
ABSTRACT
The mechanisms controlling immunosurveillance and immunoevasion often operate simultaneously to the triggering of the oncogenic signaling that results in tumor initiation. The resolution of the balance between anti-cancer immune responses and pro-tumorigenic pathways determines if a tumor cell survives and can remodel the microenvironment to reinforce immunosuppression or is eliminated by the immune system. Cancer cells must endure a toxic and metabolically challenging milieu. In its various forms, autophagy provides a way for transformed cells to survive by promoting catabolism and detoxification. Mounting evidence suggests that the boundaries between cancer immunity and mitogenic and metabolic programs are diffuse, with the same molecules likely serving several diverse roles in immunity and metabolism during tumor initiation and progression. Our recent data provide mechanistic detail and functional relevance of a new paradigm whereby the same signaling elements control immunity and autophagy in cancer.
Autophagy is a degradative process encompassing several mechanisms, including macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Much is known about how these mechanisms affect cell metabolic and detoxifying functions, and their main critical molecular players have been identified. However, how the three types of autophagy are potentially interconnected, and how their crosstalks might determine the outcome of a carcinogenic mechanism by impinging not only the metabolic but also the inflammatory characteristics of tumor cells are much less understood. This is important because identifying master regulators that orchestrate the cross-wiring of these processes might unveil unanticipated vulnerabilities to be exploited therapeutically.
Recent studies from our laboratories in hepatocellular carcinoma (HCC) have demonstrated that the atypical PRKC/PKC isoform PRKCI, in complex with SQSTM1, negatively regulates macroautophagy through the phosphorylation of MAP1LC3/LC3 at S12. In turn, macroautophagy activation by PRKCI deficiency in hepatocytes promotes HCC by feeding the OXPHOS machinery that results in the activation of a reactive oxygen species (ROS)-NFE2L2/NRF2-dependent pro-tumorigenic mechanism, and likely by disposing of the mitochondria damaged by excessive ROS production. Interestingly, in colorectal cancer, the loss of PRKCI in intestinal epithelial cells (IECs) stimulates mitogenic signaling in a cell-autonomous manner, concomitant with inflammation-driven tumorigenesis. Nonetheless, this pro-carcinogenic process is impaired by the simultaneous activation of the IFN (interferon) response that serves to activate a CD8+ T cell-mediated anti-tumor immunosurveillance pathway that prevents tumor initiation. Only the depletion of CD8+ T cells or the simultaneous shutdown of the IFN pathway by the inactivation of the other atypical PRKC, PRKCZ, unleashes the tumorigenic potential of PRKCI loss in IECs. Therefore, PRKCI suppresses tumorigenesis but also anti-cancer immunity.
Unbiased BioID approaches to identify potential partners of PRKCI revealed its well-known interactions with SQSTM1 and LC3, and other critical components of the autophagy machinery and the intracellular trafficking from the endoplasmic reticulum (ER) to the Golgi [Citation1]. This latter is relevant for anti-cancer immunity because the ER-Golgi transport of STING1 (the key protein that facilitates innate immune signaling) is an obligate step in the mechanisms leading to the activation of IRF3 and the IFN pathway. A critical PRKCI interactor identified in these unbiased BioID strategies is ULK2, which together with ULK1 are key apical activators of autophagy and have been proposed to stimulate the IFN cascade. Importantly, our data demonstrate that the phosphorylation of ULK2’s S150 by PRKCI serves to repress both the IFN pathway and macroautophagy by directly inhibiting ULK2’s enzymatic activity (). Consequently, the inactivation of PRKCI derepresses ULK2 leading to the phosphorylation of TBK1 at S172, a critical event in the STING1-mediated activation of the IFN cascade. Furthermore, we found that PRKCI curbs ULK2 function also by promoting its lysosomal degradation. Thus, our data demonstrate that the PRKCI-dependent phosphorylation of S150 in ULK2 promotes the K63-mediated ubiquitination of its K146 by the NDFIP1-NEDD4L ubiquitin ligase complex. This ubiquitination serves to target ULK2 to VPS4A-VPS4B-dependent lysosomal degradation, supportive of endosomal microautophagy. Therefore, PRKCI keeps the interferon cascade at bay by inhibiting ULK2 activity and targeting to microautophagy-mediated degradation. In this way, the activation of the IFN pathway is a safeguard to prevent tumorigenesis initiation in cells that lose PRKCI.
Figure 1. PRKCI controls the autophagy-interferon crosstalk to regulate cancer immunosurveillance. (Left panel) PRKCI phosphorylates and inactivates ULK2; ULK2 activates macroautophagy that represses the degradation of STING1 by chaperone-mediated autophagy; ULK2 also phosphorylates TBK1 at its S172 which serves to activate the STING1-mediated activation of the IFN pathway through phosphorylation of IRF3; PRKCI represses ULK2 activity by phosphorylating its S150 that also triggers the K63 ubiquitination of ULK2’s K146 through the NDFIP1-NEDD4L complex, targeting ULK2 for degradation through microautophagy; activation of macroautophagy promotes the IFN cascade through inhibition of CMA. (Right panel) The inactivation of PRKCI unleashes autophagy and the IFN pathway that serves to counteract the pro-tumorigenic effect of PRKCI deficiency, restraining cancer initiation and progression.
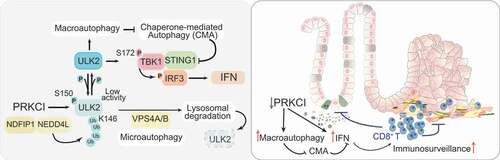
Interestingly, the unleashing of macroautophagy by PRKCI loss has another unexpected consequence. That is, one of the characteristics of PRKCI-deficient IECs is the upregulation of STING1 levels, which contributes to the hyperactivation of the IFN pathway by the unrestrained ULK2-TBK1 axis. We showed that inhibition of macroautophagy renders PRKCI-deficient cells IFN-inactive through a decrease in STING1 levels, which can be rescued, and IFN-signaling reactivated, if CMA (a pathway previously shown to contribute to STING1 degradation) is inhibited (). This observation suggests that activation of macroautophagy by PRKCI inhibition results in reduced CMA activity and the subsequent STING1 accumulation, which reinforces the existence of compensatory mechanisms between autophagic pathways, and places PRKCI at a privileged location in the crosstalk of the different autophagy mechanisms. In this regard, reduced levels of PRKCI serves to maintain metabolic and stress homeostasis (OXPHOS-ROS-NFE2L2) that promote tumorigenesis, but at the same time unleashes immunosurveillance mechanisms (IFN) required to keep tumors at bay. This new unexpected dual and antagonistic function of PRKCI in human cancer was validated by single-cell multiplex immunofluorescence and bioinformatics analysis of human CRC specimens demonstrating that low PRKCI levels correlate with enhanced IFN signaling and good prognosis.
These new data have several important corollaries. For example, even though in some systems blocking macroautophagy might prevent immunoevasion through the upregulation of MHC class I proteins, it might also impair immunosurveillance by promoting the CMA-mediated degradation of STING1, especially in tumors with low levels of PRKCI and high levels of PRKCZ. Therefore, cell context and the load of atypical PRKCs are critical questions to keep in mind when considering autophagy and immunotherapy as potential anti-cancer strategies. In this regard, our own data demonstrating in vivo that established tumors are reduced upon the inducible genetic inactivation of PRKCI, which results in a massive recruitment of CD8+ T cells to otherwise immune “cold” tumors, suggests that inhibition of PRKCI could be a viable strategy to boost immunotherapy. However, several questions need to be critically pondered when this therapeutic route is considered. First, if the tumors become immunosuppressed, inhibiting PRKCI will most likely lead to more tumorigenesis, owing to its role as a tumor suppressor. Second, the loss of PRKCI in hepatocytes results in oxidative stress and pro-tumorigenic signaling. This could be a red flag in the utilization of PRKCI inhibitors as anti-cancer drugs. However, it is possible that some of the chronic hepatic toxicity observed in the genetic mouse models in which PRKCI is lost at very early stages, could be minimized in the case of its pharmacological acute inhibition.
What is becoming apparent is that cells use the same molecules to impinge into diverse and often conflicting functions, which means that a more comprehensive understanding of the molecular and cellular mechanism of new targets is needed when considering novel cancer therapies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Linares JF, Zhang X, Martinez-Ordonez A, et al. PKCλ/ι inhibition activates an ULK2-mediated interferon response to repress tumorigenesis. Mol Cell. 2021. DOI:https://doi.org/10.1016/j.molcel.2021.08.039.