
ABSTRACT
ER-specific autophagy (reticulophagy) has emerged as a critical degradative route for misfolded secretory proteins. Our previous work showed that RTN3 (reticulon 3) drives reticulophagic clearance of disease-causing mutant prohormones. How RTN3, a protein residing on the cytosolic leaflet of the ER bilayer, recruits these lumenally-localized cargos has remained a mystery. To address this question, we used an unbiased proteomics approach to identify RTN3-interacting partners. We discovered that RTN3 recruits misfolded prohormones for lysosomal degradation through the ER transmembrane protein PGRMC1. RTN3 complexes with PGRMC1, which directly binds to misfolded prohormones via its distal ER lumenal domain. Cargos for the RTN3-PGRMC1 degradative axis include mutant POMC (proopiomelanocortin) and proinsulin, each of which oligomerizes in the ER during misfolding, entrapping their wild-type counterparts, leading to secretion defects. Although reticulophagy is thought to degrade large protein aggregates, PGRMC1 instead selectively recruits and promotes degradation of only small oligomers of the mutant prohormones. Of physiological importance, genetic or pharmacological inactivation of PGRMC1 in pancreatic β-cells expressing both wild-type and mutant proinsulin impairs mutant proinsulin turnover and promotes trafficking of wild-type proinsulin. These findings pinpoint PGRMC1 as a possible intervention point for diseases caused by ER protein retention.
Maintaining protein homeostasis is essential for cellular viability. Because the endoplasmic reticulum (ER) is a major site of protein biosynthesis, it is not surprising that the ER is endowed with elaborate protein quality control pathways, which ensure that polypeptides entering the ER are folded properly, or, for those that cannot fold, are removed efficiently. Whereas pathways such as the unfolded protein response (UPR) and ER-associated degradation (ERAD) have well-established roles in ER protein quality control, the contribution of reticulophagy is still being uncovered. To date, six ER transmembrane proteins (ATL3, CCPG1, RETREG1/FAM134B, RTN3, SEC62, and TEX264) have been identified in the targeting of substrates for reticulophagy. Each targeting complex promotes turnover of the bulk ER when autophagy is induced. In addition to this role in the recycling of general ER content, RETREG1/FAM134B and RTN3 facilitate the degradation of select ER subdomains that contain specific misfolded protein clients.
We previously identified mutant forms of proinsulin and POMC as degradative substrates for the RTN3 targeting complex. Loss of RTN3 blocks the lysosomal delivery of these prohormone mutants and causes these secretory proteins to accumulate in detergent-insoluble complexes that appear as large puncta within the ER. The relationship between mutant POMC or mutant proinsulin and RTN3 cannot be replicated by any of the other known reticulophagy receptor-targeting complexes. We therefore hypothesized that RTN3 must have the ability to recruit these degradative cargos through physical engagement.
How RTN3 recruits cargo for selective degradation has been an enigma, as RTN3 adopts a unique topology: through its two double-hairpin domains, RTN3 is restricted to the cytosol-facing leaflet of the ER membrane (). Therefore, RTN3 cannot bind directly to ER lumenal cargo. We hypothesized that RTN3 must instead form a complex with a bona fide ER transmembrane protein, which can serve as a dedicated cargo receptor, bridging the gap between RTN3 and the ER lumen. Using immunoprecipitation-mass spectrometry to identify putative RTN3-interacting partners, we uncovered PGRMC1 as an RTN3-binding partner that engages mutant POMC and proinsulin and is functionally required for their proper autophagy-dependent clearance [Citation1].
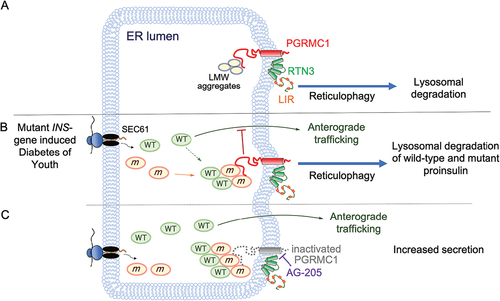
Figure 1. Proposed model for the role of PGRMC1 in proinsulin quality control and MIDY. (A) PGRMC1 links misfolded, low molecular weight reticulophagy cargo to RTN3, allowing for entry into the reticulophagy pathway. (B) A depiction of the MIDY condition, where wild-type (WT) proinsulin molecules are bound by mutant proinsulin molecules. The mutant-wild-type complexes are degraded and trafficking of wild-type proinsulin is impaired. (C) Pharmacological inactivation of PGRMC1 using AG-205 prevents the degradation of wild-type-mutant proinsulin complexes, which facilitates ER exit of wild-type proinsulin. LIR, LC3-interacting region; LMW, low molecular weight.
Loss of PGRMC1 phenocopies loss of RTN3: depletion of PGRMC1 causes mutant POMC to accumulate in detergent-insoluble fractions, which can be visualized as bright puncta under confocal microscopy. Loss of PGRMC1 specifically impairs the turnover of mutant POMC, as determined by translational shutoff experiments. To confirm that PGRMC1 operates within the RTN3 pathway (as opposed to a parallel degradative route), we conducted knockdown-rescue experiments. Mutant POMC accumulation caused by PGRMC1 knockdown can be reversed by re-expression of exogenous PGRMC1. However, when RTN3 is additionally knocked down, PGRMC1 rescue is ineffective, demonstrating that RTN3 acts downstream of PGRMC1.
Having established a requirement for PGRMC1 in RTN3-mediated reticulophagy, we sought to dissect the mechanistic basis by which PGRMC1 recognizes its substrate. Through cell-based co-immunoprecipitation experiments and an in vitro binding assay, we found that PGRMC1 physically interacts with mutant misfolded POMC. Membrane topology experiments revealed that PGRMC1 is a type II transmembrane protein with its C terminus positioned in the ER lumen, and this C-terminal region is necessary and sufficient for binding to mutant POMC. Additionally, we mapped the cargo-binding region of PGRMC1 to its extreme C terminus, as deleting the last 25 amino acids of PGRMC1 abolishes cargo interaction.
“Akita” proinsulin is the product of one of approximately 30 different INS-gene mutant alleles that can bring about the rare diabetic syndrome known as Mutant INS-gene-induced Diabetes of Youth (MIDY). We previously identified the Akita MIDY mutant to be a substrate of RTN3. As PGRMC1 is an RTN3-membrane binding partner, we asked if degradation of Akita might also be dependent on PGRMC1 and unexpectedly found that it was not. Our findings prompted us to evaluate if other MIDY mutants utilized the PGRMC1-RTN3 receptor complex for degradation.
Upon screening for six additional MIDY mutants, we found that while every proinsulin mutant tested requires RTN3 for clearance, only a subset of them requires PGRMC1. Comparison of PGRMC1-dependent versus PGRMC1-independent MIDY substrates revealed a surprising commonality: all PGRMC1-dependent substrates form relatively small oligomers, whereas PGRMC1-independent substrates form large complexes. Consistent with these results, PGRMC1 selectively interacts with mutant proinsulin that forms small oligomers. In further support of this idea, the previously described PGRMC1-dependent mutant POMC cargo also forms small oligomers. Based on these data, we concluded that PGRMC1 engages and targets smaller-sized cargos for reticulophagic clearance.
MIDY is a dominant-negative condition caused by heterozygous mutations in the INS gene. When a MIDY proinsulin undergoes oxidative folding in the ER of pancreatic β-cells, it binds and entraps the wild-type counterpart through cysteine mispairing, leading to defects in wild-type proinsulin trafficking (). To assess the physiological significance of the PGRMC1-mediated degradation pathway in the context of MIDY, we employed a cell-based MIDY model. Pancreatic β-cells expressing both wild-type and mutant proinsulin were treated with an inhibitor for PGRMC1 (AG-205). Strikingly, inactivation of PGRMC1 leads to increased wild-type proinsulin trafficking while blocking the degradation of mutant proinsulin. An outstanding question is how PGRMC1 inactivation increases wild-type proinsulin secretion under the MIDY condition. Because loss of PGRMC1 stabilizes the MIDY proinsulin (which normally binds to and entraps wild-type proinsulin in the ER), one possibility is that blocking the degradation of these mutant-wild-type proinsulin complexes increases the opportunity for wild-type proinsulin to escape the ER, thereby improving the impaired secretion that represents the hallmark of this disease ().
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Chen YJ, Knupp J, Arunagiri A, et al. PGRMC1 acts as a size-selective cargo receptor to drive ER-phagic clearance of mutant prohormones. Nat Commun. 2021;12:5991.