obtained from the China Institute of Veterinary Drug Control (Beijing, China).
Real-time PCR
Total cellular RNA was extracted using RNAiso Plus (Takara Bio, 9109) and reverse transcription was performed according to Prime Script RT reagent Kit (Takara Bio, RR047A) instructions. The real-time PCR was operated according to the TB Green™ Premix Ex Taq™ II (Takara Bio, RR820A) instruction manual, the procedure is: 95°C 10 min; 95°C 10 s, 60°C 30 s, 40 cycles. The primer sequences for real-time PCR are as follows: CSFV (F: 5′GATCCTCATACTGCCCACTTAC3′; R: 5′GTATACCCCTTCACCAGCTTG3′); ACTB/β-actin (F: 5′CAAGAACCTCTACGCCAACAC3′; R: 5′TGGAGGCGCGATGATCTT3′), ACTB/β-actin was used as internal reference.
Cell viability assay
The cell viability was measured by Cell Counting Kit-8 (CCK-8; Dojindo, CK04). In short, cells were inoculated on 96-well plates and cultured for 24–72 h. To test cell viability, the reagent (10 μL) was added in the cell culture and mixed thoroughly; then the cells were cultured at 37°C for 1 h. At this time, the color of the cell culture was changed to orange, and the absorbance values at 450 nm were measured by a microplate reader (Thermo Fisher Scientific). The cell-free treatment group served as a blank control. The concentrations of rapamycin (Sigma-Aldrich, V900930), 3-MA (Sigma-Aldrich, 189490), wortmannin (Med Chem Express, HY-10197), and CQ (Sigma-Aldrich, C6628) used in this study were determined from cell viability assay.
Western blot
The protein samples used in this study were derived from CSFV-infected or uninfected PK-15 cells. First, protein samples were obtained by lysing the cells with RIPA lysis buffer (Beyotime, P10013B) on ice, and then mixed with loading buffer (Invitrogen, NP0007) and boiled until denatured. Second, protein samples were separated by electrophoresis with 12% SAS-PAGE, and then transferred to PVDF membranes (Merck Millipore, 3010040001). Third, the membranes were blocked by skim milk at room temperature for 2 h, and then incubated with primary antibodies and secondary antibodies. Mouse anti-CSFV E2, rabbit anti-CSFV NS4B (made in our laboratory), mouse anti-CD81 (Proteintech, 66866-1-Ig), rabbit anti-BECN1 (Proteintech, 11306-1-AP), rabbit anti-LC3B (Sigma-Aldrich, L7543), rabbit anti-SQSTM1 (Proteintech, 18420-1-AP) and mouse anti-ACTB/β-actin (MultiSciences Biotech, 200–301-E69) antibodies were used in this study. Finally, an enhanced chemiluminescence (ECL; ZETA Life, 310208) was used to visualize the protein bands. Western blot analysis was repeated at least three times. ACTB/β-actin was used as an internal reference.
Indirect immunofluorescence assay and virus titration
The virus titer of CSFV was detected by indirect immunofluorescence assay. Gradient diluted virus solution (100 μL) was inoculated on a PK-15 cells monolayer grown on 96-well plates. After 72 h, cells were washed 3 times with phosphate-buffered saline (PBS, Thermo Fisher Scientific, 10010002) and fixed with 4% paraformaldehyde (Beyotime, P0099) at room temperature for 10 min; and then penetrated with 0.5% Triton-X100 (Solarbio Life Sciences, T8200) at room temperature for 15 min. After blocked with blocking buffer (Beyotime, P0260) at room temperature for 2 h, the cells were incubated with diluted mouse anti-CSFV E2 antibody (1:200) at 4°C for 12 h, followed by incubated with fluorescently labeled goat anti-mouse IgG (Yeasen, 33206ES60) at room temperature for 2 h. Nuclei were colored using 4′,6-diamidino-2-phenylindole (DAPI, Beyotime, C1006). Finally, the liquid in the culture plate was replaced with ultra-pure water. The results were observed using fluorescence microscope (Nikon) and recorded for statistical analysis. The virus titer as half of the tissue culture infection dose.
Transwell coculture assay
Transwell coculture system (polycarbonate membrane, 0.4 and 3.0 μm pore sizes, 24-well; Corning, 3413 and 3415) was assembled using CSFV-infected and uninfected PK-15 cells. PK-15 cells were infected with CSFV (MOI = 1) for 72 h, and then digested and seeded in transwells; uninfected PK-15 cells were seeded in 24-well plates. Then the transwells were moved into the corresponding wells of 24-well plates to create the coculture system. The system was cultured in the medium supplemented with neutralizing antibodies (10× ND50) which replaced every 24 h and incubated at 37°C for 72 h.
Fluorescence microscopy
For confocal assay, cells were transfected with pCMV-mCherry-GFP-LC3B (Beyotime, D2816) or labeled with specific antibodies using indirect immunofluorescence. The primary antibodies specific for ATG5 (10181-2-AP) and LAMP2 (27823-1-AP) were obtained from Proteintech. Mouse anti-LC3B (ab243506) was purchased from Abcam. The Alexa Fluor 488-conjugated goat anti-mouse (33206ES60) and Alexa Fluor 594-conjugated goat anti-rabbit (33112ES60) antibodies were obtained from Yeasen. The labeled cells were imaged with laser confocal scanning microscopy (Nikon, 100×).
Electron microscopy
When the cells grow to logarithmic phase (60%−80% confluence), CSFV (MOI = 1) was inoculated. After 12 h, cells were collected and centrifuged into a cell mass. First, the cell mass was fixed in cold glutaraldehyde (2.5%, pH 7.4; Sigma-Aldrich, G6257) at 4°C overnight, and then fixed in osmium tetroxide (1%, prepared with PBS; Ted Pella, 18456) at 4°C for 2 h. Second, the fixed cell mass was gradient dehydrated in different concentrations of ethanol (Sigma-Aldrich, E7023) and acetone (Sigma-Aldrich, 650501), and infiltrated in resin (SPI Supplies, 90529-77-4). The resin was embedded and solidified in an oven. Third, the sample was sequentially sliced with an ultra-thin slicer (Leica), and stained with 2% uranyl acetate (Zhongjingkeyi Technology, GZ02625) and lead citrate (Zhongjingkeyi Technology, GA10701). Finally, prepared samples were observed and imaged with an electron microscope (FEI, 80kV).
Isolation of EVs
PK-15 cells (about 1 × 109) were inoculated with CSFV of 10 MOI and cultured at 37°C; the cell culture medium was replaced by serum-free DMEM at 12 hpi. After 24 h, the culture supernatant (about 200 mL) was collected for differential centrifugation at 4°C. The steps of centrifugation were as follow: first at 500 g for 10 min, then at 2,500 g for 20 min, and last at 10,000 g for 30 min; the supernatant was discarded, and the pellet was resuspended in 50 mL PBS and centrifuged at 10,000 g for 30 min; finally, the pellet of EVs was resuspended in 1 mL PBS.
Size-exclusion chromatography
The Sepharose CL-2B (Cytiva, 17014001) of 10 mL was washed with PBS and filled into affinity columns (12 mL; Solarbio Life Sciences, DS0110) to form a column with diameter of 1.5 cm and height of 4.5 cm. The EVs isolated by differential centrifugation was loaded to the column and eluted with PBS, and the eluate was collected in 30 sequential fractions of 0.5 mL.
Statistical analysis
All data were expressed as mean ± standard deviation (SD). The data were analyzed by one-way ANOVA and unpaired t-test with Excel and GraphPad Prism 6 software. P < 0.05 was considered significant.
Supplemental Material
Download MS Word (3.3 MB)Acknowledgments
This work was supported by the National Natural Science Foundation of China under Grant [Project No.31772765]. We thank the Life Science Research Core Services, NWAFU (Kerang Huang and Yanqing Wang) for technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Lindenbach BD, Murray CL, Thiel HJ. Flaviviridae. In: Knipe DM, Howley PM, Cohen JI, et al., editors. Field’s virology. vol. 2. 6th ed. Philadelphia (PA): Lippincott Williams & Wilkins; 2013. p. 712–746.
- Tu C, Lu Z, Li H, et al. Phylogenetic comparison of classical swine fever virus in China. Virus Res. 2001;81(1–2):29–37.
- Lefkowitz EJ, Dempsey DM, Hendrickson RC, et al. Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018;46:D708–D717.
- Thiel HJ, Stark R, Weiland E, et al. Hog cholera virus: molecular composition of virions from a pestivirus. J Virol. 1991;65(9):4705–4712.
- Coronado L, Bohórquez JA, Muñoz-González S, et al. Investigation of chronic and persistent classical swine fever infections under field conditions and their impact on vaccine efficacy. BMC Vet Res. 2019;15(1):247.
- Zhong P, Agosto LM, Munro JB, et al. Cell-to-cell transmission of viruses. Curr Opin Virol. 2013;3(1):44–50.
- Mothes W, Sherer NM, Jin J, et al. Virus cell-to-cell transmission. J Virol. 2010;84(17):8360–8368.
- Chen YH, Du W, Hagemeijer MC, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160(4):619–630.
- Aydin H, Cook JD, Lee JE. Crystal structures of beta- and gammaretrovirus fusion proteins reveal a role for electrostatic stapling in viral entry. J Virol. 2014;88(1):143–153.
- Bose S, Song AS, Jardetzky TS, et al. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J Virol. 2014;88(8):3925–3941.
- Johnson DC, Huber MT. Directed egress of animal viruses promotes cell-to-cell spread. J Virol. 2002;76(1):1–8.
- Maas S, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 2017;27(3):172–188.
- Pant S, Hilton H, Burczynski ME. The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol. 2012;83(11):1484–1494.
- Wang T, Fang L, Zhao F, et al. Exosomes mediate intercellular transmission of porcine reproductive and respiratory syndrome virus. J Virol. 2018;92:e01734–17.
- Yang Y, Han Q, Hou Z, et al. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol Immunol. 2017;14(5):465–475.
- Pei J, Zhao M, Ye Z, et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy. 2014;10(1):93–110.
- Lin J, Wang C, Liang W, et al. Rab1A is required for assembly of classical swine fever virus particle. Virology. 2018;514:18–29.
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3(3):181–206.
- Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–398.
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455.
- Brimacombe CL, Grove J, Meredith LW, et al. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J Virol. 2011;85(1):596–605.
- Lee CY, Kam YW, Fric J, et al. Chikungunya virus neutralization antigens and direct cell-to-cell transmission are revealed by human antibody-escape mutants. PLoS Pathog. 2011;7(12):e1002390.
- Favoreel HW, Van Minnebruggen G, Adriaensen D, et al. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc Natl Acad Sci U S A. 2005;102(25):8990–8995.
- Narasimhulu V, Bellamy-McIntyre AK, Laumaea AE, et al. Distinct functions for the membrane-proximal ectodomain region (MPER) of HIV-1 gp41 in cell-free and cell-cell viral transmission and cell-cell fusion. J Biol Chem. 2018;293(16):6099–6120.
- Dutartre H, Clavière M, Journo C, et al. Cell-free versus cell-to-cell infection by human immunodeficiency virus type 1 and human T-lymphotropic virus type 1: exploring the link among viral source, viral trafficking, and viral replication. J Virol. 2016;90(17):7607–7617.
- Shahid MA, Abubakar M, Hameed S, et al. Avian influenza virus (H5N1); effects of physico-chemical factors on its survival. Virol J. 2009;6(1):38.
- Tkach M, Kowal J, Théry C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos Trans R Soc London B: Biol Sci. 2018;373(1737):20160479.
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–228.
- Boulanger CM, Loyer X, Rautou PE, et al. Extracellular vesicles in coronary artery disease. Nature reviews. Cardiology. 2017;14:259–272.
- Muralidharan-Chari V, Clancy J, Plou C, et al. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol. 2009;19(22):1875–1885.
- Sinha S, Hoshino D, Hong NH, et al. Cortactin promotes exosome secretion by controlling branched actin dynamics. J Cell Biol. 2016;214(2):197–213.
- Haraszti RA, Didiot MC, Sapp E, et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J Extracell Vesicles. 2016;5(1):32570.
- Böing AN, Van Der Pol E, Grootemaat AE, et al. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J Extracell Vesicles. 2014;3(1):10.3402.
- Momen-Heravi F. Isolation of extracellular vesicles by ultracentrifugation. Methods Mol Biol. 2017;1660:25–32.
- Ford T, Graham J, Rickwood D. Iodixanol: a nonionic iso-osmotic centrifugation medium for the formation of self-generated gradients. Anal Biochem. 1994;220(2):360–366.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12.
- Choi Y, Bowman JW, Jung JU. Autophagy during viral infection – a double-edged sword. Nat Rev Microbiol. 2018;16(6):341–354.
- Viret C, Rozières A, Faure M. Autophagy during early virus-host cell interactions. J Mol Biol. 2018;430(12):1696–1713.
- Zhu E, Wu H, Chen W, et al. Classical swine fever virus employs the PERK- and IRE1-dependent autophagy for viral replication in cultured cells. Virulence. 2021;12(1):130–149.
- Pei J, Deng J, Ye Z, et al. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy. 2016;12(10):1738–1758.
- Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116.
- Robinson SM, Tsueng G, Sin J, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10(4):e1004045.
- Jackson WT, Giddings TH Jr, Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3(5):e156.
- Phillips DM. The role of cell-to-cell transmission in HIV infection. AIDS. 1994;8(6):719–732.
- Nolte-’t Hoen E, Cremer T, Gallo RC, et al. Extracellular vesicles and viruses: are they close relatives? Proc Natl Acad Sci U S A. 2016;113(33):9155–9161.
- Criscuolo E, Castelli M, Diotti RA, et al. Cell-to-cell spread blocking activity is extremely limited in the sera of herpes simplex virus 1 (HSV-1)- and HSV-2-infected subjects. J Virol. 2019;93(11):e00070–19.
- Chahar HS, Bao X, Casola A. Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses. 2015;7(6):3204–3225.
- Kouwaki T, Fukushima Y, Daito T, et al. Extracellular vesicles including exosomes regulate innate immune responses to hepatitis B virus infection. Front Immunol. 2016;7:335.
- Kloss M, Decker P, Baltz KM, et al. Interaction of monocytes with NK cells upon toll-like receptor-induced expression of the NKG2D ligand MICA. The Journal of Immunology. 2008;181(10):6711–6719.
- Ebihara T, Masuda H, Akazawa T, et al. Induction of NKG2D ligands on human dendritic cells by TLR ligand stimulation and RNA virus infection. Int Immunol. 2007;19(10):1145–1155.
- Giannecchini S. Evidence of the mechanism by which polyomaviruses exploit the extracellular vesicle delivery system during infection. Viruses. 2020;12(6):E585.