
ABSTRACT
Macroautophagy/autophagy, a fundamental cell process for nutrient recycling and defense against pathogens (termed xenophagy), is crucial to human health. ATG16L2 (autophagy related 16 like 2) is an autophagic protein and a paralog of ATG16L1. Both proteins are implicated in similar diseases such as cancer and other chronic diseases; however, most autophagy studies to date have primarily focused on the function of ATG16L1, with ATG16L2 remaining uncharacterized and understudied. Overexpression of ATG16L2 has been reported in various cancers including colorectal, gastric, and prostate carcinomas, whereas altered methylation of ATG16L2 has been associated with lung cancer formation and poorer response to therapy in leukemia. In addition, ATG16L2 polymorphisms have been implicated in a range of other diseases including inflammatory bowel diseases and neurodegenerative disorders. Despite this likely role in human health, the function of this enigmatic protein in autophagy remains unknown. Here, we review current studies on ATG16L2 and collate evidence that suggests that this protein is a potential modulator of autophagy as well as the implications this has on pathogenesis.
Abbreviations: ATG5: autophagy related 5; ATG12: autophagy related 12; ATG16L1: autophagy related 16 like 1; ATG16L2: autophagy related 16 like 2; CD: Crohn disease; IBD: inflammatory bowel diseases; IRGM: immunity related GTPase M; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; PE: phosphatidylethanolamine; RB1CC1: RB1 inducible coiled-coil 1; SLE: systemic lupus erythematosus; WIPI2B: WD repeat domain, phosphoinositide interacting 2B
Introduction
Autophagy is the critical cellular self-degradative pathway for nutrient recycling, removal of damaged organelles and defective proteins. There are three types of autophagy characterized: macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy (herein referred to as autophagy) is the most well-studied and has been comprehensively reviewed in [Citation1-3]. Xenophagy, a form of selective autophagy, is an important component of the innate immune system capable of selectively tagging and degrading intracellular pathogens such as bacteria, viruses and fungi (see reviews on xenophagy [Citation4, Citation5] and other forms of selective autophagy [Citation6, Citation7]. Autophagy involves the initial formation and expansion of a phagophore, which is then joined to create a double-membrane vesicle (autophagosome) enclosing the sequestered cytoplasmic content. The autophagosome is then fused with lysosomes to degrade the enclosed material [Citation1, Citation8, Citation9].
Autophagy is highly conserved in all eukaryotic organisms and is mediated by >30 proteins called autophagy-related (ATG) proteins [Citation10]. It plays a critical role in maintaining homeostasis, stress response, innate immunity, regulation of inflammation and tumor suppression. Dysregulated autophagy has been implicated in a wide range of cancers [Citation11] and chronic inflammatory diseases [Citation12] including inflammatory bowel diseases (IBD) and systemic lupus erythematosus (SLE) (reviewed in [Citation13]). Therefore, uncovering the complex role of autophagy is essential to understanding carcinogenesis and has broad implications for other human diseases.
ATG16L2 (autophagy related 16 like 2) is an autophagic protein and a paralog of ATG16L1, another key autophagic protein for autophagosome formation. ATG16L2 has been increasingly associated with growth [Citation14] and ageing [Citation15], various autoimmune diseases, neurodegenerative diseases and cancers [Citation16], as well as their response to chemotherapy [Citation17, Citation18]. Despite being associated with human diseases, the exact function of ATG16L2 remains unknown, precluding a complete understanding of autophagy. Therefore, in this review, we examine current research on ATG16L2 and provide insights into its role in autophagy as well as discuss its importance in disease etiology and therapeutic interventions.
Structural differences between ATG16L1 and ATG16L2
ATG16L2 contains 18 exons and has a 38.83% amino acid similarity with ATG16L1, which contains 19 exons. There are also two isoforms of ATG16L2 (α and β) compared to three in ATG16L1 (α, β and γ). ATG16L2α is missing exon 8, while ATG16L2β contains all 18 exons and is the main ATG16L2 isoform expressed, in contrast to ATG16L1 where isoform expression is tissue-specific [Citation19].
Unlike ATG16L1, which is present in all eukaryotes, ATG16L2 was initially thought to have evolved relatively recently and was only primarily present and conserved in all mammals [Citation19]. However additional genome sequencing has identified ATG16L2 homologs in other animals including zebrafish (Danio rerio), western claw frog (Xenopus tropicalis) and spotted gar (Lepisosteus oculatus) (). Additionally, an ATG-16.2 homolog is also found in Caenorhabditis elegans although it is distantly related and forms a separate branch to vertebrate-specific ATG16L2 [Citation19]. In C. elegans, both ATG-16.1 and ATG-16.2 are required for autophagy and have partially redundant roles in recruiting lipidated LGG-1 (GABARAPL2/Atg8 homolog in C. elegans) as deletion of either corresponding gene impairs autophagy, while a double deletion further exacerbates this impairment [Citation20]. While ATG-16.2 is required for autophagy in C. elegans, it was found to be more similar to mammalian ATG16L1 than ATG16L2 [Citation19], suggesting a different role for ATG16L2 in mammals and other vertebrates.
Figure 1. The phylogenetic relationship, domain structure comparison and tissue gene expression of ATG16L1 and ATG16L2. (A) The phylogenetic relationship of ATG16L1 and ATG16L2 sequences in selected species from UniProt. The maximum likelihood tree was constructed using Mega X and illustrates two distinct ATG16L branches corresponding to ATG16L1 (blue) and ATG16L2 (green). Bootstrap values are shown. UniProt identifiers are shown in brackets. (B) Comparison of the domain structures in human ATG16L1 and ATG16L2. The UniProt canonical isoform for each protein (ATG16L1β and ATG16L2β) is depicted with the major domains and SNPs highlighted. The three ATG16L regions (N, M and C) are shown. Known domains and interaction sites are highlighted in colored blocks (coiled coil, yellow; ATG5 binding site, green; RB1CC1, blue; and WIPI2, magenta). ATG16L1 and RAB33B interact with ATG16L1 and L2 in the M-region as shown. ATG16L2 rs11235604 (R220W) and ATG16L1 rs2241880 (T300A) are displayed in red. (C) Heatmap illustrating the expression of ATG16L1 and ATG16L2 in different tissues. The color indicates the relative expression, in transcripts per million (TPM), of ATG16L1 and ATG16L2. The gene expression data shown in this manuscript were obtained from the Genotype-Tissue Expression (GTEx) Multi gene query[Citation31] on 31/08/2021.
![Figure 1. The phylogenetic relationship, domain structure comparison and tissue gene expression of ATG16L1 and ATG16L2. (A) The phylogenetic relationship of ATG16L1 and ATG16L2 sequences in selected species from UniProt. The maximum likelihood tree was constructed using Mega X and illustrates two distinct ATG16L branches corresponding to ATG16L1 (blue) and ATG16L2 (green). Bootstrap values are shown. UniProt identifiers are shown in brackets. (B) Comparison of the domain structures in human ATG16L1 and ATG16L2. The UniProt canonical isoform for each protein (ATG16L1β and ATG16L2β) is depicted with the major domains and SNPs highlighted. The three ATG16L regions (N, M and C) are shown. Known domains and interaction sites are highlighted in colored blocks (coiled coil, yellow; ATG5 binding site, green; RB1CC1, blue; and WIPI2, magenta). ATG16L1 and RAB33B interact with ATG16L1 and L2 in the M-region as shown. ATG16L2 rs11235604 (R220W) and ATG16L1 rs2241880 (T300A) are displayed in red. (C) Heatmap illustrating the expression of ATG16L1 and ATG16L2 in different tissues. The color indicates the relative expression, in transcripts per million (TPM), of ATG16L1 and ATG16L2. The gene expression data shown in this manuscript were obtained from the Genotype-Tissue Expression (GTEx) Multi gene query[Citation31] on 31/08/2021.](/cms/asset/fa4c57e0-a39a-4c49-b466-e13bbd84e908/kaup_a_2042783_f0001_oc.jpg)
Both ATG16L2 and ATG16L1 share many structural motifs (). This includes the N-terminus region, M region and C terminus region. The N-terminus region in ATG16L1 and ATG16L2 binds to ATG12 (autophagy related 12)–ATG5 [Citation19]. The C terminus region contains seven WD40 repeats. In ATG16L1, these WD40 repeats are not required for canonical autophagy; however, they are important for the recruitment of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3), a homolog of Atg8, for single membrane lipidation in non-canonical autophagy [Citation21]. Whether WD40 repeats in ATG16L2 are also important in non-canonical autophagy is unknown. Finally, the M region contains a coiled-coil (CC) domain, which in ATG16L1, interacts with several important proteins including other ATG16L1 and ATG16L2 proteins, RAB33B (a small GTPase), WIPI2B (WD repeat domain, phosphoinositide interacting 2) and RB1CC1/FIP200 (RB1 inducible coiled-coil 1) [Citation22-25]. The primary structural difference between ATG16L2 and ATG16L1 lies within the M region [Citation19], and this has important implications for functional differences between ATG16L2 and ATG16L1.
ATG16L2 is a potential autophagic inhibitor
ATG16L1 is required for autophagosome formation and together with ATG5 and ATG12, forms an E3-like ubiquitin ligase complex (reviewed in [Citation26]). During expansion of the phagophore membrane, ATG16L1 forms a homodimer and recruits the ATG12–ATG5 complex to the phagophore, creating a complex (ATG12–ATG5-ATG16L1) [Citation27]. This complex determines the site for lipidation of LC3 to phosphatidylethanolamine (PE), an essential step for fusion of the phagophore membrane and formation of the autophagosome [Citation27] ().
Figure 2. Schematic overview summarizing the role of ATG16L1 and ATG16L2 in autophagy. (A) ATG16L1 (purple circle) complexes with ATG12–ATG5 (red and blue circles, respectively), leads to LC3 lipidation and autophagosome formation. (B) Effect of ATG16L2 overexpression. ATG16L2 (orange circle) is a putative dominant negative inhibitor of ATG16L1, therefore, ATG16L2 competes with ATG16L1 to bind with ATG12–ATG5 complexes. ATG16L2 lacks WIPI2 and RB1CC1 interaction sites, hence ATG12–ATG5-ATG16L2 complexes are not recruited to the phagophore membrane resulting in reduced LC3 lipidation and autophagy.
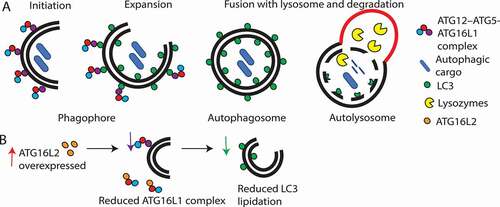
In humans, ATG16L2 can form a homodimer with itself or a heterodimer with ATG16L1 [Citation19]. Furthermore, the ATG16L2 N terminus region can also bind to ATG12–ATG5 with similar affinity as ATG12–ATG5-ATG16L1 [Citation19]. Despite these similarities, in most studies mammalian-specific ATG16L2 was not identified to be required for autophagy or LC3 lipidation to PE [Citation19]. Deletion of ATG16L1 impairs autophagy, leads to accumulation of LC3-I and results in increased pro-inflammatory cytokines (IL1B/IL-1B and IL18) [Citation27]. In mouse pancreatic acinar cells, ATG16L2 knockdown also appears to increase LC3-II accumulation [Citation28]. However, in a separate study, knockdown of ATG16L2 in mouse embryonic fibroblasts did not affect autophagy or lead to increased LC3-I and IL1B [Citation19]. It is possible that ATG16L2 may have different functions in different cell types, leading to the reported differences. It is also possible that the differences may be due to off-target effects from siRNA knockdown. A follow up study found that atg16l2 knockout in mice does not result in embryonic lethality in contrast to knockout of Atg16l1 [Citation29]. Furthermore, atg16l2 knockout mice follow mendelian ratios and show no defects in autophagy in various cell types [Citation29]. Double deletions or knockdowns of Atg16l2 and Atg16l1 also do not lead to greater impairment or accumulation of LC3-I and IL1B production [Citation19, Citation29]. Together, these studies demonstrate that ATG16L2 does not compensate or rescue the loss of ATG16L1 nor does its depletion exacerbate autophagy impairment. Thus, ATG16L2 does not appear to be required for LC3 lipidation to PE during autophagosome formation.
In addition to ATG12–ATG5, ATG16L1 also interacts with three other proteins (RAB33B, RB1CC1, WIPI2B) in its M-region. This interaction recruits the ATG12–ATG5-ATG16L1 complex to the phagophore membrane for LC3 lipidation [Citation22-25]. While ATG16L2 can bind RAB33B, albeit at a slightly lower affinity [Citation19], one key difference between ATG16L1 and ATG16L2 is that ATG16L2 cannot interact with RB1CC1 [Citation23] or WIPI2B [Citation22] due to differences in its M region. This important difference means that ATG12–ATG5-ATG16L2 complexes are not recruited to the phagophore membrane for LC3 lipidation [Citation22, Citation24]. Indeed, ATG16L2 was found to not localize to the phagophore membrane and remained in the cytosol [Citation19]. Interestingly, a recent study in prostate adenocarcinoma cells suggested that ATG16L2 may act as a competitive inhibitor of ATG16L1. Overexpression of ATG16L2 reduced ATG12–ATG5-ATG16L1 complexes and LC3 lipidation [Citation30] (). It is suggested that binding of ATG16L2 to ATG12–ATG5 displaces and prevents ATG16L1 from binding, leading to ATG16L1 polyubiquitination and its proteasomal degradation [Citation30]. Therefore, it is possible that ATG16L2 may function as a potential dominant negative competitive inhibitor of ATG16L1.
However, despite showing that overexpression of ATG16L2 reduces ATG12–ATG5-ATG16L1 complexes and LC3 lipidation, questions still remain regarding whether at endogenous levels, ATG16L2 is also an inhibitor of ATG16L1, LC3 lipidation and autophagy, as this has not yet been demonstrated. It is also unclear why ATG16L2 knockdowns and knockouts have not resulted in increased autophagy. One possible explanation may be that endogenous expression of ATG16L2 under basal conditions is insufficient to inhibit ATG16L1 and alter autophagy, thus ATG16L2 knockdown/knockout appears to have no effect.
Both ATG16L2 and ATG16L1 are constitutively expressed in all cell types including at the developmental stages [Citation15, Citation29] (). This suggests that ATG16L2 may be a relevant player in regulating autophagy as a potential dominant negative inhibitor of ATG16L1. Although ubiquitously expressed, each cell type has varying levels of ATG16L2 and ATG16L1 expression, which may imply tissue-specific functions for ATG16L2 and ATG16L1 as suggested by Khor et al. [Citation29]. For example, Atg16l2 expression is higher in spleen, blood, lung and skin, Atg16l1 is higher in the brain (cerebellar), skeletal muscle and testis, while the kidney and gastrointestinal tract have similar expression levels of both genes [Citation31] (). It is possible that expression levels of both Atg16 genes need to be delicately balanced and reflect the need for autophagy in these different tissue types. It is unclear what determinants regulate the levels of ATG16L2 in different cell types, however, evidence has shown that ATG16L2 can be regulated post-transcriptionally by microRNAs (miRNA) such as Mir885-3p [Citation32]. It has been reported that Mir885-3p binds to the 3ʹUTR of Atg16l2 and blocks translation. This reduces ATG16L2 and induces autophagy, which may further support ATG16L2 as a negative inhibitor of autophagy [Citation32]. Determining how ATG16L2 is modulated in different cell types will have important implications for understanding autophagy.
Beyond LC3 lipidation, ATG16L1 is a key hub protein and is involved in other pathways including cell death (apoptosis and necroptosis), inflammation, and xenophagy. ATG16L1 has been found to interact with as many as 15 other proteins including six proteins (TMEM59 [transmembrane protein 59], TMEM74 [transmembrane protein 74], EVA1A/TMEM166 [eva-1 homolog A, regulator of programmed cell death], C3 [complement C3], ubiquitin, ATP6V0C [ATPase H+ transporting V0 subunit c]) that recruit ATG16L1 to the site of bacterial infections during xenophagy, and NOD1 and NOD2 to suppress inflammatory cytokines [Citation33]. Whether ATG16L2 can also interact with these other proteins and what effect it has on inflammation remains to be clarified. One protein that interacts with ATG16L2 but not ATG16L1 is CHUK/IKKα, an NFKB/NF-κβ inhibitor. However, what effect this interaction has on autophagy and inflammation is yet to be determined [Citation28].
Because expression of ATG16L2 is highest in spleen and blood cells, the role of ATG16L2 in erythropoiesis, innate immune function and B and T cell function has also been examined. Loss of ATG16L2 was not identified to affect any of these functions [Citation29]. However, given our new understanding that ATG16L2 functions as a potential dominant negative inhibitor, the effect of ATG16L2 overexpression on these functions should be re-examined.
Role of ATG16L2 in xenophagy
Xenophagy forms a key component of innate immunity through selective autophagic degradation of intracellular pathogens. It is important for the clearance of a diverse range of human pathogens and pathobionts including influenza virus [Citation34], Salmonella typhimurium [Citation35], Helicobacter pylori [Citation36], Campylobacter concisus [Citation37] and Candida albicans [Citation38]. However, several pathogens have also evolved mechanisms to block xenophagy clearance [Citation39] and even hijack xenophagy for replication and/or survival such as Chlamydia trachomatis [Citation40] and Coxiella burnetii [Citation41] To date, there have been very few studies which have investigated the role of ATG16L2 in xenophagy (or selective autophagy). ATG16L1 is essential for colocalizing LC3 to intracellular bacteria such as Salmonella and Listeria for xenophagy-mediated bacterial clearance. Deletion of Atg16l1 abolished antibacterial autophagic killing, leading to systemic infection and increased inflammation in mice [Citation42]. In contrast, deletion of Atg16l2 had no effect on xenophagy or levels of intracellular Listeria and Salmonella [Citation29]. Additionally, ATG16L1 rs2241880 increases the risk of H. pylori infection43, however, no association was found between ATG16L2 rs11235604 and Mycobacterium tuberculosis infection [Citation44] (). Interestingly, it has been found that ATG16L1 deletion leads to resistance to Citrobacter rodentium infection and colitis in mice [Citation45]. While Atg16l2 deletion had no effect on Citrobacter infection, a double deletion of Atg16l1 and Atg16l2 reversed the Citrobacter resistance conferred by single Atg16l1 deletion [Citation29]. This suggests that there may be important ATG16L2-ATG16L1 interactions affecting xenophagy and bacterial clearance or that there may be additional functions of ATG16L2 beyond inhibiting ATG16L1 which require further investigation. Deletion of different autophagy proteins have contrasting effects on resistance to Citrobacter infection. For example, lineage-specific deletion of Atg5 in mouse eosinophils leads to increased resistance to C. rodentium [Citation46] while deletion of Atg7 (involved in conjugating ATG5 to ATG12) in intestinal mouse epithelial cells led to increased susceptibility [Citation47]. Therefore, it is conceivable that deletion of ATG16L1 and ATG16L2 may impact protein-protein interactions with one of their partners leading to the observed differences in susceptibility to Citrobacter infection. One potential interaction partner may be IRGM (immunity related GTPase M). Although IRGM has not yet been shown to interact with ATG16L2, IRGM has been shown to bind directly to ATG16L1 via the WD40 repeats and plays a key role in recruiting NOD2-ATG16L1 complexes to the site of bacterial infection [Citation48]. Deletion of IRGM in mice was shown to affect apoptosis in several myeloid cell types, only when infected with C. rodentium. This led to increased susceptibility to Citrobacter infection [Citation49]. Since ATG16L2 expression is highest in haemopoietic cells, it is possible that deletion of Atg16l2 may also affect myeloid function and susceptibility to Citrobacter infection in a context and lineage specific manner.
Table 1. ATG16L2 SNPs implicated in human disease
The role of ATG16L2 in cancer
Autophagy plays a critical but complex role in carcinogenesis. It can act as a tumor suppressor or tumor enhancer depending on the stage of the disease and the tumor type [Citation11]. During the initial stages of carcinogenesis, autophagy acts as a tumor suppressor in non-malignant cells by degrading damaged organelles and oncogenic proteins while in the latter stages, autophagy assists malignant cells to reprogram metabolic pathways to survive hypoxic and metabolic stress. Furthermore, the antimicrobial role of xenophagy is also important in limiting oncogenic bacteria, such as H. pylori, which promote inflammation and tumorigenesis [Citation43].
ATG16L2 has been linked with several types of cancers. rs10751215, an ATG16L2 intron variant, was shown to be a protective SNP against clear cell renal carcinoma [Citation50] (). Furthermore, overexpression of ATG16L2 has been reported in gastric [Citation51], renal [Citation52] and prostate carcinoma [Citation30] (). In colorectal carcinoma and clear cell renal carcinoma, ATG16L2 overexpression was a prognostic indicator for high risk of relapse [Citation52, Citation53] (). Overexpression of ATG16L2 in colorectal carcinoma has also been associated with higher survival and decreased lymph node metastasis [Citation16]. However, in lung cancer and melanoma, decreased levels of ATG16L2 compared to adjacent normal tissues were reported () [Citation54, Citation55]. No differences in ATG16L1 expression were reported throughout these studies [Citation16, Citation30, Citation51-55]. In addition, the methylation status of ATG16L2 has also been associated with different cancers. ATG16L2 hypomethylation was associated with lung cancer [Citation56] while hypermethylation was associated with renal and chronic myeloid leukemia [Citation17]. The role of ATG16L2 and how expression/methylation levels affect oncogenesis is unknown. In a recent study published by Tang et al. [Citation16] the authors showed that stable overexpression of ATG16L2 in a colorectal cancer cell line decreased cell proliferation but had no effect on cell migration. When these ATG16L2 overexpressed cells were subcutaneously xenografted into mice, they displayed slower tumor growth compared to wild type cells. The authors hypothesized that this phenotypic difference may be associated with the favorable prognosis observed in colorectal tumors with high ATG16L2 expression. Thus, ATG16L2 appears to play a role in cancer biology, however, the mechanism behind how ATG16L2 leads to decreased tumor growth needs to be explored further. Given the complex role of autophagy in cancer biology, it is likely that ATG16L2 is also context, cancer-type, and stage specific.
Table 2. ATG16L2 SNPs associated with cancer and efficacy of cancer treatments
Figure 3. Heat map illustrating significant upregulation or downregulation of ATG16L2 reported in different cancer and neurodegenerative diseases from published studies and/or Gene expression profiling interactive analysis (GEPIA) [Citation84]. Color is indicative of significant upregulation (red) or downregulation (blue) not the magnitude of the fold-change. For the specified cancers and neurodegenerative diseases, no significant differences in ATG16L1 expression were reported in the published studies and/or GEPIA.
![Figure 3. Heat map illustrating significant upregulation or downregulation of ATG16L2 reported in different cancer and neurodegenerative diseases from published studies and/or Gene expression profiling interactive analysis (GEPIA) [Citation84]. Color is indicative of significant upregulation (red) or downregulation (blue) not the magnitude of the fold-change. For the specified cancers and neurodegenerative diseases, no significant differences in ATG16L1 expression were reported in the published studies and/or GEPIA.](/cms/asset/5598b57d-5b43-4c11-98db-1022459c60b3/kaup_a_2042783_f0003_oc.jpg)
Given the current evidence that ATG16L2 may be a blocker of autophagy and antagonist of ATG16L1, it is conceivable that upregulation of ATG16L2 during the early stages of carcinogenesis inhibits ATG16L1 and decreases autophagy. This provides a more favorable environment for cancer formation as oncogenic proteins and/or microbes are not degraded. However, during the latter stages of carcinogenesis, it may be more favorable for ATG16L2 to be downregulated/inactivated to allow tumors to survive hypoxic and metabolic stress.
Autophagy also affects the efficacy of cancer treatment in a context dependent manner. Some studies have reported that induction of autophagy during therapy was cytotoxic leading to cancer cell death while in other studies increased autophagy during therapy was cytoprotective and led to chemoresistance [Citation57]. ATG16L2 has been associated with response to cancer therapy. ATG16L2 hypermethylation was associated with poorer response to imatinib, a tyrosine kinase inhibitor, in chronic myeloid leukemia [Citation17]. The ATG16L2 SNP, rs11235604, was associated with poorer progression free survival and overall survival in lung adenocarcinoma treated with the EGFR inhibitor, gefitinib [Citation58] (). Another SNP, rs10898880, located in the promotor of ATG16L2 and associated with increased ATG16L2 mRNA levels, was linked to increased overall survival following radiotherapy in non-small cell lung carcinoma and nasopharyngeal carcinoma [Citation55, Citation59] (). However, rs10898880 was also associated with increased toxicity such as radiation-induced pneumonitis, oral mucositis, and myelosuppression [Citation55, Citation59] (). rs10898880 may lead to increased inflammation which is beneficial against cancer cells but toxic to normal tissues. This suggests a dual role for ATG16L2 in cancer treatment outcomes.
The potential role of ATG16L2 in affecting treatment outcomes is further highlighted in breast cancer where cisplatin treatment upregulated ATG16L2 mRNA and autophagy [Citation60]. Cisplatin-induced autophagy was found to be cytotoxic as inhibition of autophagy with 3-Methyladenine (3-MA) reduced cisplatin anti-tumor activity. In a separate study, cisplatin treatment of squamous cell carcinoma also reduced cell viability, however in contrast, this was associated with reduced ATG16L2 protein levels [Citation32]. Differences between ATG16L2 expression in breast and squamous cell carcinoma may point to differences in cancer cell types. It is also possible that in breast cancer, ATG16L2 may be upregulated due to negative feedback to control autophagy, as several other genes which induce autophagy (BECN1/Beclin1, ULK1 (Unc-51 Like Autophagy Activating Kinase 1), MAP1LC3B, ATG5, etc.) are also increased [Citation60].
It is clear that ATG16L2 and autophagy play an essential role in cancer biology, and understanding the role of this protein could lead to new treatment options for patients.
Implication of ATG16L2 in other diseases
SNPs in autophagy genes such as ATG16L1 and IRGM are associated with a range of chronic inflammatory diseases, and some have been shown to play important roles in pathogenesis [Citation61]. For example, ATG16L1 rs2241880, a non-synonymous mutation which changes threonine to alanine (T300A), increases the susceptibility of ATG16L1 to caspase-3 cleavage. This reduces autophagy, inhibits bacterial clearance, and increases proinflammatory cytokine production, contributing to Crohn disease (CD) pathogenesis [Citation62, Citation63].
To date, two ATG16L2 SNPs (rs11235667 and rs11235604) have been linked to autoimmune diseases in the Asian population (excluding South Asian) including IBD/CD [Citation18, Citation64-66] and systemic lupus erythematosus (SLE) [Citation67, Citation68] (). The first SNP, rs11235667, is located ~300 kb downstream of ATG16L2. eQTL has linked rs11235667 with ATG16L2 expression [Citation64]. The second SNP, rs11235604, is a non-synonymous SNP in ATG16L2 which alters arginine to tryptophan (R220W) (). rs11235604 is therefore the potential biologically plausible variant as both SNPs are in strong linkage disequilibrium (D’=1, r2= 0.9339, p <0.0001). Interestingly, both ATG16L2 SNPs are monomorphic in non-Asian populations. While ATG16L1 rs2241880 is a significant risk factor for CD in various populations including Caucasians, this effect was not observed in Asian populations [Citation69]. This suggests that there are important population specific differences in the contribution of ATG16L1 and ATG16L2 to autophagy and the pathogenesis of autoimmune diseases like CD and SLE. Whereas ATG16L1 rs2241880 () leads to increased CASP3 (caspase 3) cleavage and reduces autophagy, the functional consequence of ATG16L2 rs11235604 remains undetermined. rs11235604 has been associated with a slight reduction in ATG16L2 expression [Citation59]. However, given that ATG16L2 is a potential inhibitor of autophagy, it appears unlikely that increased IBD risk is due to decreased ATG16L2 expression as ATG16L2 deletion has no effect on LC3 lipidation and autophagy [Citation19, Citation29]. rs11235604 is located in the M region (exon 6) which is disordered in ATG16L2. It is possible that rs11235604 alters the binding affinity of ATG16L2 to other autophagic proteins, however, this remains to be investigated.
In addition, while ATG16L2 rs11235667 and rs11235604 were associated with increased risk of CD, these SNPs were significant protective factors against SLE (). This inverse relation between ATG16L2 SNPs in CD and SLE is consistent with other IBD-SLE shared loci including PTPN22, IRF8, FCGR2A and IKZF1 [Citation70, Citation71]. The opposing effect of these SNPs may point to differences in the role of ATG16L2, autophagy and/or other branches of the immune system in the pathogenesis of these diseases. It suggests that while decreased autophagy is associated with CD, activated or excessive autophagy is associated with the pathogenesis of SLE [Citation72].
Besides CD and SLE, ATG16L2 rs11235604 has also been identified as a protective factor against coronary heart disease [Citation73] (). In rheumatoid arthritis, ATG16L2 rs11235604 was also a risk factor, however, only when combined with SNPs in ATG16L1 (rs2241880 or rs6758317) [Citation74] (). This suggests that there is a cumulative effect and an interaction between the two paralogs in determining disease susceptibility.
ATG16L2 has also been implicated in several neurodegenerative diseases. Significant increases in ATG16L2 expression were reported in patients or representative mouse models of amyotrophic lateral sclerosis [Citation75], Alzheimer disease [Citation76] and Machado-Joseph disease [Citation77] while decreased expression were observed in multiple sclerosis [Citation78, Citation79] (). In studies which also examined the expression of ATG16L1 [Citation76, Citation79], changes in ATG16L2 expression were not accompanied by altered ATG16L1 expression. This suggests that any potential inhibitory effects from increased ATG16L2 expression are not being compensated for by increased ATG16L1 levels. Importantly, these expression changes were observed in both Asian [Citation78] and European populations [Citation75, Citation77, Citation79]. This indicates that ATG16L2 involvement in disease is not just specific to the Asian population but is of broad relevance to multiple ethnicities. The reason for differences in ATG16L2 expression in neurodegenerative diseases has not been determined, however, ATG16L2 expression in amyotrophic lateral sclerosis was recently linked to rs2282613 [Citation75], an ATG16L2 intron variant located between exons 3 and 4. This SNP is polymorphic in multiple populations and has not yet been investigated in other populations or diseases.
Conclusion and future directions
ATG16L2 is an important but currently underappreciated gene. Recent studies have provided new insights into ATG16L2 and revealed its role as a potential competitive dominant inhibitor of ATG16L1 and autophagy. To date, most studies on ATG16L2 have mainly focused on loss of function through genetic deletion or silencing, subsequently determining its effect on relevant pathways such as autophagy, inflammation, and pathogen defense. Given the latest insight, the role of ATG16L2 on these pathways should be revisited and more studies are needed to explore the impact of ATG16L2 overexpression. Additional studies are also required to determine whether endogenous levels of ATG16L2 can inhibit ATG16L1 and autophagy or whether there are specific circumstances where expression of ATG16L2 is increased to inhibit autophagy when it is not required.
One similarity between ATG16L1 and ATG16L2 is that both proteins contain the seven WD40 repeats in the C-terminus region. In ATG16L1, the WD40 domains are important for single membrane lipidation in non-canonical autophagy [Citation21]. Therefore, it would be interesting to investigate whether ATG16L2 is also involved in non-canonical autophagy. Furthermore, to establish infection, many bacterial species have developed strategies to block or exploit xenophagy [Citation4]. Since ATG16L2 may be a potential autophagy inhibitor, it will be important to determine what role, if any, ATG16L2 has in xenophagy, and whether intracellular pathogens can manipulate ATG16L2 to block xenophagy-mediated clearance and aid survival. Besides autophagy, ATG16L1 also has important molecular functions in apoptosis [Citation80], necroptosis [Citation81], and endoplasmic reticulum stress [Citation82 Citation83] thus it will be important to determine whether ATG16L2 also affects the functions of other cellular pathways.
ATG16L2 is increasingly seen to play an important role in the pathogenesis of various cancers, autoimmune diseases, and neurodegenerative disorders. Many of the current studies available have identified an association between ATG16L2 expression and disease but have not yet directly identified mechanistically whether or how ATG16L2 is involved in disease. Thus, a renewed focus on elucidating the function of ATG16L2 and how it contributes to the underlying pathology of these diseases will allow us to elucidate the potential ofATG16L2 as a future drug target or disease marker. As a potential modulator of autophagy, the future development of ATG16L2 mimetics may represent a new class of autophagy blockers.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nature reviews Molecular cell biology 2018; 19:349–364.
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell research 2014; 24:24–41.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018; 14:207–215.
- Kimmey JM, Stallings CL. Bacterial Pathogens versus Autophagy: Implications for Therapeutic Interventions. Trends in molecular medicine 2016; 22:1060–1076.
- Mitchell G, Isberg RR. Innate Immunity to Intracellular Pathogens: Balancing Microbial Elimination and Inflammation. Cell host & microbe 2017; 22:166–175.
- Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nature cell biology 2018; 20:233–242.
- Lamark T, Johansen T. Mechanisms of Selective Autophagy. Annual review of cell and developmental biology 2021; 37:143–169.
- Mizushima N. Autophagy: process and function. Genes & development 2007; 21:2861–2873.
- Nishimura T, Tooze SA. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell discovery 2020; 6:32.
- Ktistakis NT, Tooze SA. Digesting the Expanding Mechanisms of Autophagy. Trends in cell biology 2016; 26:624–635.
- Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Frontiers in oncology 2020; 10:578418.
- Yang Z, Goronzy JJ, Weyand CM. Autophagy in autoimmune disease. Journal of molecular medicine 2015; 93:707–717.
- Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: A comprehensive review. Biomedicine & pharmacotherapy 2018; 104:485–495.
- Pinto G, Shtaif B, Phillip M, Gat-Yablonski G. Growth attenuation is associated with histone deacetylase 10-induced autophagy in the liver. The Journal of nutritional biochemistry 2016; 27:171–180.
- Gerovska D, Araúzo-Bravo MJ. Does mouse embryo primordial germ cell activation start before implantation as suggested by single-cell transcriptomics dynamics? Molecular human reproduction 2016; 22:208–225.
- Tang J, Wang D, Shen Y, Xue F. ATG16L2 overexpression is associated with a good prognosis in colorectal cancer. Journal of gastrointestinal oncology 2021; 12:2192–2202.
- Dunwell T, Hesson L, Rauch TA, Wang L, Clark RE, Dallol A, et al. A genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Molecular cancer 2010; 9:44.
- Ma T, Wu S, Yan W, Xie R, Zhou C. A functional variant of ATG16L2 is associated with Crohn’s disease in the Chinese population. Colorectal disease 2016; 18:O420–O426.
- Ishibashi K, Fujita N, Kanno E, Omori H, Yoshimori T, Itoh T, et al. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12–5-16L2 complex. Autophagy 2011; 7:1500–1513.
- Zhang H, Wu F, Wang X, Du H, Wang X, Zhang H. The two C. elegans ATG-16 homologs have partially redundant functions in the basal autophagy pathway. Autophagy 2013; 9:1965–1974.
- Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. The EMBO journal 2018; 37.
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Molecular cell 2014; 55:238–252.
- Gammoh N, Florey O, Overholtzer M, Jiang X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nature structural & molecular biology 2013; 20:144–149.
- Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO reports 2013; 14:284–291.
- Pantoom S, Konstantinidis G, Voss S, Han H, Hofnagel O, Li Z, et al. RAB33B recruits the ATG16L1 complex to the phagophore via a noncanonical RAB binding protein. Autophagy 2020:1–15.
- Gammoh N. The multifaceted functions of ATG16L1 in autophagy and related processes. Journal of cell science 2020; 133.
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264–268.
- Li N, Wu X, Holzer RG, Lee JH, Todoric J, Park EJ, et al. Loss of acinar cell IKKα triggers spontaneous pancreatitis in mice. The Journal of clinical investigation 2013; 123:2231–2243.
- Khor B, Conway KL, Omar AS, Biton M, Haber AL, Rogel N, et al. Distinct Tissue-Specific Roles for the Disease-Associated Autophagy Genes ATG16L2 and ATG16L1. Journal of immunology 2019; 203:1820–1829.
- Wible DJ, Chao HP, Tang DG, Bratton SB. ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell discovery 2019; 5:42.
- Stanfill AG, Cao XJBRFN. Enhancing Research Through the Use of the Genotype-Tissue Expression (GTEx) Database. Biological research for nursing 2021:1099800421994186.
- Huang Y, Chuang AY, Ratovitski EA. Phospho-ΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell cycle 2011; 10:3938–3947.
- Xiong Q, Li W, Li P, Yang M, Wu C, Eichinger L. The Role of ATG16 in Autophagy and The Ubiquitin Proteasome System. Cells 2018; 8.
- Feizi N, Mehrbod P, Romani B, Soleimanjahi H, Bamdad T, Feizi A, et al. Autophagy induction regulates influenza virus replication in a time-dependent manner. Journal of medical microbiology 2017; 66:536–541.
- Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. The Journal of biological chemistry 2006; 281:11374–11383.
- Wang YH, Wu JJ, Lei HY. The autophagic induction in Helicobacter pylori-infected macrophage. Experimental biology and medicine 2009; 234:171–180.
- Burgos-Portugal JA, Mitchell HM, Castaño-Rodríguez N, Kaakoush NO. The role of autophagy in the intracellular survival of Campylobacter concisus. FEBS Open Bio 2014; 4:301–309.
- Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nature communications 2015; 6:5779.
- Campbell-Valois FX, Sachse M, Sansonetti PJ, Parsot C. Escape of Actively Secreting Shigella flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA. mBio 2015; 6:e02567–14.
- Al-Younes HM, Al-Zeer MA, Khalil H, Gussmann J, Karlas A, Machuy N, et al. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy 2011; 7:814–828.
- Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS pathogens 2014; 10:e1004286.
- Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 2013; 145:1347–1357.
- Castaño‐Rodríguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. Autophagy in Helicobacter pylori infection and related gastric cancer. Helicobacter 2015; 20:353–369.
- Songane M, Kleinnijenhuis J, Alisjahbana B, Sahiratmadja E, Parwati I, Oosting M, et al. Polymorphisms in autophagy genes and susceptibility to tuberculosis. PloS one 2012; 7:e41618.
- Marchiando AM, Ramanan D, Ding Y, Gomez LE, Hubbard-Lucey VM, Maurer K, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell host & microbe 2013; 14:216–224.
- Germic N, Hosseini A, Stojkov D, Oberson K, Claus M, Benarafa C, et al. ATG5 promotes eosinopoiesis but inhibits eosinophil effector functions. Blood 2021; 137:2958–2969.
- Inoue J, Nishiumi S, Fujishima Y, Masuda A, Shiomi H, Yamamoto K, et al. Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Archives of biochemistry and biophysics 2012; 521:95–101.
- Chauhan S, Mandell MA, Deretic V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Molecular cell 2015; 58:507–521.
- Taylor GA, Huang HI, Fee BE, Youssef N, Jewell ML, Cantillana V, et al. Irgm1-deficiency leads to myeloid dysfunction in colon lamina propria and susceptibility to the intestinal pathogen Citrobacter rodentium. PLoS pathogens 2020; 16:e1008553.
- Santoni M, Piva F, De Giorgi U, Mosca A, Basso U, Santini D, et al. Autophagic Gene Polymorphisms in Liquid Biopsies and Outcome of Patients with Metastatic Clear Cell Renal Cell Carcinoma. Anticancer research 2018; 38:5773–5782.
- Wang M, Jing J, Li H, Liu J, Yuan Y, Sun L. The expression characteristics and prognostic roles of autophagy-related genes in gastric cancer. PeerJ 2021; 9:e10814.
- Wan B, Liu B, Yu G, Huang Y, Lv C. Differentially expressed autophagy-related genes are potential prognostic and diagnostic biomarkers in clear-cell renal cell carcinoma. Aging 2019; 11:9025–9042.
- Mo S, Dai W, Xiang W, Li Y, Feng Y, Zhang L, et al. Prognostic and predictive value of an autophagy-related signature for early relapse in stages I-III colon cancer. Carcinogenesis 2019; 40:861–870.
- Ren M, Wei CY, Wang L, Deng XY, Lu NH, Gu JY. Integration of individual prediction index based on autophagy-related genes and clinical phenomes in melanoma patients. Clinical and translational medicine 2020; 10:e132.
- Wen J, Liu H, Wang L, Wang X, Gu N, Liu Z, et al. Potentially Functional Variants of ATG16L2 Predict Radiation Pneumonitis and Outcomes in Patients with Non-Small Cell Lung Cancer after Definitive Radiotherapy. Journal of thoracic oncology 2018; 13:660–675.
- Bai H, He Y, Lin Y, Leng Q, Carrillo JA, Liu J, et al. Identification of a novel differentially methylated region adjacent to ATG16L2 in lung cancer cells using methyl-CpG binding domain protein-enriched genome sequencing. Genome 2021; 64:533–546.
- Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer research 2014; 74:647–651.
- Yuan J, Zhang N, Yin L, Zhu H, Zhang L, Zhou L, et al. Clinical Implications of the Autophagy Core Gene Variations in Advanced Lung Adenocarcinoma Treated with Gefitinib. Scientific reports 2017; 7:17814.
- Yang Z, Liu Z. Potentially functional variants of autophagy-related genes are associated with the efficacy and toxicity of radiotherapy in patients with nasopharyngeal carcinoma. Molecular genetics & genomic medicine 2019; 7:e1030.
- Shen M, Duan WM, Wu MY, Wang WJ, Liu L, Xu MD, et al. Participation of autophagy in the cytotoxicity against breast cancer cells by cisplatin. Oncology reports 2015; 34:359–367.
- Palomino-Morales RJ, Oliver J, Gómez-García M, López-Nevot MA, Rodrigo L, Nieto A, et al. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: a meta-analysis approach. Genes and immunity 2009; 10:356–364.
- Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014; 506:456–462.
- Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proceedings of the National Academy of Sciences 2014; 111:7741–7746.
- Yang SK, Hong M, Zhao W, Jung Y, Baek J, Tayebi N, et al. Genome-wide association study of Crohn’s disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut 2014; 63:80–87.
- Fuyuno Y, Yamazaki K, Takahashi A, Esaki M, Kawaguchi T, Takazoe M, et al. Genetic characteristics of inflammatory bowel disease in a Japanese population. Journal of gastroenterology 2016; 51:672–681.
- Luu LDW, Popple G, Tsang SPW, Vinasco K, Hilmi I, Ng RT, et al. Genetic variants involved in innate immunity modulate the risk of inflammatory bowel diseases in an understudied Malaysian population. Journal of gastroenterology and hepatology 2021; n/a.
- Lessard CJ, Sajuthi S, Zhao J, Kim K, Ice JA, Li H, et al. Identification of a Systemic Lupus Erythematosus Risk Locus Spanning ATG16L2, FCHSD2, and P2RY2 in Koreans. Arthritis & rheumatology 2016; 68:1197–1209.
- Molineros JE, Yang W, Zhou XJ, Sun C, Okada Y, Zhang H, et al. Confirmation of five novel susceptibility loci for systemic lupus erythematosus (SLE) and integrated network analysis of 82 SLE susceptibility loci. Human molecular genetics 2017; 26:1205–1216.
- Grigoras CA, Ziakas PD, Jayamani E, Mylonakis E. ATG16L1 and IL23R variants and genetic susceptibility to crohn’s disease: mode of inheritance based on meta-analysis of genetic association studies. Inflammatory bowel diseases 2015; 21:768–776.
- Ramos PS, Criswell LA, Moser KL, Comeau ME, Williams AH, Pajewski NM, et al. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS genetics 2011; 7:e1002406.
- Richard-Miceli C, Criswell LA. Emerging patterns of genetic overlap across autoimmune disorders. Genome medicine 2012; 4:6.
- Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Annals of the rheumatic diseases 2015; 74:912–920.
- Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nature genetics 2020; 52:669–679.
- Mo JJ, Zhang W, Wen QW, Wang TH, Qin W, Zhang Z, et al. Genetic association analysis of ATG16L1 rs2241880, rs6758317 and ATG16L2 rs11235604 polymorphisms with rheumatoid arthritis in a Chinese population. International immunopharmacology 2021; 93:107378.
- Saez-Atienzar S, Bandres-Ciga S, Langston RG, Kim JJ, Choi SW, Reynolds RH, et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Science advances 2021; 7.
- Caberlotto L, Nguyen TP, Lauria M, Priami C, Rimondini R, Maioli S, et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Scientific reports 2019; 9:3965.
- Sittler A, Muriel MP, Marinello M, Brice A, den Dunnen W, Alves S. Deregulation of autophagy in postmortem brains of Machado-Joseph disease patients. Neuropathology 2018; 38:113–124.
- Yin L, Liu J, Dong H, Xu E, Qiao Y, Wang L, et al. Autophagy-related gene16L2, a potential serum biomarker of multiple sclerosis evaluated by bead-based proteomic technology. Neuroscience letters 2014; 562:34–38.
- Igci M, Baysan M, Yigiter R, Ulasli M, Geyik S, Bayraktar R, et al. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene 2016; 588:38–46.
- Liu TC, Kern JT, VanDussen KL, Xiong S, Kaiko GE, Wilen CB, et al. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. The Journal of clinical investigation 2018; 128:5110–5122.
- Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. The Journal of experimental medicine 2017; 214:3687–3705.
- Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut 2014; 63:1081–1091.
- Molineros JE, Yang W, Zhou X-j, Sun C, Okada Y, Zhang H, et al. Confirmation of five novel susceptibility loci for systemic lupus erythematosus (SLE) and integrated network analysis of 82 SLE susceptibility loci. Human molecular genetics 2017; 26:1205–1216.
- Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research 2017; 45:W98–W102.