
ABSTRACT
The STX17-SNAP29-VAMP8 SNARE complex mediates autophagosome-lysosome fusion. Our recent study showed that MTOR directly phosphorylates VAMP8ʹs T48 residue in nutrient-rich conditions. Phosphorylated VAMP8 inhibits autophagosome-lysosome fusion by blocking STX17-SNAP29-VAMP8 SNARE complex formation. Our study also showed that SCFD1 is a previously unrecognized macroautophagy/autophagy regulatory protein, which can be recruited by VAMP8 (in its non-phosphorylated form) to autolysosomes, where it promotes STX17-SNAP29-VAMP8 complex assembly – and consequently promotes autophagosome-lysosome fusion. Moreover, we observed that mice harboring a phosphomimic VAMP8 variant accumulate aberrantly high lipid levels in their livers. VAMP8 phosphorylation can disrupt autophagosome-lysosome fusion in the liver and thereby dysregulate lipid metabolism. Beyond providing insights into the molecular mechanisms of autophagosome maturation, our study suggests that modulating autophagic SNARE function may help treat liver lipid disorders.
Autophagy is a highly conserved intracellular degradative process in which targeted substrates are sequestered by phagophores, which mature into autophagosomes, and then fuse with lysosomes for degradation. The STX17-SNAP29-VAMP8 SNARE complex functions in autophagosome-lysosome fusion, and recent studies have demonstrated impacts from post-translational modification of SNARE proteins – as regulated by various upstream signals – in regulating autophagy. For example, SNARE complex formation is inhibited by the acetylation of STX17 and by the O-GlcNAcylation of SNAP29, whereas the phosphorylation of STX17 promotes autophagy initiation.
In our recent study [Citation1], we detected phosphorylated VAMP8 in HEK293T cells, and noted that VAMP8 phosphorylation was dramatically decreased upon autophagy induction (via EBSS or MTORC1 inhibition). Consistently, VAMP8 phosphorylation was also decreased in mouse livers when MTORC1 activity was attenuated by fasting. Given that both MTORC1 and VAMP8 are resident proteins on lysosomes, we hypothesized that the kinase MTORC1 may phosphorylate VAMP8. To assess this hypothesis, we knocked down RPTOR/Raptor (a subunit of MTORC1) and detect reduced VAMP8 phosphorylation. Moreover, in vitro kinase assays and mass spectrometry confirmed that VAMP8ʹs T48 residue is a direct substrate of MTORC1.
Our study explored autophagy-related impacts of this VAMP8 phosphorylation using a variety of assays. Autophagy flux assays performed in VAMP8-knockdown U2OS cells completed with wild-type VAMP8, phosphomimic VAMP8, or dephosphomimic VAMP8 show an elevated SQSTM1/p62 and LC3 level in cells expressing the VAMP8 phosphomimic variant, indicating that VAMP8 phosphorylation impairs autophagy flux. The VAMP8-knockdown cells also show reduced colocalization of the autophagosome marker LC3 with the lysosome marker LAMP2, and this reduction is rescued by VAMP8[2A] (the dephosphorylation mimic variant) but not VAMP8[2D] (the phosphorylation mimic variant). mRFP-GFP-LC3 assays revealed that cells expressing VAMP8[2D] have more intense yellow puncta (mRFP+, GFP+) than cells with WT VAMP8, indicating an accumulation of autophagosomes in the former. Finally, when we reconstituted WT VAMP8, VAMP8[2A], and VAMP8[2D] liposomes and mimicked autophagosome-lysosome fusion in vitro, we observe a dramatic decrease in VAMP8[2D]-liposome (mimicked autophagosome) fusion, and an increase for VAMP8[2A]. These results collectively established that VAMP8 phosphorylation inhibits autophagosome-lysosome fusion ().
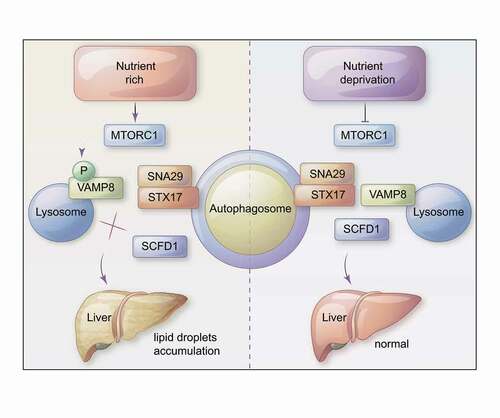
Figure 1. A model showing a role of MTOR in hepatic lipid metabolism. In nutrient-rich conditions (left panel), MTORC1 directly phosphorylates VAMP8, thereby inhibiting autophagosome-lysosome fusion by blocking STX17-SNAP29-VAMP8 SNARE complex formation. Upon nutrient deprivation, MTORC1 activity is inhibited (right panel) and SCFD1 accelerates STX17-SNAP29-VAMP8 complex formation. We also determined that differential VAMP8 phosphorylation of SCFD1 can cause dysregulation of lipid metabolism in the livers of mice.
We then explored the mechanism through which VAMP8 phosphorylation inhibits autophagosome-lysosome fusion. We initially noted that MTORC1 inhibition (Torin treatment) increased the strength of VAMP8-STX17 and VAMP8-SNAP29 interactions. Subsequent work confirmed that VAMP8 phosphorylation mimic variants weaken SNARE formation. Immunofluorescence assays then showed that VAMP8 phosphorylation decreases the colocalization of STX17 and LC3, supporting the idea that the observed inhibitory effect of VAMP8 phosphorylation on autophagosome-lysosome fusion is mediated through weakening of autophagic SNARE formation.
Our study also included tandem affinity purification (TAP) assays in Tet-on Flag-VAMP8 HEK293T cells to identify VAMP8 interactors. We found that the SM-like protein SCFD1 interacts with VAMP8, and coimmunoprecipitation assays later confirmed that SCFD1 interacts with VAMP8 and can also interact with STX17. Intrigued, we experimentally pursued potential autophagy-related impacts of SCFD1 and ultimately found that SCFD1 knockdown leads to LC3 puncta accumulation in U2OS cells. Consistently, both mRFP-GFP-LC3 and autophagy flux assays confirmed that SCFD1 knockdown blocks autophagosome-lysosome fusion. We then narrowed our focus and examined the effects of SCFD1 on STX17-SNAP29-VAMP8 SNARE complex formation. The interaction of VAMP8 and STX17 is dramatically decreased in SCFD1 knockdown cells. Further, we found that MTOR activation decreases the strength of the SCFD1-VAMP8 interaction, thus implicating VAMP8 phosphorylation in SCFD1ʹs regulation of autophagic SNARE formation. Thus, SCFD1 is a previously unrecognized autophagy regulatory protein, which can be recruited by VAMP8 (in its non-phosphorylated form) to autolysosomes, where it promotes STX17-SNAP29-VAMP8 complex assembly – and consequently autophagosome-lysosome fusion.
There is accumulating evidence to support the idea that autophagy serves as a quality control mechanism in maintaining hepatic lipid homeostasis. Inspired by these findings, we worked with a mouse model and used AAV9 virus injection to induce liver-specific VAMP8 (and mutant variants) expression. Briefly, hepatic autophagy flux assays revealed the expected reduced autophagy in livers expressing the VAMP8[2D] (phosphomimic variant), and we found that these VAMP8[2D] livers have dramatically elevated accumulation of lipids. These results extended our basic insights about VAMP8 into broader physiological relevance, demonstrating that VAMP8 phosphorylation can disrupt autophagosome-lysosome fusion in the liver and thereby dysregulate lipid metabolism.
In conclusion, our study demonstrates a mechanism in which differential phosphorylation of the autophagic SNARE component VAMP8 mediates autophagosome-lysosome fusion. The kinase MTOR phosphorylates VAMP8, and this modification inhibits STX17-SNAP29-VAMP8 SNARE complex formation. Moreover, our study establishes SCFD1 as a regulator of autophagosome-lysosome fusion, showing that VAMP8 phosphorylation disrupts SCFD1ʹs role in accelerating STX17-SNAP29-VAMP8 complex formation. In mice, a dephosphomimic variant of VAMP8 dramatically rescues an aberrant lipid droplet accumulation phenotype that we initially observed in the livers of mice carrying a phosphomimic VAMP8 variant (). Given that hepatic lipid metabolism disorders are linked to liver disease including nonalcoholic fatty liver disease/NAFLD and nonalcoholic steatohepatitis/NASH, among others, our study suggests that targeting the autophagic SNARE VAMP8 to reduce its phosphorylation may enable innovative treatments for several liver diseases.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
Reference
- Huang H, Ouyang QQ, Zhu M, et al. mTOR-mediated phosphorylation of VAMP8 and SCFD1 regulates autophagosome maturation. Nat Commun. 2021;12(1). DOI:https://doi.org/10.1038/s41467-021-26824-5