
ABSTRACT
A variety of disturbances such as starvation, organelle damage, heat stress, hypoxia and pathogen infection can influence the autophagic process. However, how the macroautophagy/autophagy machinery is regulated intrinsically by structural damage of the cell remains largely unknown. In this work, we utilized the C. elegans epidermis as the model to address this question. Our results showed that structural damage by mechanical wounding exerted proximal inhibitory effect and distant promotional effect on autophagy within the same epidermal cell. By disrupting individual mechanical supporting structures, we found that only damage of the basal extracellular matrix or the underlying muscle cells activated a distinct autophagic response in the epidermis. On the contrary, structural disruption of the epidermal cells at the apical side inhibited autophagy activation caused by different stress factors. Mechanistic studies showed that the basal promotional effect of structural damage on epidermal autophagy was mediated by a mechanotransduction pathway going through the basal hemidesmosome receptor and LET-363/MTOR, while the apical inhibitory effect was mostly carried out by activation of calcium signaling. Elevated autophagy in the epidermis played a detrimental rather than a beneficial role on cell survival against structural damage. The results obtained from these studies will not only help us better understand the pathogenesis of structural damage- and autophagy-related diseases, but also provide insight into more generic rules of autophagy regulation by the structural and mechanical properties of cells across species.
Abbreviations : ATG: autophagy related; BLI-1: BLIstered cuticle 1; CeHDs: C. elegans hemidesmosomes; COL-19: COLlagen 19; DPY-7: DumPY 7; ECM: extracellular matrix; EPG-5: ectopic PGL granules 5; GFP: green fluorescent protein; GIT-1: GIT1 (mammalian G protein-coupled receptor kinase InTeractor 1) homolog; GTL-2: Gon-Two Like 2 (TRP subfamily); HIS-58, HIStone 58; IFB-1: Intermediate Filament, B 1; LET: LEThal; LGG-1: LC3, GABARAP and GATE-16 family 1; MTOR: mechanistic target of rapamycin; MTORC1: MTOR complex 1; MUP-4: MUscle Positioning 4; NLP-29: Neuropeptide-Like Protein 29; PAT: Paralyzed Arrest at Two-fold; PIX-1: PIX (PAK (p21-activated kinase) Interacting eXchange factor) homolog 1; RFP: red fluorescent protein; RNAi: RNA interference; SQST-1: SeQueSTosome related 1; UNC: UNCoordinated; UV: ultraviolet; VAB-10: variable ABnormal morphology 10; WT: wild type.
Introduction
The epithelial tissues form the physical barriers of most of our organs and constantly face the challenges of physical injury and structural damage. However, whether and how the structural damage of the epithelial cells affects macroautophagy/autophagy cell-autonomously is still not fully understood. Evidence suggests that multiple types of mechanical supporting structures within the epithelial cells are involved in regulating the autophagy process. First of all, autophagy can be induced when the epithelial layer breaks away from the adhesive support of the extracellular matrix (ECM), a process termed as “detachment induced autophagy” [Citation1–4]. Such phenomena are mostly caused by the destruction of cellular adhesion structures or changes of extracellular matrix components [Citation5]. Secondly, cytoskeletal fibers such as actin filaments and microtubules not only directly participate in the formation and transportation of autophagic vesicles, but also regulate their functions by interacting with BECN1/Beclin 1, BCL2L11/BIM and other key autophagic proteins [Citation5,Citation6]. Although intermediate filaments are not directly involved in autophagosome formation, some studies have found that it can inhibit autophagy by physical association with BECN1 [Citation7]. Thirdly, the change of mechanical tension can directly affect the autophagic process. For instance, applying mechanical pressure upon in vitro cultured epithelial cells induces autophagosome formation [Citation8]. Another example shows that the renal epithelial cell layer can sense the shear stress generated by fluid flow and activate autophagy to regulate its own cell volume [Citation9]. Together, these findings indicate that structural integrity of the epithelial cells may have complex influences on the autophagic process. However, the above mentioned studies are rather fragmented and were mostly performed on in vitro cultured cells, which cannot faithfully reproduce the intact three-dimensional architecture of the epithelial cell layer in vivo. Therefore, comprehensive investigations of the autophagic response to structural damage in epithelial cells of live organisms are required to reveal the detailed regulatory mechanisms and in vivo physiological significance.
C. elegans is an ideal model organism to study the biological issues regarding epithelial structural damage and autophagy. The C. elegans epidermis is mostly composed of a large syncytium named hyp7, whose structural components are organized into a highly organized, three-dimensionally supported architecture [Citation10,Citation11]. The main anchoring structures, namely the C. elegans hemidesmosomes (CeHDs), connect the epidermis apically with the cuticle exoskeleton, and basally with the body-wall muscles through the basement membrane [Citation12]. The experimental methods for applying different types of mechanical insults to the C. elegans epidermis have been well established and widely used in studies regarding wound healing, injury-induced immune response and cross-talks between epidermal damage and other biological processes [Citation11,Citation13,Citation14]. However, the regulatory mechanisms of autophagy in response to epidermal structural damage and its physiological significance remain to be elucidated.
In this study, we utilized the C. elegans epidermis as the model and systemically investigated the relationship between structural damage and the epithelial autophagic process. We found that mechanical wounding simultaneously caused autophagic elevation and repression in different regions of the hyp7 epidermal cell. Further investigation identified that basal and apical structural damage exerted opposite effects on epidermal autophagic response. Our data also suggested that autophagy played a detrimental rather than a beneficial role under the stress of structural damage. Our findings reveal that autophagy is bidirectionally regulated by different structural components of the epithelial cells, and that the ultimate effect on the epithelial autophagic response depends on the specific types of subcellular structures that are damaged. In a broad sense, this work may help us better understand the mechanisms of various diseases involving epithelial structural damage, and the effects of epithelial structural integrity on autophagy-related biological processes.
Results
Mechanical injury has both promotional and inhibitory effects on autophagy in the C. elegans epidermis
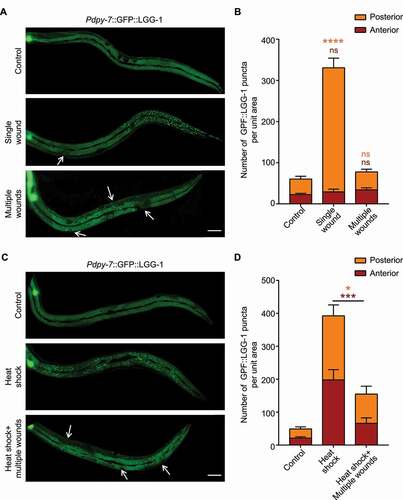
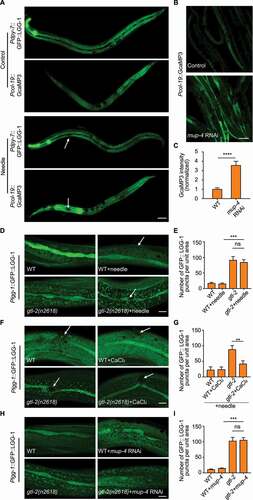
To investigate how autophagy is regulated in the C. elegans epidermis after injury, we monitored the autophagic process using an epidermal-specific promoter-driven GFP-tagged LGG-1/Atg8 reporter, which visualizes autophagosomes as fluorescent puncta [Citation15]. Mechanical wounding of the epidermis was performed by micrometer-scale needle puncture. Intriguingly, a single mechanical wound in the anterior region of the giant hyp7 epidermal syncytium caused a remarkable increase of the GFP::LGG-1 puncta in the posterior part but not the anterior part of the epidermis (, 1B and S1A). Single wounding in the posterior region of the epidermis resulted in a similar increase of the GFP::LGG-1 puncta in the anterior part (Figure S1A and S1B). Timelapse imaging showed that the distant up-regulated LGG-1 puncta away from the injury site appeared as quickly as 10 min after needle wounding (Figure S1C). However, when multiple wounds were introduced and dispersed along the hyp7 epidermal cell, no increase of LGG-1 puncta was observed in the entire epidermis ( and 1B). GFP::LGG-1 driven by the endogenous lgg-1 promoter displayed identical autophagic induction patterns as LGG-1 driven by the epidermal-specific dpy-7 promoter after needle wounding, suggesting that transcriptional regulation of lgg-1 was not involved (Figure S1A and S1B). These observations could be explained by the hypothesis that mechanical injury inhibits the formation of autophagosomes in the vicinity of the wounded area but promotes autophagosome formation in the subcellular regions distant from the injury site. Thus, the induction of autophagosomes by one wound could be blocked by another wound under multiple wounding conditions. To confirm the proximal inhibitory effect of mechanical injury on autophagosome formation, we induced autophagy by using heat shock treatment, and then applied multiple wounds to the epidermis. Consistent with our hypothesis, multiple wounds abolished the formation of LGG-1 puncta induced by heat shock ( and 1D) [Citation16]. Taken together, we propose that mechanical injury has proximal inhibitory effect and distant promotional effect on autophagy within the same epidermal cell.
Figure 1. Mechanical injury has both promotional and inhibitory effects on autophagy in the C. elegans epidermis. (A) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by the epidermal-specific Pdpy-7 promoter in the adults with an intact epidermis (control), a single wound or multiple wounds created by needle puncture. (B) Quantification of the number of GFP::LGG-1 puncta per unit area in the anterior or posterior part of the epidermis in wounded animals compared with control, corresponding to results in (A). n ≥ 6 per condition. (C) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by the epidermal-specific Pdpy-7 promoter in adult animals subjected to heat shock treatment with or without multiple needle wounding. (D) Quantification of the number of GFP::LGG-1 puncta per unit area in the anterior or posterior part of the epidermis with indicated treatments, corresponding to results in (C). n ≥ 8 per condition. Arrows point to sites of wounding. Scale bars: 40 μm. Error bars represent mean ± SEM. ns, not significant; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (two-tailed, unpaired t-test).
Structural damage at the basal side of the epidermis increases the autophagic response
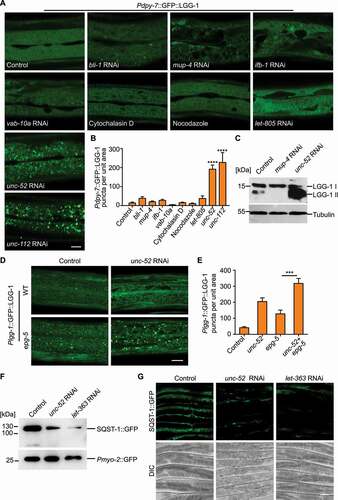
To dissect the differential effect of epidermal structural damage on autophagy, we disrupted individual epidermal supporting structures by RNA interference (RNAi) or drug treatment and analyzed their effects on autophagic response using the GFP::LGG-1 reporter driven by the epidermal-specific promoter Pdpy-7. The epidermal supporting structures include struts of the apical exoskeleton (bli-1 RNAi), apical CeHDs (mup-4 RNAi), the plakin cytolinker (vab-10a RNAi), intermediate filaments (ifb-1 RNAi), basal CeHDs (let-805 RNAi), actin filaments (cytochalasin D), microtubules (nocodazole), basal ECM (unc-52 RNAi) and the adjacent muscle quadrants (unc-112 RNAi) [Citation17–25]. Our results showed that, surprisingly, most types of structural damage did not induce LGG-1 puncta formation. Only damage of the basal extracellular matrix, namely the basement membrane, or the underlying muscle cells dramatically increased LGG-1 puncta in the epidermis ( and 2B). Similar results were obtained using the GFP::LGG-1 reporter driven by the lgg-1 promoter (Figure S2).The increase in the amount of autophagosomes caused by basal structural damage was further confirmed by analyzing the level of the lipidated LGG-1 (LGG-1-II) (). On the contrary, inactivation of the apical CeHD receptor MUP-4 reduced the level of LGG-1-II, indicating that apical CeHD disruption might inhibit autophagic activity (). The elevated number of autophagosomes could result from either an enhanced autophagosome formation process or a suppressed autophagosome turnover process. To distinguish between these two possibilities, we used an epg-5 loss-of-function mutant to assess the autophagy flux. In epg-5(tm3425) mutant, the formation of degradative autolysosomes is blocked, resulting in an accumulation of LGG-1 puncta ( and 2E) [Citation26]. An increase in autophagosome formation, but not a block of autophagosome turnover, aggravates the accumulation of LGG-1 puncta in epg-5 mutants [Citation27,Citation28]. As shown in and 2E, basal structural damage caused by unc-52 inactivation enhanced the number of epidermal GFP::LGG-1 puncta in epg-5 mutants ( and 2E). This observation suggests that epidermal autophagosome formation is indeed increased upon basal structural damage. Consistent with this notion, fluorescence reporter and western blot analysis revealed that the amount of the autophagic substrate SQST-1/SQSTM1 under the control of the epidermal-specific promoter Pcol-19 was also decreased by unc-52 inactivation (). Taken together, these results indicate that disruption of the basal epidermal supporting structures alone promotes autophagosome formation in the epidermis.
Figure 2. Structural damage at the basal side of the epidermis increases the autophagic response. (A) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by the epidermal-specific Pdpy-7 promoter in the young adults with intact (control) or damaged struts (bli-1), apical CeHDs (mup-4), the plakin cytolinker (vab-10a), cytoskeletons (ifb-1, cytochalasin D and nocodazole), basal CeHDs (let-805), basal ECM (unc-52) or adjacent muscle quadrants (unc-112). Scale bar: 10 μm. (B) Quantification of results in (A) showing the number of GFP::LGG-1 puncta per unit area in the epidermis of worms after disruption of various epidermal supporting structures compared with control. n ≥ 8 per condition. (C) Western blot analysis of the protein levels of LGG-1-I (unlipidated form) and LGG-1-II (lipidated form) in wild-type animals treated with the empty vector L4440 (control), mup-4 RNAi, or unc-52 RNAi. Tubulin serves as the loading control. (D) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by its own promoter in the epidermis of wild-type (WT) or epg-5(tm3425) young adults treated with L4440 (control) or unc-52 RNAi. Scale bar: 10 μm. (E) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis of animals with the indicated genotypes, corresponding to results in (D). n ≥ 10 per condition. (F) Western blot analysis of the protein levels of SQST-1::GFP driven by the epidermal-specific promoter Pcol-19 in the adults treated with L4440 (control), unc-52 RNAi or let-363 RNAi (positive control). Co-injected Pmyo-2::GFP serves as the internal control. (G) Representative confocal images showing the protein levels of SQST-1::GFP in L4440 (control), unc-52 RNAi or let-363 RNAi-treated animals. Scale bar: 40 μm. Error bars represent mean ± SEM. ***, P < 0.001; ****, P < 0.0001 (two-tailed, unpaired t-test).
Structural disruption at the apical side of the epidermis inhibits autophagy activation
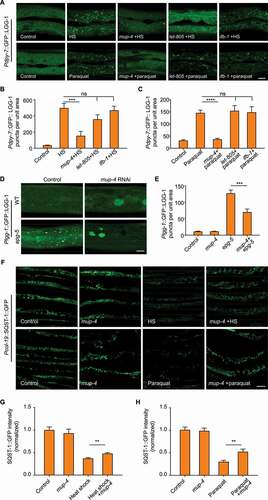
Because analysis of LGG-1 lipidation suggested that disruption of the apical CeHD receptor MUP-4 might inhibit baseline autophagy activity (), we next wanted to investigate the negative impact of CeHDs on autophagy in more detail. Firstly, we employed two autophagy-inducing conditions in the C. elegans epidermis, namely heat shock and paraquat treatment [Citation16,Citation29]. Next, we utilized RNAi to inactivate the major components of CeHDs, including the apical receptor MUP-4, the basal receptor LET-805 and the intermediate filament protein IFB-1. The results showed that only loss of MUP-4 caused a substantial decrease of LGG-1 puncta induced by different causes (), suggesting that autophagy induction was impaired specifically by disruption of certain apical epidermal supporting structures. It was recently reported that lysosomes could be activated by CeHD disruption [Citation30], raising another possibility that reduced autophagosomes could be due to accelerated autolysosome turnover caused by lysosome activation. However, we found that mup-4 RNAi treatment still reduced the number of LGG-1 puncta when the autolysosome turnover was blocked by epg-5 inactivation ( and 3E), therefore ruling out the influence of lysosome activation [Citation26–28]. Analysis of the autophagy degradation substrate SQST-1 also confirmed that apical CeHD damage indeed decreased the autophagic activity in the epidermis (-3 H). These observations altogether suggest that disruption of apical epidermal attachment structures has an inhibitory effect on the autophagic process.
Figure 3. Apical epidermal structural damage inhibits autophagy activation. (A) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by the epidermal-specific Pdpy-7 promoter in sham-treated adults fed with bacteria carrying the empty vector L4440 (control), heat shock (HS) or paraquat-treated adults fed with L4440, mup-4 RNAi, let-805 RNAi or ifb-1 RNAi clones. Scale bar: 10 μm. (B) Quantification of results in (A) showing the number of GFP::LGG-1 puncta per unit area in the epidermis of worms treated with heat shock after damage of CeHD components compared with control. n ≥ 7 per condition. HS, heat shock. (C) Quantification of results in (A) showing the number of GFP::LGG-1 puncta per unit area in the epidermis of worms treated with paraquat after damage of CeHD components compared with control. n ≥ 11 per condition. (D) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by its own promoter in the epidermis of wild-type (WT) or epg-5(tm3425) young adults treated with L4440 (control) or mup-4 RNAi. Scale bar: 10 μm. (E) Quantification of the number of GFP::LGG-1 puncta in the epidermis of animals with the indicated genotypes, corresponding to results in (D). n ≥ 7 per condition. (F) Representative confocal images showing the protein levels of SQST-1::GFP driven by the epidermal-specific promoter Pcol-19 in sham-treated adults fed with L4440 (control) or mup-4 RNAi, heat shock (HS) or paraquat-treated animals fed with L4440 or mup-4 RNAi clones. Scale bar: 40 μm. (G and H) Quantification of the relative Pcol-19::SQST-1::GFP abundance with the indicated treatments, corresponding to results in (F). n ≥ 26 per condition. Error bars represent mean ± SEM. ns, not significant; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed, unpaired t-test).
Basement structural damage promotes epidermal autophagy through a mechanotransduction pathway mediated by the basal CeHD receptor and LET-363/MTOR
Based on the finding that both the disruption of basement membrane and the damage of adjacent muscle structure induced autophagy in the epidermis (, 2B and S2), we speculated that elevated autophagy could be due to loss of muscle tension to the epidermal cells. Consistent with this hypothesis, epidermal autophagy was also induced by disruption of other muscle structural components or the molecule directly controlling muscle contraction (, S3A and S3B) [Citation31–34]. Furthermore, inhibiting the contraction of body-wall muscles by using gamma-aminobutyric acid type A (GABAA) receptor agonist muscimol greatly increased the number of GFP::LGG-1 puncta in the epidermal cells ( and 4C). It supports the notion that loss of muscle tension alone is sufficient to stimulate an autophagic response in the epidermis. The fact that unc-52 RNAi and muscimol treatment elicited autophagy specifically in the epidermis but not other tissues ruled out the possibility that autophagy induced by basal structural damage and tension loss was due to secondary effects such as starvation, GABAergic signaling alteration or systemic stress (Figure S3C). Moreover, muscimol treatment did not further augment autophagy in unc-52 inactivated worms, which indicates that basal structural damage induces epidermal autophagy via the same pathway as tension loss ( and 4C). Together, these results suggest that increased epidermal autophagic activity upon basal structural damage is caused by loss of mechanical tension.
Figure 4. Basal structural damage promotes epidermal autophagy through a mechanotransduction pathway mediated by the basal CeHD receptor and LET-363/MTOR. (A) Representative confocal images showing the distribution patterns of GFP::LGG-1 driven by the epidermal-specific Pdpy-7 promoter in the young adults treated with the empty vector L4440 (control), unc-97 RNAi, pat-6 RNAi or pat-2 RNAi. (B) Representative confocal images showing the distribution patterns of Pdpy-7::GFP::LGG-1 puncta in the young adults treated with L4440 or unc-52 RNAi in the presence or absence of muscimol. (C) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis with the indicated treatments, corresponding to results in (B). n = 6 per condition. (D) Representative confocal images showing the distribution patterns of Pdpy-7::GFP::LGG-1 puncta in the young adults treated with L4440 or let-805 RNAi in the presence or absence of muscimol. (E) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis with the indicated treatments, corresponding to results in (D). n = 7 per condition. (F) Representative confocal images showing the distribution patterns of Pdpy-7::GFP::LGG-1 puncta in the young adults treated with L4440 or let-363 RNAi in the presence or absence of muscimol. (G) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis with the indicated treatments, corresponding to results in (F). n ≥ 10 per condition. (H) Western blot analysis showing the total protein level and the phosphorylation level of LET-363/MTOR on serine 2448 in wild-type (WT) and unc-52(e1421) young adults. Tubulin was used as the loading control. Scale bars: 10 μm. Error bars represent mean ± SEM. ns, not significant; ***, P < 0.001 (two-tailed, unpaired t-test).
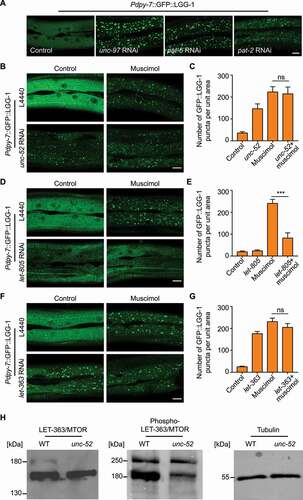
Because LET-805 is the basal CeHD receptor that connects the muscle cells with the epidermis, we asked whether muscle tension goes through LET-805 to regulate the autophagic process [Citation19]. Consistent with our hypothesis, inactivation of let-805 expression significantly inhibited epidermal autophagosome formation induced by tension loss (). Interestingly, loss of let-805 did not inhibit autophagic induction caused by other factors such as heat shock or paraquat treatment (). It suggests that LET-805 specifically functions as the direct link between mechanical signals and the epidermal autophagic process. MTOR (mechanistic target of rapamycin kinase) is a critical regulator not only of autophagy induction but also of mechanotransduction [Citation9,Citation35,Citation36], prompting us to investigate the involvement of MTOR in autophagy activation by tension loss following basal structural damage. As shown in , muscimol treatment did not induce more autophagosome formation in the let-363 (the C. elegans ortholog of MTOR)-inactivated epidermis, suggesting that tension loss and MTOR inhibition promotes autophagy through the same pathway. In support of this hypothesis, unc-52 inactivation down-regulated the phosphorylation level of LET-363/MTOR on serine 2448 (), which is an established read-out of MTOR activation [Citation37]. Taken together, we propose that the epidermis with basal structural damage could convert loss of mechanical signal to the autophagy response through the LET-805-MTOR signaling axis.
Apical epidermal structural disruption inhibits the autophagy process via activation of calcium influx
We next explored the mechanism by which structural damage inhibits autophagy in the epidermal cells. It was previously reported that mechanical wounding of the C. elegans epidermis triggers an increase of Ca2+ signal around the injury site [Citation13]. We noticed that the distribution area of Ca2+ was complementary to that of the LGG-1 puncta after needle wounding. That is to say, wherever the autophagic activity is low, the Ca2+ level is high ( and S4). Similar as needle wounding, disruption of the apical CeHD receptor MUP-4 also elevated cytosolic Ca2+ level ( and 5C). Therefore, it appears that autophagy inhibition by epidermal structural damage always accompanies Ca2+ activation. We hence hypothesized that the surge of Ca2+ influx following epidermal structural damage could be the cause of autophagy inhibition. It was previously reported that TRPM2 (transient receptor potential cation channel subfamily M member 2)-dependent Ca2+ influx has inhibitory effect on the induction of early autophagy in certain circumstances [Citation38]. To test whether it is the same case in autophagy inhibition by structural damage, we examined the influence of C. elegans TRPM-family channel GTL-2 on autophagy in the epidermal cells [Citation13]. In agreement with our hypothesis, gtl-2 inactivation resulted in elevated autophagic process, as shown by the rise of LGG-1 puncta numbers in the epidermis ( and 5E). Moreover, the autophagic activation in gtl-2 mutants can by suppressed by external Ca2+ but not needle wounding (). Loss of function in gtl-2 also abolished the suppressive effect of mup-4 inactivation on autophagy ( and 5I). Taken together, these results suggest that structural damage indeed inhibits autophagy in a Ca2+-dependent manner.
Figure 5. Epidermal structural damage inhibits the autophagy process via activation of calcium influx. (A) Representative confocal images showing the distribution patterns of epidermal GFP::LGG-1 puncta and GCaMP3 fluorescence in the adults with an intact epidermis (control) or a single wound caused by needle puncture. Arrows point to sites of wounding. Scale bar: 40 μm. (B) Representative confocal images showing the fluorescent levels of epidermal GCaMP3 in the animals treated with the empty vector L4440 (control) or mup-4 RNAi. Scale bar: 40 μm. (C) Quantification of the relative GCaMP3 fluorescence in the epidermis with the indicated treatments, corresponding to results in (B). n = 12 per condition. (D) Representative confocal images showing the distribution patterns of Plgg-1::GFP::LGG-1 puncta in wild-type (WT) or gtl-2(n2618) young adults treated with or without needle wounding. Arrows point to sites of wounding. Scale bar: 10 μm. (E) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis with the indicated treatments, corresponding to results in (D). n ≥ 10 per condition. (F) Representative confocal images showing the distribution patterns of Plgg-1::GFP::LGG-1 puncta in wild-type (WT) or gtl-2(n2618) young adults soaked with CaCl2 or control solution for 1 h after needle wounding. Arrows point to sites of wounding. Scale bar: 10 μm. (G) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis with the indicated treatments, corresponding to results in (F). n ≥ 7 per condition. (H) Representative confocal images showing the distribution patterns of Plgg-1::GFP::LGG-1 puncta in wild-type (WT) or gtl-2(n2618) young adults treated with or without mup-4 RNAi. Scale bar: 10 μm. (I) Quantification of the number of GFP::LGG-1 puncta per unit area in the epidermis with the indicated treatments, corresponding to results in (H). n ≥ 16 per condition. Error bars represent mean ± SEM. ns, not significant; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed, unpaired t-test).
Elevated autophagy plays a detrimental role during epidermal structural damage response
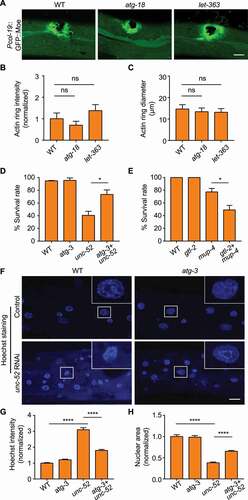
To investigate the physiological significance of autophagy during structural damage, we first analyzed the effect of autophagy on epidermal wound repair using GFP::Moe (Moesin; the C-terminal end of the sole Drosophila ERM protein) as the marker for actin-mediated wound closure process [Citation13]. The results showed that neither autophagy inhibition by atg-3 or atg-18 inactivation nor autophagy enhancement by let-363 RNAi significantly affected the formation of actin rings around the wounded site (, 6B, S5A and S5B) [Citation39,Citation40]. The diameters of the wounded sites were likewise unchanged, suggesting that the process of wound closure was unaffected by elevated or reduced autophagic activity ( and S5C). However, autophagy appeared to influence the survival of the organisms under the stress of structural damage. Suppressing the autophagic process by loss-of-function mutation of atg-3 or atg-18 significantly rescued the survival rate of unc-52-inactivated animals with defective basement membrane ( and S5D). Conversely, elevating autophagy by gtl-2 mutation increased the death rate of mup-4-inactivated worms (). These data suggest that excessive autophagic activity could reduce the tolerance of the animals to epidermal structural damage. Considering the fact that autophagy could trigger apoptosis under certain circumstances [Citation41,Citation42], we examined whether apoptosis is involved in the detrimental effect of autophagy on structural damage control. Hoechst staining showed that the epidermis of unc-52 RNAi-treated animals displayed brighter fluorescence signal and smaller nuclei, reflecting chromatin condensation characteristic of apoptosis (-6 H). Suppressing the autophagic process by atg-3 or atg-18 mutation ameliorated the apoptotic features of epidermal nuclei induced by unc-52 RNAi (-6 H, S5E-S5G). The data obtained from the mCherry fusion of the chromatin-associating protein HIS-58 were consistent with the Hoechst staining results (Figure S5H-S5J) [Citation43]. These observations suggest that autophagy could exert detrimental effects on the organism with damaged epidermal structure through aggravating apoptotic cell death. Given that the innate immune response is induced upon structural damage and is important for epidermal survival, we wondered whether damage-induced autophagy affects the innate immune response [Citation11,Citation14]. qPCR analysis showed that the autophagy inhibition indeed increased the expression of the antimicrobial peptide nlp-29 upon disruption of the basement membrane (Figure S5K). This data suggests that autophagy could play a suppressive role in damage-induced innate immune response, which could be a contributing factor for the detrimental effect of autophagy on structural damage.
Figure 6. Elevated autophagy plays a detrimental role during epidermal structural damage. (A) Representative confocal images showing the formation of actin rings visualized by Pcol-19::GFP::Moe in wild-type (WT), atg-18(gk378) and let-363 RNAi-treated adults after needle wounding. (B) Quantification of the relative intensities of the actin rings, corresponding to results in (A). n ≥ 9 per condition. (C) Quantification of the average diameter of the actin rings, corresponding to results in (A). n ≥ 7 per condition. (D) Survival rates of wild type (WT) and the atg-3(bp412) single mutant combined with or without unc-52(e1421) mutation (three biological replicates, n = 30 per condition). (E) Survival rates of wild type (WT) and the gtl-2(n2618) single mutant treated with or without mup-4 RNAi (three biological replicates, n = 30 per condition). (F) Hoechst staining showing epidermal nuclei of wild type (WT) and the atg-3(bp412) single mutant combined with or without unc-52 RNAi. Insets correspond to boxed areas in each image. (G) Quantification of the relative intensities of nuclear Hoechst staining in the epidermis of animals with the indicated genotypes, corresponding to results in (F). n ≥ 8 per condition. (H) Quantification of the relative sizes of the nuclei in the epidermis of animals with the indicated genotypes, corresponding to results in (F). n ≥ 8 per condition. Scale bars: 10 μm. Error bars represent mean ± SEM. ns, not significant; *, P < 0.05; ****, P < 0.0001 (two-tailed, unpaired t-test).
Discussion
In conclusion, we propose the following model for the regulation of epidermal autophagy by structural damage: the destruction of epidermal supporting structures can induce and inhibit autophagy simultaneously. Autophagy induction is mediated through the weakening of mechanical tension caused by basal structural damage, while autophagy inhibition is mainly achieved through up-regulation of calcium signaling pathway triggered by apical structural damage. Furthermore, elevated autophagy level appears to be not beneficial but detrimental for the C. elegans epidermis to cope with structural damage (Figure S6). We believe that the injured epidermal cells may release autophagy-inhibiting signals in order to counteract the damage caused by the up-regulation of autophagy. Our findings also indicate that the ultimate effect of mechanical injury on autophagy in a single epithelial cell is likely dependent on the type, location and the damage extent of the supporting structures. These discoveries may help us better understand the damage response of the autophagy machinery, and provide insights into the pathogenesis of various diseases related to autophagy or epithelial cell structural damage.
Although it is uncertain whether the bidirectional regulation of autophagy by mechanical injury also occurs in mammalian epithelial cells, several published works suggest that this phenomenon is likely to be evolutionarily conserved. For example, a scratch-wound healing assay using HeLa cells showed that autophagy is inhibited in the cells close to the wound edge but increased in the cells located at the rear [Citation44]. This observation suggests that autophagy is regulated both positively and negatively in the mammalian epithelial cells after mechanical injury. However, whether autophagy is promoted and inhibited in the same damaged cell remains to be further confirmed. In addition, the regulatory mechanisms of autophagy by components of supporting structures in the C. elegans epidermis may also exist in mammalian epithelial cells. For example, we found that destruction of the apical CeHDs inhibited autophagy induced by different factors, while damage of the basal CeHDs specifically inhibited autophagy induction caused by tension loss, indicating that C. elegans hemidesmosomes in general play a positive rather than negative role in autophagy regulation. Consistent with this finding, antibody-mediated blockade of the hemidesmosome receptor ITGB4/integrin β4 or ITGA6/integrin α6 does not induce autophagy in the human mammary epithelial cells, but inhibits the formation of autophagosomes induced by growth factor starvation in the prostate epithelial cells [Citation2,Citation45]. Therefore, we speculate that the positive regulation of hemidesmosomes on autophagy might be evolutionarily conserved. Moreover, similar to the negative regulation of the C. elegans ECM protein UNC-52 on epidermal autophagy, the deficiency of HSPG2/perlecan (the mammalian homolog of UNC-52) also enhances autophagy in the muscle cells [Citation46], although the effect of HSPG2 on autophagy in mammalian epidermal cells has not been reported. Interestingly, a recent study using C. elegans neuronal cells as the model found that after axonal transection by laser, autophagic vesicles are detected in the cell bodies distant from the injury site but not in the axons close to the wounded area [Citation47]. This observation is similar to the spatially-restricted distribution of autophagosomes distant from the wounding site in the C. elegans epidermis after puncture injury, suggesting that the proximal inhibitory effect and distal promotional effect of physical injury on autophagy may also exist in nerve cells. It is worth noting that different types of mechanical injuries to mammalian nerve tissues often lead to different effects on the autophagy flux in nerve cells. For example, traumatic injury by weight-drop upon the mouse brain increases autophagy flux in the injured cortical neurons [Citation48]. Crush injury of rat optic nerve axons by a surgical suture also induces autophagy in the optic nerve cells [Citation49]. On the contrary, the autophagy flux is inhibited in the cortical neurons, activated microglia and oligodendrocytes of the mouse cortex injured by a pneumatic impactor with tip [Citation50]. Contusion injury of the rat spinal cord caused by weight-drop also inhibits the autophagy flux of motor neurons, activated microglia and oligodendrocytes in the injury site [Citation51]. The fact that different damage methods have different effects on the autophagic process in nerve cells implies that the bidirectional regulatory mechanisms by different structural components might also exist in nerve cells.
Our results suggest that hemidesmosomes inhibit the activity of MTOR and induce autophagy after receiving the signal of tension change caused by destruction of the basal epidermal supporting structures. In accordance with this finding, deficiency of the hemidesmosome receptor ITGB4 in human keratinocytes activates MTOR through a PTK2/FAK (protein tyrosine kinase 2)-class I phosphoinositide 3-kinase (PI3K)-AKT/protein kinase B (AKT serine/threonine kinase) signaling pathway [Citation52], suggesting that the negative regulation of MTOR signaling by hemidesmosomes may be evolutionarily conserved. However, it is necessary to further verify whether hemidesmosomes also inhibit MTOR activity through the PTK2-PI3K-AKT signal pathway in the case of epidermal structural damage. In addition, a recent report found that MTOR complex 1 (MTORC1) can localize to the focal adhesions and its activity is regulated by the focal adhesions [Citation53]. Whether there is a similar spatial association between MTORC1 and hemidesmosomes, and whether this direct interaction can regulate the activity of MTORC1 in the case of structural damage is worthy of further investigation. This report also identified interaction of MTORC1 with GIT2 (G protein-coupled receptor kinase interactor 2) and ARHGEF7/β-PIX (Rho guanine nucleotide exchange factor 7) using a proximity labeling and proteomics approach [Citation53]. Interestingly, the C. elegans homolog for GIT2 and ARHGEF7, GIT-1 and PIX-1, are localized in the hemidesmosomes and are involved in the mechanotransduction mediated by hemidesmosomes during embryonic development [Citation54]. These results indicate that GIT-1 and PIX-1 may also be involved in the regulation of autophagy mediated by hemidesmosomes and MTOR.
In most cases, autophagy is induced to render epithelial cells more resistant to a variety of stressed conditions including ultraviolet (UV) radiation, heat/cold shock, oxidative stress and ECM detachment [Citation1,Citation55–65]. However, some reports also found that elevated autophagy level can play a cytotoxic role in epithelial cells under certain stressed conditions. For example, inhibition of autophagy by 3-methyladenine and ATG5 RNAi prevents the death of normal human epidermal keratinocytes (NHEK) induced by oxidative stress [Citation66]. Induction of autophagy by rapamycin reduces the thermotolerance of human epidermal keratinocytes HaCaT [Citation67]. RIPK1 (receptor interacting serine/threonine kinase 1)-mediated mitophagy reduces the survival of the human non-small cell lung cancer cell line H460 during ECM detachment [Citation68]. Inhibition of autophagy by bafilomycin A1 or BECN1 RNAi reverses the anoikis of ovarian cancer cells induced by overexpression of sialidase NEU2 (neuraminidase 2) [Citation69]. Inhibition of autophagy by 3-methyladenine also reduces the death rate of suspended lung cancer cells caused by loss of CDCP1 (CUB domain containing protein 1) [Citation70]. In this study, we found that autophagy also promoted the apoptosis of the C. elegans epidermis under the stress of epidermal structural damage. The reason for this phenomenon may be that high level of autophagy results in excessive degradation of damaged supporting structural components and other molecules involved in damage response and repair, which may further amplify the apoptotic signal. For example, it was reported that intermediate filaments with abnormal dynamics can be degraded by autophagy [Citation71,Citation72], and that the depletion of certain intermediate filament components increases the sensitivity of epithelial cells to apoptosis induced by different factors [Citation73,Citation74]. Therefore, it is possible that structural damage of epithelial cells leads to the disruption of intermediate filaments, which are subjected to degradation by elevated autophagy. The deficiency of intermediate filaments might render the epidermal cells more susceptible to damage-induced cell death. In addition, our data suggest that autophagy might suppress the innate immune response induced by structural damage, which is consistent with previous reports showing that autophagy-deficiency leads to up-regulation of pro-inflammatory cytokines in higher organisms [Citation75–78]. Therefore, compromised innate defense might also contribute to elevated cell death caused by excessive autophagy. The specific roles and regulatory mechanisms of autophagy on cell survival during different types of injury remain to be further studied.
Materials and methods
Worm strains and maintenance
C. elegans strains were maintained following standard procedures [Citation79]. The worms were grown at 20°C and fed with OP50 E. coli (Caenorhabditis Genetics Center; CGC) unless noted otherwise. Wildtype N2 strain and strains carrying adIs2122[Plgg-1::GFP::lgg-1+ rol-6(su1006)], juIs319[Pcol-19::GcaMP3+ Pcol-19::tdTomato], juls352[Pcol-19::GFP::moe], atg-18(gk378), unc-52(e1421) or gtl-2(n2618) were obtained from CGC (DA2123, CZ13896, CZ14748, VC893, CB1421 and CZ9957), which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40OD010440). epg-5(tm3425) was generated by the International C. elegans Gene Knockout Consortium. atg-3(bp412);him-5(e1490) was kindly provided by Dr. Hong Zhang (Chinese Academy of Sciences, China). yqIs25 [Plgg-1::GFP::lgg-1] was a gift from Dr. Chonglin Yang (Yunnan University, China). Strains carrying sdaIs12[Pdpy-7::GFP::lgg-1+ Pmyo-2::GFP], sdaIs16[Pcol-19::sqst-1::GFP+Pdpy-7::tdTomato], sdaEx316[Pdpy-7::RFP::lgg-1+ Pmyo-2::GFP] and sdaEx309[Peft-3::FRT::mCherry::his-58::FRT::GFP::his-58+ Pmyo-2::mCherry] were generated for this study.
Molecular cloning and plasmid construction
All DNA fragments were amplified by PrimeSTAR GXL Premix (TaKaRa, R051B). Fusion plasmids were constructed using the ClonExpress II One Step Cloning Kit (Vazyme Biotech, C112-02) or the ClonExpress MultiS One Step Cloning Kit (Vazyme Biotech, C113-02). The Pdpy-7::GFP::lgg-1 plasmid was created by inserting a 650-bp dpy-7 promoter and the entire lgg-1 coding sequence into the pPD95.75 (Addgene, 1494; Andrew Fire) vector whose stop codon after GFP was removed by Q5 Site-Directed Mutagenesis Kit (NEB, E0552S). The Pdpy-7::RFP::lgg-1 plasmid was created by inserting a 650-bp dpy-7 promoter, RFP and the entire lgg-1 coding sequence into the pPD95.75 backbone using KpnI and EcoRI. The Pcol-19::sqst-1::GFP plasmid was constructed by inserting a 910-bp col-19 promoter and the entire sqst-1 coding sequence in the pPD95.75 backbone. The Peft-3::FRT::mCherry::his-58::FRT::GFP::his-58 plasmid was created by replacing the hsp-16.41 promoter of pBN154 (Addgene, 86718; Peter Askjaer) with a 600-bp eft-3 promoter. All plasmids were confirmed by sequencing before microinjection.
Strain construction and transgenesis
Transgenic strains were obtained by microinjection of fusion constructs (10 ng/μl) with Pmyo-2::GFP or Pmyo-2::mCherry (20 ng/μl) as co-injection markers. Microinjection was carried out following established protocols [Citation80]. Extrachromosomal arrays of Pdpy-7::GFP::lgg-1 and Pcol-19::sqst-1::GFP were integrated into the C. elegans genome by UV irradiation as previously described [Citation81]. These integrated transgenic strains were outcrossed for at least 4 times prior to analysis.
Mechanical wounding of the epidermis
We performed needle wounding of the C. elegans epidermis as described previously [Citation11,Citation82]. Briefly, a microinjection needle was used by hand to make one wound to the anterior or posterior body or three wounds to the anterior, middle and posterior body of adult animals immobilized on the pre-cooled NGM plates (3 g/L NaCl [Sangon Biotech, A100241], 2.5 g/L peptone [Solarbio, P8450], 20 g/L agar [CaLbiochem, 12,177], 25 mM K2HPO4/KH2PO4 [Sangon Biotech, A501728, A501211; pH 6.0], 1 mM CaCl2 [Sangon Biotech, A501330], 1 mM MgSO4 [Sangon Biotech, A610329], 5 mg/L cholesterol [Sangon Biotech, A100433]). Wounded animals were transferred to new plates for recovery and collected 1 h later for fluorescent imaging of GFP::LGG-1 and RFP::LGG-1 or 2 h later for that of GFP::Moe.
Fluorescence imaging and analysis
Animals were immobilized by 0.625 mM levamisole (Sangon Biotech, A506644) in M9 buffer (3 g/L KH2PO4, 6 g/L Na2HPO4 [Sangon Biotech, A607793], 5 g/L NaCl, 1 mM MgSO4) on 2% agarose pad and were imaged using Zeiss LSM800 confocal microscope (Zeiss, Germany) equipped with a 20x (for multiple worms) or 63x (for single worms) objective. All image quantifications were performed using the ImageJ software (https://imagej.nih.gov/ij/). The number of LGG-1 puncta per unit area (500 μm2) was calculated as the total area occupied by LGG-1 puncta divided by the mean area of one C. elegans autophagosome (0.126 μm2) [Citation83]. For each experiment, images from the cytoplasmic regions of the epidermis in at least 6 individual animals were used for quantification. To analyze SQST-1::GFP abundance in the epidermis, the average intensities of Pcol-19::SQST-1::GFP fluorescent signal in at least 26 individual animals were measured. For the quantification of calcium signaling in the epidermis, the average intensities of Pcol-19::GcamP3 fluorescent signal in 12 individual animals were measured. In order to quantify actin ring intensity, we measured the average intensities of the GFP::Moe fluorescence signal in the wounded area and an intact area of the epidermis of at least 9 animals. The difference of signal intensity between the two regions was calculated as the relative intensity of an actin ring. To quantify the diameter of an actin ring, the plot profile tool of ImageJ was used to extract the fluorescence intensity along the long axis of the actin ring, in which the two peaks represent the fluorescence intensities of actin at the wound edge. The distance between the two peaks was defined as the diameter of the actin ring. To visualize condensed, apoptotic nuclei, worms were stained with 2.5 mg/ml Hoechst 33342 (Sigma-Aldrich, 14533) for 2 h at 20°C in M9 buffer with bacteria and then washed three times with M9 buffer. To analyze nuclear chromatin condensation, images from at least 8 individuals were used and the average fluorescence intensities and the total area occupied by each nucleus labeled by Hoechst 33342 or mCherry::HIS-58 in the epidermis were measured.
RNA interference
Most RNAi clones used in this study were obtained from the MRC library, after verifying the sequence of the corresponding inserts. vab-10a RNAi clone was constructed by inserting a 544-bp fragment within exon17 of vab-10 into the HindIII site of vector L4440 (Addgene, 1654; Andrew Fire) as described before [Citation11]. For bli-1, unc-52, unc-112, unc-97, pat-2 and let-363 RNAi, worms were fed with RNAi against the corresponding genes from the L1 stage until young adult stage for analysis. For pat-6 and unc-45 RNAi, worms were fed with RNAi from the L4 stage to get paralyzed young-adult F1 for analysis. For mup-4, ifb-1, vab-10a and let-805 RNAi, worms were fed with RNAi against those genes from the L2/L3 stage to prevent premature lethality.
Drug and heat shock treatment
For disruption of microtubule bundles, young adult worms were soaked in 60 μg/ml nocodazole (Sangon Biotech, A606391) dissolved in M9 for 2 h, and then recovered in M9 buffer for 2 h before collection. For disruption of actin cytoskeleton, young adult worms were soaked in 6 μg/ml cytochalasin D (Aladdin, c102396) dissolved in M9 for 2 h, and then recovered in M9 buffer for 2 h before collection. Control groups were soaked in M9 buffer for 4 h before analysis.
Paraquat treatment was carried out based on previously established method [Citation29]. Briefly, 400 μl 486 mM paraquat (Sigma-Aldrich, 36541) or H2O solvent was spread over each 6-cm NGM plate seeded with OP50 killed by UV irradiation (0.5 J/cm2, 15 min). The plates were allowed to dry in the dark for overnight at room temperature. Young adult worms were transferred onto control or paraquat plates for 8 h and were then collected for confocal imaging.
To remove muscle contraction, 50 μl 100 mM muscimol (Abcam, ab120094) or H2O solvent mixed with 50 μl OP50 was added to each 6-cm NGM plate. The plates were allowed to dry in the dark for overnight at room temperature. Young adult worms were transferred onto control or muscimol plates for 1 h and were then collected for confocal imaging. For induction of autophagy by heat shock, young adult worms were placed at 37°C for 0.5 h on plates seeded with OP50 and were collected after 1 h recovery for confocal imaging of GFP::LGG-1. Young adult worms were placed at 37°C for 2 h on plates seeded with OP50 and were then were collected after 8 h recovery for confocal imaging of SQST-1::GFP.
To perform external Ca2+ supplement, 2 μl 1 M CaCl2 or H2O solvent was mixed with 1 ml S buffer (129 ml 0.05 M K2HPO4, 871 ml 0.05 M KH2PO4, 5.85 g NaCl). Wounded worms were incubated in 100 μl CaCl2 or control solution for 1 h and then were immobilized by 0.625 mM levamisole in CaCl2 or control solution on 2% agarose pad for confocal imaging.
Western blot analysis
For western blot analysis, about 500 young adults were collected and washed three times with M9 buffer. After two times snap-freezing in liquid nitrogen, the dry pellets were mixed with equal-volume 2X SDS-PAGE Sample Loading Buffer (Sangon Biotech, C508321) supplemented with cOmplete EDTA-free Protease Inhibitor Cocktail (ROCHE, 04693132001). The lysates were boiled for 10 min and centrifuged for SDS-PAGE analysis. Dilution factors for primary antibodies are: anti-GFP (Santa Cruz Biotechnology, sc-9996; 1:1000); anti-MTOR (Cell Signaling Technology, 2972; 1:1000); anti-phospho-MTOR (Ser2448) (Cell Signaling Technology, 2971; 1:1000); anti-tubulin (DSHB, AA4.3; 1:1000); anti-LGG-1, 1:1000. The LGG-1 polyclonal antibody was raised against the peptide expressed by the plasmid pET21b-lgg-1, which was obtained from Xiaochen Wang lab (Chinese Academy of Sciences, China). The secondary antibodies used in were HRP goat anti-rabbit antibody (Bioworld Technology, BS13278; 1:10,000) and HRP goat anti-mouse antibody (Bioworld Technology, BS12478; 1:10,000). Immobilon Western Chemiluminescent HRP Substrate (ECL) (Millipore, WBKLS0500) was used for detection. The ChemiDoc Touch imaging system (BIO-RAD, U.S.A.) was used for the documentation of the western results.
Survival assay
30 L4 larvae per genotype were cultured at standard conditions for 144 h to score the viability of wild type (WT), atg-3(bp412) and atg-18(gk378) single mutant combined with or without unc-52(e1421) mutation or for 24 h to score the viability of WT and the gtl-2(n2618) single mutant treated with or without mup-4 RNAi. Failure to respond to touch was defined as death. The survival assay was replicated in three independent experiments.
Quantitative RT-PCR (qPCR)
For qPCR experiments, total RNA of young adults was extracted by RNA isolater Total RNA Extraction Reagent (Vazyme, R401-01) and reverse transcribed by HiScript III All-in-one RT SuperMix (Vazyme, R333-01). Real-time PCR was performed with ChamQ SYBR Color qPCR Master Mix (Vazyme, Q421-02) on Lightcycler 96 system (Roche, Switzerland). Each experiment was repeated four times using independently collected RNA samples, and each reaction was run in quadruplicate. The expression levels of target genes were normalized using the reference gene act-1. Primers used in qPCR reactions are: act-1-F, TCCTTACCGAGCGTGGTTAC; act-1-R, GTTTCCGACGGTGATGACTT; nlp-29-F, TCCTTCTCGCCTGCTTCA; nlp-29-R, CTTTCCCCATCCTCCATACA.
Statistical analysis
Statistical analysis of all assays was performed using the Student’s t test (unpaired for most experiments or paired for qPCR, two-tailed) on GraphPad Prism 7.0 software (https:/www.graphpad.com:443/). Error bars, P value, and sample sizes are indicated in the figure legends. Significance was accepted as P < 0.05.
Supplemental Material
Download MS Word (35.6 MB)Acknowledgments
We are grateful to Hong Zhang, Xiaochen Wang, Chonglin Yang, the Caenorhabditis Genetics Center and the International C. elegans Gene Knockout Consortium for reagents. This work was supported by funds from the National Natural Science Foundation of China [31871384, 32170690 and 31900545], Natural Science Foundation of Jiangsu Province of China [BK20160009], Jiangsu Provincial Innovative Research Team, the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT-IRT1075) and the Priority Academic Program Development of Jiangsu Higher Education Institutions [PAPD]. Some strains were provided by CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Fung C, Lock R, Gao S, et al. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008 Mar;19(3):797–806.
- Chen N, Debnath J. IκB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy. 2013 Aug;9(8):1214–1227.
- Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, et al. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 2013 Oct 10 32(41):4932–4940.
- Yoo BH, Zagryazhskaya A, Li Y, et al. Upregulation of ATG3 contributes to autophagy induced by the detachment of intestinal epithelial cells from the extracellular matrix, but promotes autophagy-independent apoptosis of the attached cells. Autophagy. 2015;11(8):1230–1246.
- Vlahakis A, Debnath J. The interconnections between autophagy and integrin-Mediated cell adhesion. J Mol Biol. 2017 Feb 17; 429(4):515–530.
- Kast DJ, Dominguez R. The cytoskeleton-autophagy connection. Curr Biol. 2017 Apr 24; 27(8):R318–r326.
- Wang RC, Wei Y, An Z, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science (New York, NY). 2012 Nov 16 338(6109):956–959.
- King JS, Veltman DM, Insall RH. The induction of autophagy by mechanical stress. Autophagy. 2011 Dec;7(12):1490–1499.
- Orhon I, Dupont N, Zaidan M, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016 Jun;18(6):657–667.
- Chisholm AD, Xu S. The Caenorhabditis elegans epidermis as a model skin. II: differentiation and physiological roles. WIRES Dev Biol. 2012 Nov-Dec;1(6):879–902.
- Zhang Y, Li W, Li L, et al. Structural damage in the C. elegans epidermis causes release of STA-2 and induction of an innate immune response. Immunity. 2015 Feb 17 42(2):309–320.
- Zhang H, Labouesse M. The making of hemidesmosome structures in vivo. Dev Dyn. 2010 May;239(5):1465–1476.
- Xu S, Chisholm AD. A Gαq-Ca2; signaling pathway promotes actin-mediated epidermal wound closure in C. elegans. Curr Biol. 2011 Dec 6; 21(23):1960–1967.
- Pujol N, Cypowyj S, Ziegler K, et al. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr Biol. 2008 Apr 8 18(7):481–489.
- Zhang H, Chang JT, Guo B, et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy. 2015;11(1):9–27.
- Kumsta C, Chang JT, Schmalz J, et al. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat Commun. 2017 Feb 15;8:14337.
- Bosher JM, Hahn BS, Legouis R, et al. The caenorhabditis elegans vab-10 spectraplakin isoforms protect the epidermis against internal and external forces. J Cell Biol. 2003 May 26 161(4):757–768.
- Hong L, Elbl T, Ward J, et al. MUP-4 is a novel transmembrane protein with functions in epithelial cell adhesion in Caenorhabditis elegans. J Cell Biol. 2001 Jul 23 154(2):403–414.
- Hresko MC, Schriefer LA, Shrimankar P, et al. Myotactin, a novel hypodermal protein involved in muscle-cell adhesion in caenorhabditis elegans. J Cell Biol. 1999 Aug 9 146(3):659–672.
- Mullen GP, Rogalski TM, Bush JA, et al. Complex patterns of alternative splicing mediate the spatial and temporal distribution of perlecan/UNC-52 in caenorhabditis elegans. Mol Biol Cell. 1999 Oct;10(10):3205–3221.
- Rogalski TM, Mullen GP, Gilbert MM, et al. The UNC-112 gene in caenorhabditis elegans encodes a novel component of cell-matrix adhesion structures required for integrin localization in the muscle cell membrane. J Cell Biol. 2000 Jul 10 150(1):253–264.
- Woo WM, Goncharov A, Jin Y, et al. Intermediate filaments are required for C. elegans epidermal elongation. Dev Biol. 2004 Mar 1 267(1):216–229.
- Costa M, Draper BW, Priess JR. The role of actin filaments in patterning the caenorhabditis elegans cuticle. Dev Biol. 1997 Apr 15; 184(2):373–384.
- Wang S, Wu D, Quintin S, et al. NOCA-1 functions with γ-tubulin and in parallel to Patronin to assemble non-centrosomal microtubule arrays in C. elegans. eLife. 2015 Sep 15;4:e08649.
- Lints R, Hall DH. The cuticle. WormAtlas. 2009. 10.3908/wormatlas.1.12
- Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010 Jun 11 141(6):1042–1055.
- Wang Z, Miao G, Xue X, et al. The vici syndrome protein EPG5 Is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016 Sep 1 63(5):781–795.
- Guo B, Liang Q, Li L, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014 Dec;16(12):1215–1226.
- Palikaras K, Tavernarakis N. Assessing mitochondrial selective autophagy in the nematode caenorhabditis elegans. Methods Mol Biol. 2017;1567:349–361.
- Miao R, Li M, Zhang Q, et al. An ECM-to-Nucleus signaling pathway activates lysosomes for C. elegans larval development. Dev Cell. 2020 Jan 6 52(1):21–37.e5.
- Norman KR, Cordes S, Qadota H, et al. UNC-97/PINCH is involved in the assembly of integrin cell adhesion complexes in caenorhabditis elegans body wall muscle. Dev Biol. 2007 Sep 1 309(1):45–55.
- Lin X, Qadota H, Moerman DG, et al. C. elegans PAT-6/actopaxin plays a critical role in the assembly of integrin adhesion complexes in vivo. Curr Biol. 2003 May 27 13(11):922–932.
- Moerman DG, Williams BD. Sarcomere assembly in C. elegans muscle The C. elegans Research Community, WormBook . 2006 Jan;16:1–16.
- Lee CF, Melkani GC, Bernstein SI. The UNC-45 myosin chaperone: from worms to flies to vertebrates. Int Rev Cell Mol Biol. 2014;313:103–144.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011 Feb;13(2):132–141.
- Dalla Costa AP, Clemente CF, Carvalho HF, et al. FAK mediates the activation of cardiac fibroblasts induced by mechanical stress through regulation of the mTOR complex. Cardiovasc Res. 2010 Jun 1 86(3):421–431.
- Rosner M, Siegel N, Valli A, et al. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids. 2010 Jan;38(1):223–228.
- Wang Q, Guo W, Hao B, et al. Mechanistic study of TRPM2-Ca(2+)-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy. 2016 Aug 2 12(8):1340–1354.
- Chin RM, Fu X, Pai MY, et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014 Jun 19 510(7505):397–401.
- Zheng H, Yuan C, Zhang H, et al. The tissue- and developmental stage-specific involvement of autophagy genes in aggrephagy. Autophagy. 2020 Apr;16(4):589–599.
- Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006 Aug;116(8):2161–2172.
- Chen Z, Nie SD, Qu ML, et al. The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 2018 Jul 10 9(7):771.
- Lant B, Derry WB. Methods for detection and analysis of apoptosis signaling in the C. elegans germline. Methods (San Diego. Calif). 2013 Jun 1;61(2):174–182.
- Tuloup-Minguez V, Hamaï A, Greffard A, et al. Autophagy modulates cell migration and β1 integrin membrane recycling. cell cycle (Georgetown, Tex). Cell Cycle (Georgetown, Tex.). 2013 Oct 15 12(20):3317–3328.
- Edick MJ, Tesfay L, Lamb LE, et al. Inhibition of integrin-mediated crosstalk with epidermal growth factor receptor/Erk or Src signaling pathways in autophagic prostate epithelial cells induces caspase-independent death. Mol Biol Cell. 2007 Jul;18(7):2481–2490.
- Ning L, Xu Z, Furuya N, et al. Perlecan inhibits autophagy to maintain muscle homeostasis in mouse soleus muscle. Matrix Biol. 2015 Oct;48:26–35.
- Ko SH, Apple EC, Liu, Z et al. Age-dependent autophagy induction after injury promotes axon regeneration by limiting NOTCH. Autophagy. 2020 Nov;16(11):2052–2068.
- Luo CL, Li BX, Li QQ, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011 Jun 16;184:54–63.
- Knöferle J, Koch JC, Ostendorf T, et al. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010 Mar 30;107 (13):6064–6069.
- Sarkar C, Zhao Z, Aungst S, et al. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10(12):2208–2222.
- Liu S, Sarkar C, Dinizo M, et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015 Jan 8 6(1):e1582.
- Wang W, Zuidema A, Te Molder L, et al. Hemidesmosomes modulate force generation via focal adhesions. J Cell Biol. 2020 Feb 3;219(2). 10.1083/jcb.201904137.
- Rabanal-Ruiz Y, Byron A, Wirth A, et al. mTORC1 activity is supported by spatial association with focal adhesions. J Cell Biol. 2021 May 3;220(5). 10.1083/jcb.202004010.
- Zhang H, Landmann F, Zahreddine H, et al. A tension-induced mechanotransduction pathway promotes epithelial morphogenesis. Nature. 2011 Mar 3 471(7336):99–103.
- Zhao Y, Zhang CF, Rossiter H, et al. Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J Invest Dermatol. 2013 Jun;133(6):1629–1637.
- Song X, Narzt MS, Nagelreiter IM, et al. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biol. 2017Apr;11:219–230.
- Wang Q, Ye Y, Liu W, et al. Dual effects of silibinin treatment on autophagy-regulated dermal apoptosis retardation and epidermal apoptosis up-regulation in UVB-induced skin inflammation. J Asian Nat Prod Res. 2012;14(7):688–699.
- Wang Q, Liu W, Zeng H, et al. p53-mediated autophagy adjustment is involved in the protection of silibinin against murine dermal inflammation and epidermal apoptosis induced by UVB irradiation. J Asian Nat Prod Res. 2013;15(2):117–129.
- Qiang L, Wu C, Ming M, et al. Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem. 2013 Jan 18 288(3):1603–1611.
- Chen YL, Tao J, Zhao PJ et al. Adiponectin receptor PAQR-2 signaling senses low temperature to promote C. elegans longevity by regulating autophagy. Nat Commun. 2019 Jun 13; 10(1):2602.
- Yang Y, Wang H, Wang S, et al. GSK3β signaling is involved in ultraviolet B-induced activation of autophagy in epidermal cells. Int J Oncol. 2012 Nov;41(5):1782–1788.
- Moriyama M, Moriyama H, Uda J, et al. BNIP3 upregulation via stimulation of ERK and JNK activity is required for the protection of keratinocytes from UVB-induced apoptosis. Cell Death Dis. 2017 Feb 2 8(2):e2576.
- Mou K, Liu W, Han D, et al. HMGB1/RAGE axis promotes autophagy and protects keratinocytes from ultraviolet radiation-induced cell death. J Dermatol Sci. 2017 Mar;85(3):162–169.
- Misovic M, Milenkovic D, Martinovic T, et al. Short-term exposure to UV-A, UV-B, and UV-C irradiation induces alteration in cytoskeleton and autophagy in human keratinocytes. Ultrastruct Pathol. 2013 Aug;37(4):241–248.
- Chen Y, Leboutet R, Largeau C, et al. Autophagy facilitates mitochondrial rebuilding after acute heat stress via a DRP-1-dependent process. J Cell Biol. 2021 Apr 5;220(4). 10.1083/jcb.201909139.
- Deruy E, Gosselin K, Vercamer C, et al. MnSOD upregulation induces autophagic programmed cell death in senescent keratinocytes. PloS one. 2010 Sep 14 5(9):e12712.
- Wu S, Pei Q, Ni W, et al. HSPA1A Protects Cells from Thermal Stress by Impeding ESCRT-0-Mediated Autophagic Flux in Epidermal Thermoresistance. J Invest Dermatol. 2021 Jan;141(1):48–58.e3.
- Hawk MA, Gorsuch CL, Fagan P, et al. RIPK1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells. Nat Cell Biol. 2018 Mar;20(3):272–284.
- Satyavarapu EM, Nath S, Mandal C. Desialylation of Atg5 by sialidase (Neu2) enhances autophagosome formation to induce Anchorage-dependent cell death in ovarian cancer cells. Cell Death Discov. 2021 Feb 1; 7(1):26.
- Uekita T, Fujii S, Miyazawa Y, et al. Suppression of autophagy by CUB domain-containing protein 1 signaling is essential for Anchorage-independent survival of lung cancer cells. Cancer Sci. 2013 Jul;104(7):865–870.
- Harada M, Hanada S, Toivola DM, et al. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008 Jun;47(6):2026–2035.
- Kongara S, Kravchuk O, Teplova I, et al. Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol Cancer Res. 2010 Jun;8(6):873–884.
- Gilbert S, Loranger A, Daigle N, et al. Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J Cell Biol. 2001 Aug 20 154(4):763–774.
- Caulin C, Ware CF, Magin TM, et al. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol. 2000 Apr 3 149(1):17–22.
- Wang C, Mendonsa GR, Symington JW, et al. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jul 3;109 (27):11008–11013.
- Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proceedings of the National Academy of Sciences of the United States of America. 2009 Dec 8;106 (49):20842–20846.
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008 Nov 13 456(7219):264–268.
- Martin PK, Marchiando A, Xu R, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol. 2018 Oct;3(10):1131–1141.
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974 May;77(1):71–94.
- Mello CC, Kramer JM, Stinchcomb D, et al. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991 Dec;10(12):3959–3970.
- Mariol MC, Walter L, Bellemin S, et al. A rapid protocol for integrating extrachromosomal arrays with high transmission rate into the C. elegans genome. J vis exp. 2013 Dec;9(82):e50773.
- Xu S, Chisholm AD. Methods for skin wounding and assays for wound responses in C. elegans. J vis exp. 2014 Dec 3;(94 51959).
- Meléndez A, Levine, B. Autophagy in C. elegans. 2009 24 Aug 2009; The C. elegans Research Community, WormBook, http://dx.doi.org/10.1895/wormbook.1.147.1