
ABSTRACT
Impaired mitophagy is a primary pathogenic event underlying diverse aging-associated diseases such as Alzheimer and Parkinson diseases and sarcopenia. Therefore, augmentation of mitophagy, the process by which defective mitochondria are removed, then replaced by new ones, is an emerging strategy for preventing the evolvement of multiple morbidities in the elderly population. Based on the scaffold of spermidine (Spd), a known mitophagy-promoting agent, we designed and tested a family of structurally related compounds. A prototypic member, 1,8-diaminooctane (VL-004), exceeds Spd in its ability to induce mitophagy and protect against oxidative stress. VL-004 activity is mediated by canonical aging genes and promotes lifespan and healthspan in C. elegans. Moreover, it enhances mitophagy and protects against oxidative injury in rodent and human cells. Initial structural characterization suggests simple rules for the design of compounds with improved bioactivity, opening the way for a new generation of agents with a potential to promote healthy aging.
Introduction
Morbidities associated with aging, such as Parkinson and Alzheimer diseases, congestive heart failure and sarcopenia, occur in non-dividing tissues where cells are exposed to continuous wear and tear throughout the organism’s lifespan. In particular, intracellular reactive oxygen species (ROS), generated as part of ongoing metabolism, can compromise mitochondrial function by damaging its DNA and membranes. Indeed, the prevalence of damaged mitochondria is greater in post-mitotic tissues than in mitotically active ones [Citation1]. To negate it, a high-fidelity surveillance system removes such defective mitochondria via macroautophagy/autophagy, a process known as mitophagy, which is complemented by the biogenesis of new mitochondria [Citation2,Citation3].
Mitophagy is evolutionarily conserved, from yeast to humans [Citation4]. In this process, a phagophore interacts with the mitochondrial portion destined to removal from the healthy mitochondrial network. The resulting autophagosome, containing the excluded mitochondrion, is fused with the lysosome, in which mitochondria are degraded [Citation4,Citation5]. Key proteins that drive the mitophagy process include PINK1 (PTEN induced kinase 1) and the E3 ubiquitin ligase PRKN, as well as the mitophagy receptors BNIP3 and BNIP3L [Citation4]. These proteins may collaborate along the mitophagy pathway; e.g., in the nematode Caenorhabditis elegans (C. elegans), the autophagy receptor DCT-1 (BNIP3/BNIP3L ortholog) is ubiquitinated in a PINK-1-PDR-1 (orthologs of PINK1 and PRKN, respectively)-dependent manner, to facilitate mitophagy [Citation2].
As mitophagy declines with age [Citation6], malfunctioning mitochondria are accumulated with the ensuing energy-supply failure. It is hence suggested that impaired mitophagy is an underlying mechanism behind multiple aging-associated diseases, including Alzheimer [Citation7] and Parkinson diseases [Citation8,Citation9], and sarcopenia [Citation10,Citation11]. Thus, the identification of effective means to promote mitophagy might have far-reaching consequences to the individual’s wellness and to public health.
The polyamine spermidine (Spd) demonstrated mitophagy enhancing properties [Citation12–16]. Moreover, Spd was shown to extend the health and lifespan in several model organisms, including yeast, C. elegans, flies, and mice [Citation13,Citation17,Citation18]. However, Spd catabolism can produce toxic metabolites like acrolein [Citation19,Citation20] and, therefore, its use may be limited. This study explores the utility of polyamine-mimetic compounds in mitophagy induction, healthspan, lifespan, and degenerative-disease models. We set out three criteria for identifying successful mitophagy activating compounds (MACs): (1) Robust mitophagy induction; (2) Protection against oxidative injury; (3) Healthspan extension in wild-type and age-related disease models in worms. We discovered that a defined set of synthetic linear diamines could induce mitophagy in C. elegans and protect the whole organism against oxidative damage, in a superior manner, compared to Spd. In particular, we show that a prototypic MAC, 1,8 diaminoctane (hereafter referred to as VL-004), fulfills all the above criteria in C. elegans and protects rat and human cells against oxidative injury and senescence. We further identified key transcription factors and mitophagy-related proteins required for VL-004 function, thus providing molecular insights regarding its mechanism of action. Finally, we leverage our expertise to generate novel MACs that demonstrate improved potency. Such compounds can assist in sorting out the role of mitophagy in the wellbeing of the aging organism and provide a structural template that might enable the development of effective therapy for diseases affecting the global elderly population.
Results
VL-004 is a potent mitophagy inducer in C. elegans
We used IR1631 C. elegans [Citation2] to screen for mitophagy inducers. This strain expresses the mito-Rosella sensor in body wall muscles. The sensor comprises a mitochondria targeting sequence (TOMM-20), followed by pH-insensitive red and pH-sensitive green fluorescence proteins (). At neutral pH, the red and green fluorescence are comparable, while the acidic environment of the autolysosome lowers the green fluorescence. Thus, decreased green to red fluorescence ratio indicates an increased mitophagy.
Figure 1. VL-004 induces robust mitophagy in C. elegans. (A) Mito-Rosella sensor function. Under neutral pH the mito-Rosella green and red fluorescence intensities are comparable, while the acidic environment of the autolysosome causes a green fluorescence suppression. Therefore, a decrease in the green to red fluorescence ratio indicates mitophagy. (B) The effect of polyamine compounds on mitophagy in the body wall muscles (BWM) of C. elegans. Transgenic mito-Rosella worms were treated for 16 h with 1 mM of Spd, 1-methyl-spermidine (MetSpd), VL-002, VL-003, VL-004, and VL-005, whose structure is shown. N ≥ 10 worms/per treatment, n ≥ 3. (C) Colocalization of mitochondria and lysosome. Arrowheads indicate representative colocalization. The inset represents a nine-fold enlargement – scale bar: 20 μm. (D) Quantification of colocalization. N = 40 worms/per treatment, n ≥ 3. (E) Mitochondrial to nuclear DNA ratio (mtDNA:nDNA). n = 8. In (B) and (D and E) asterisks indicate significance compared to vehicle. *p < 0.05, **p < 0.01, ***p < 0.001, NS = non-significant.
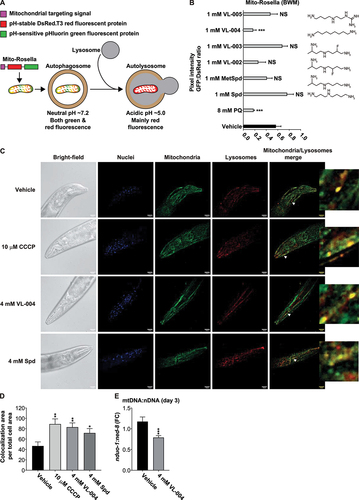
As a primary screen, we tested the mitophagy activity of five polyamine-related compounds at 1 mM: Spd, 1-methyl-spermidine (MetSpd), VL-004, and 3 new Spd-derivatives (VL-002, VL-003, and VL-005) ( and Figure S1). Also, we tested the effect of the potent superoxide inducer paraquat (PQ [Citation21],). The treatment was given for 24 h, from the fourth larval stage (L4) to the adult worm. Only VL-004 induced significant mitophagy among those compounds, comparable to the one induced by PQ ( and Figure S1).
We further examined the mitophagy-inducing properties of VL-004, by exploring its activity in neurons and testing its function using two alternative assays. First, we generated transgenic worms expressing a pan-neuronal mito-Rosella-sensor (EVG1414 strain), targeted to mitochondria via the mitochondrial chaperone HSP-6 [Citation22]. VL-004 and Spd induced significant mitophagy after 48 h incubation (L1 to young adults, 4 mM) compared to vehicle controls (Figure S2A-B), indicating that VL-004 is a potent mitophagy inducer in both muscles and neurons.
We measured the colocalization of mitochondria and lysosomes in the head body wall muscles using organelle-specific dyes (Cytopainter, ab139487). Notably, in this experiment, we measured mitophagy in day 6 worms (six days post L1), as described in [Citation23], treated with either 4 mM VL-004, 4 mM Spd, or vehicle control, from the L1 stage. As a positive control, we used the mitochondrial uncoupler CCCP [Citation24] (given on day 5, for 16 h). Both Spd and VL-004, at 4 mM, induced significant mitophagy in wild-type worms (N2 laboratory strain), as did CCCP (). Finally, we measured the ratio of mitochondrial DNA (mtDNA) per nuclear genome (nDNA). We performed these experiments just prior to the appearance of eggs in post-L4 hermaphrodites and used the nduo-1 and ned-8 loci to quantify mtDNA:nDNA. VL-004 decreased the mtDNA:nDNA ratio (), as expected from a mitophagy-inducing agent [Citation25]. Together, these results point out that VL-004 is a potent mitophagy inducer in C. elegans and thus fulfills the first criterion we set for an efficient MAC.
VL-004 protects from oxidative injury in a dose-dependent manner
Mitophagy protects against oxidative stress [Citation26,Citation27]. Therefore, we hypothesized that VL-004 should provide superior protection against oxidative injury compared to the compounds tested above. We treated N2 worms (L4 stage) with 1 mM of the above 6 compounds for 24 h and measured survival after 3, and 6 h in 200 mM PQ, a concentration that induces acute oxidative stress [Citation28]. Only VL-004 provided significant protection against PQ, agreeing with its mitophagic potency ().
Figure 2. VL-004 protects against oxidative injury. Stacked bar graphs showing worms’ Survival in 200 mM PQ after 3 h and 6 h. (A) Protection against oxidative injury by VL-004 vs. Spd, MetSpd, VL-002, VL-003, and VL-005, at 1 mM. Dose-response of VL-004 (B), Spd (C), and MetSpd (D). (E) Mediators of the mitophagy pathway. The mitophagy genes dct-1 (F) and pink-1 (G) are essential for VL-004 protection. (H) Upper panel: PDR-1 is not essential for VL-004 activity in oxidative stress – lower panel: Schematic presentation of pdr-1 alternative transcripts (in gray). The relative positions of the gk448, tm598, tm396 deletion mutations are indicated by dark rectangular. For (A-D) and (F-G) graphs, n = 6. For (H), n = 9. Total number of worms in each experiment: (A) ≥ 134; (B-D) ≥ 112; (F and G) ≥ 133; (H ≥ 189). Asterisks indicate significance compared with vehicle/PQ treated worms. (I) VL-004 effect on autophagy/mitophagy gene expression. N2 worms were treated with 4 mM VL-004 for 48 h. Gene expression was measured by qPCR. n ≥ 4. *p < 0.05, **p < 0.01, ****p < 0.0001, NS = non-significant.
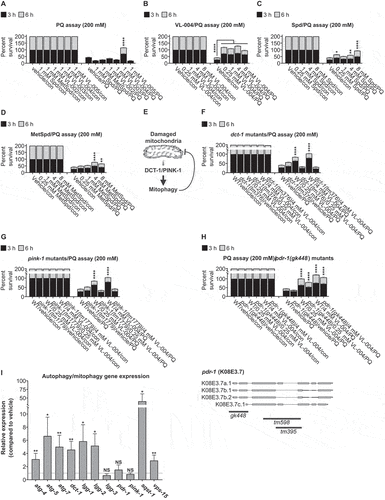
We next explored whether VL-004, Spd, or MetSpd protect against oxidative injury in a dose-dependent manner at concentrations that do not affect control worms’ viability (). VL-004 provided significant protection against PQ at all concentrations, with decreased activity at 8 mM () and was superior to Spd and MetSpd (). Notably, although Spd is inferior to VL-004, it provides significant protection in defending against PQ-induced oxidative damage (). This result agrees with a previous study suggesting that Spd induces mitophagy [Citation15]. However, it is not consistent with our primary screen results (). To address this, we tested the mitophagy activity of Spd at several concentrations, using a larger animal sample size (≥39 per Spd concentration), in a new mitophagy strain we generated (EVG1421); the EVG1421 strain expresses a body wall muscle mito-Rosella sensor that targets the mitochondria via the Tom70 MTS (a bona fide outer mitochondria marker [Citation29]). Spd induced significant mitophagy at 1,4, and 8 mM (after 16 h, L4 stage to the adult worm, Figure S2C, and S3), confirming previous findings and our colocalization-mitophagy experiments ().
The anti-diabetic agent metformin affects C. elegans lifespan by changing both bacterial and worms’ metabolism [Citation30]. We, therefore, tested whether VL-004 activity depends on bacteria viability by repeating the PQ assay using heat-killed vs. live bacteria. As shown in Figure S2D, bacterial viability did not affect either VL-004 potency or the sensitivity of vehicle-treated worms to PQ. Thus, this set of experiments shows that VL-004 function is independent of bacteria metabolism.
Collectively, these results demonstrate that VL-004 protects against oxidative injury in a dose-dependent manner and fulfills the second criterion we set.
The mitophagy pathway genes dct-1 and pink-1 are required for VL-004 activity
So far, we have shown that VL-004 induces mitophagy in C. elegans and protects against oxidative injury (). To determine whether it relates to typical mitophagy, we explored the involvement of the mitophagy proteins DCT-1, and PINK-1 (). We performed PQ-survival experiments with dct-1(tm376) and pink-1(tm1779) mutants, using N2 worms as controls. As expected, VL-004 at 0.25 and 4 mM concentrations protected against PQ toxicity in wild-type worms (). However, it failed to protect the dct-1 and pink-1 mutants (), indicating that VL-004 protection operates in a mitophagy-dependent manner. To further investigate the link between VL-004 activity and canonical mitophagy, we asked whether mitophagy-induction by VL-004 is PINK-1-dependent. To explore this, we introduced the EVG1421 muscle mito-Rosella sensor into pink-1(tm1779) mutants and measured mitophagy induction after 6 h incubation with 4 mM VL-004 (in young adults). In addition, we measured mitophagy induction in EVG1421 worms as a control. Consistent with the PQ experiment data, VL-004 did not induce mitophagy in pink-1(tm1779) mutants (Figure S4A-B). Furthermore, it did not induce mitophagy in dct-1(tm376) mutants bearing the same mito-Rosella sensor (Figure S4C, same experimental conditions as S4A-B), further supporting our conclusion that VL-004 activity is mitophagy-dependent.
PDR-1 is not required for VL-004 activity
A previous study suggested that PDR-1 (the C. elegans PRKN homolog) cooperates with PINK-1 and DCT-1 to regulate mitophagy [Citation2]. Therefore, we asked whether PDR-1 is necessary for VL-004 activity in oxidative stress. To test this, we performed PQ-survival experiments with animals bearing the pdr-1(gk448) deletion allele (), using N2 worms as controls. Unexpectedly, VL-004 significantly protected the pdr-1 mutants against PQ (at both 0.25 mM and 4 mM concentrations), suggesting that PDR-1 is not required for VL-004 activity in oxidative stress. To further explore this, we repeated the experiment with two other pdr-1 mutants, bearing the pdr-1(tm395) and pdr-1(tm598) deletion alleles (). Again, VL-004 significantly increased the resistance of these mutants to PQ (Figure S4D, E).
That previous study suggested that PDR-1 is essential for mitophagy induction by a toxic mitochondrial uncoupler and paraquat [Citation2]. Therefore, we hypothesized that PDR-1 is required for mitophagy induced by mitochondrial toxins, but not VL-004. To directly examine this hypothesis, we generated transgenic pdr-1(gk448) expressing the muscle mito-Rosella sensor and tested mitophagy induction by 5 μM of the mitochondrial uncoupler FCCP and 4 mM VL-004 after 5 h and 6 h incubation, respectively. In addition, we tested the mitophagy response of wild-type worms carrying the same sensor, i.e., EVG1421 worms. Both FCCP and VL-004 induced mitophagy in wild-type worms (Figure S4F). Strikingly, FCCP did not induce mitophagy in pdr-1 mutants. However, VL-004 induced a robust mitophagy. These results show that although PDR-1 is required for mitophagy induction by mitochondrial toxins, it is dispensable for VL-004 induced mitophagy, suggesting that the latter operates via PDR-1 independent mitophagy-pathway.
VL-004 induces the expression of autophagy/mitophagy genes
To further characterize VL-004ʹs activity, we asked whether it induces the expression of autophagy/mitophagy genes. Indeed, VL-004 significantly induced the expression of dct-1 (). However, it did not affect the expression of pink-1 and pdr-1. In addition, VL-004 induced the expression of key autophagy genes, including (a) the atg genes atg-4, atg-5, and atg-7 [Citation31]; (b) the LC3- and GABARAP-related genes lgg-1 and lgg-2, respectively [Citation32]; (c) the autophagy receptor sqst-1 [Citation33]; (d) the class III phosphatidylinositol 3-kinase gene vps-15 [Citation34] (). Together, these results imply that VL-004 enhances the expression of key mitophagy/autophagy genes.
Autophagy and mitophagy share some similar autophagosome/autolysosome proteins [Citation35,Citation36]. Our results reveal that VL-004 affects the expression of autophagy genes (). Therefore, we asked whether VL-004 also induces autophagy. We measured autophagosome formation in transgenic worms expressing intestinal LGG-1 tagged with an N-terminal mCherry [Citation37]. Four hours of starvation of young adult worms (a bona fide autophagy stimulus) significantly increased the formation of mCherry-LGG-1 puncta, indicating LGG-1 incorporation into autophagosome structures (). Moreover, 4 mM VL-004 treatment (from the L1 to the young adult stage) induced significant autophagy compared to the vehicle controls. However, lesser than starvation.
Figure 3. VL-004 induces bulk autophagy. (A) Transgenic mCherry::LGG-1 worms exposed to starvation, 4 mM VL-004, or vehicle. The inset represents a 40-fold enlargement. Scale bar: 50 μm. (B) Quantification of LGG-1 puncta. ****p < 0.0001. N ≥ 32 worms/per treatment, n = 3. (C-G) Stacked bar graphs displaying the survival of the atg-4.2(ola316), atg-16.2(ok3224), atg-18(gk378), and unc-51(e1189) autophagy mutants in 200 mM PQ after 3 h and 6 h. For (C-G) graphs, n = 6. Total number of worms in each experiment: (C) ≥ 138; (D) ≥ 144; (E and F) ≥ 145. Asterisks indicate significance compared with vehicle/PQ treated worms. ****p < 0.0001.
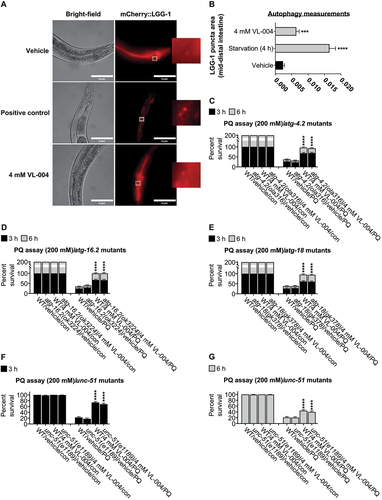
To further explore the link between VL-004 and autophagy, we tested whether the autophagy genes atg-4.2, atg-16.2, atg-18, and unc-51 are required for VL-004 activity in oxidative stress. VL-004 significantly protected atg-4.2(ola316), atg-16.2(ok3224), atg-18(gk378), and unc-51(e1189) mutants against PQ (), suggesting that these genes are not essential for its protective function.
Canonical aging pathway genes are required for VL-004 activity
The mitophagy process in C. elegans is regulated by aging pathway genes, including the sole insulin/IGF-1 family member daf-2, its downstream FOXO transcription factor target daf-16, and the NFE2L2/NRF2 C. elegans ortholog skn-1 [Citation2]. Therefore, we examined whether the protective effect of VL-004 against PQ depends on the following aging-pathway genes: daf-16, daf-2, eat-2, hif-1, hlh-30, sqst-1, pha-4, and skn-1.
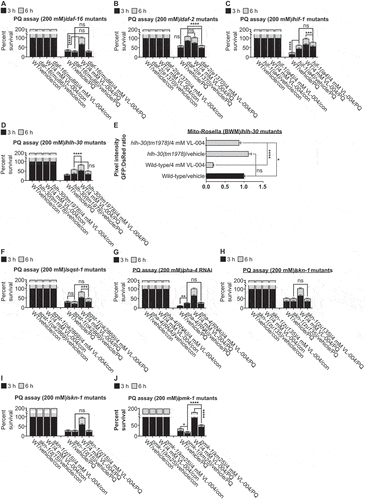
VL-004 was unable to protect worms bearing the daf-16(mu86) null allele [Citation38] (), suggesting that DAF-16 is vital for VL-004 function. DAF-16 regulates dct-1 mitophagy activity [Citation3]. Therefore, this result further illustrates that VL-004 acts through the mitophagy pathway.
Figure 4. Role of canonical aging genes in VL-004 activity. (A-D) and (F-J) Stacked bar graphs showing worms’ Survival in 200 mM PQ after 3 h and 6 h, n = 6. Total number of worms was as followed: (A) ≥ 127; (B) ≥ 219; (C) ≥ 140; (D and F) ≥ 141; hlh-30 and sqst-1 mutants’ experiments were performed together, therefore shared same wild-type controls; (G) ≥ 124; (H) ≥ 142; (I) ≥ 155; (J) ≥175. (E) Bar graph quantifying mitophagy. Data represent an average of n = 30 animals/per treatment, N = 3. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = non-significant.
daf-2 mutants show pronounced mitophagy induction under normal growth conditions [Citation2], consistent with elevated DAF-16 activity in these worms [Citation39]. As expected from previous studies [Citation40], daf-2(e1370) mutants showed significant resistance to PQ (p < 0.0001) (). Intriguingly, the survival rate of daf-2(e1370) mutants treated with the vehicle was similar to N2 worms treated with 4 mM VL-004. Moreover, VL-004 significantly decreased daf-2 mutants’ survival in PQ, to the level of vehicle-treated N2 worms in PQ.
A possible explanation for this observation is that mitophagy is chronically activated in daf-2 mutants, and VL-004 enhances this process to a toxic level. To examine this, we introduced the muscle mito-Rosella sensor into daf-2(e1370) mutants and measured mitophagy in vehicle and VL-004 treated worms (L1 to young adults, 4 mM). Vehicle-treated daf-2 mutants displayed enhanced mitophagy (Figure S5A, B), significantly higher than wild-type worms treated with VL-004. As hypothesized, VL-004 significantly increased mitophagy in daf-2 mutants, probably beyond the “healthy range.”
Is daf-2 mutants’ resistance to oxidative stress mitophagy-dependent? To answer this, we generated daf-2 double mutants bearing the dct-1(tm376) and pink-1(tm1779) deletion mutations and tested their resistance to PQ. In addition, we examined whether VL-004 affects the PQ tolerance in these mutants. Impairment of either DCT-1 or PINK-1 activity significantly decreased the resistance of daf-2 mutants to PQ (Figure S5C), suggesting that healthy mitophagy is required for optimal defense against oxidative stress. Moreover, VL-004 did not enhance protection against PQ in daf-2(e1370); dct-1(tm376) and pink-1(tm1779); daf-2(e1370) mutants, further supporting the conclusion that VL-004 activity is mediated through these mitophagy genes.
Dietary restriction (DR) extends the lifespan of most organisms, including C. elegans [Citation41]. Worms bearing the eat-2(ad1116) mutation have defective pharyngeal-pumping-rate and, as a result, consume less food and show DR phenotype, including extended lifespan [Citation42]. Moreover, eat-2 mutants are better protected against oxidative stress due to higher SOD (superoxide dismutase) and CTL (catalase) activity [Citation43]. VL-004 significantly increased the survival of eat-2(ad1116) mutants in PQ (Figure S5D), suggesting that it acts parallel to the DR pathway.
Notably, the body length of DR wild-type worms is shorter than that of well-fed animals [Citation44]. On the other hand, VL-004 significantly increased it (worms were treated with VL-004 from the L1 stage for 11 days, i.e., day 11 – Figure S5E), further supporting the conclusion that the DR pathway does not drive VL-004 activity.
HIF-1 (Hypoxia Inducible Factor 1) is a regulator of mitophagy [Citation45] and lifespan in C. elegans [Citation46]. Animals bearing the hif-1(ia4) deletion mutation [Citation47] showed significant PQ resistance compared to wild-type animals (). However, VL-004 did not increase their survival in PQ, indicating that HIF-1 is required for the VL-004 function.
The increased resistance of hif-1 mutants to PQ prompted us to ask whether mitophagy is increased in these worms. To examine this, we introduced the muscle mito-Rosella sensor into hif-1(ia4) mutants and measured mitophagy in vehicle and VL-004 treated worms (4 mM, 6 h). Indeed, the mitophagy magnitude was similar between VL-004-treated wild-type worms and vehicle-treated hif-1 mutants (Figure S4A, G). Intriguingly, VL-004 significantly decreased mitophagy in hif-1 mutants (p < 0.01). This result is unexpected since the addition of VL-004 to hif-1 mutants did not affect their PQ resistance ().
HLH-30, the C. elegans ortholog of mammalian TFEB (transcription factor EB), regulates autophagy genes and longevity [Citation48]. Similar to hif-1 mutants, vehicle-treated worms bearing the hlh-30(tm1978) deletion allele were more resistant to PQ than wild-type worms (). However, VL-004 did not increase their PQ resistance further, suggesting that HLH-30 is required for VL-004 activity. To further explore this, we introduced the muscle mito-Rosella sensor into hlh-30(tm1978) mutants and measured mitophagy after 6 h incubation with 4 mM VL-004. VL-004 induced mitophagy in hlh-30 mutants (). However, the magnitude was significantly lesser than wild-type controls (p < 0.0001), suggesting that HLH-30 is required for efficient VL-004-induced mitophagy.
SQST-1 extends lifespan in an autophagy-dependent manner [Citation33]. VL-004 treatment did not increase the survival of animals bearing the sqst-1(ok2869) deletion allele (), indicating that SQST-1 is required for VL-004 activity. The fact that VL-004 induces sqst-1 expression, together with other autophagy genes (), and that SQST-1 is mandatory for VL-004 activity, implies that it acts via the autophagy pathway.
The PHA-4/FOXA transcription factor mediates the autophagy response in DR worms and promotes longevity [Citation49]. Treatment with RNAi against the coding sequence of pha-4 increased the resistance of wild-type worms to PQ (). However, pha-4 RNAi prevented the beneficial effect of VL-004, indicating that PHA-4 is needed for VL-004 activity. To further validate PHA-4 requirement for VL-004 function, we performed RNAi against the 3’ UTR region of pha-4. The results recapitulated those of RNAi targeting the coding region of pha-4 (Figure S5F), indicating that PHA-4 is vital for VL-004 activity.
VL-004 did not affect the survival of animals bearing the skn-1(zu135) loss-of-function allele [Citation50] (), signifying that SKN-1 is also crucial for VL-004 activity. Like DAF-16, SKN-1 regulates dct-1 activity in mitophagy [Citation3], further supporting our conclusion that DCT-1, hence mitophagy, is essential for VL-004 function. We repeated the PQ assay with animals bearing another skn-1 loss of function mutation, skn-1(zj15) [Citation51], and obtained similar results (), supporting a key role for SKN-1 in VL-004 activity.
The mitogen-activated protein kinase PMK-1 regulates SKN-1 activity in oxidative stress [Citation52]. Moreover, its neuroprotection activity is mediated through mitophagy [Citation53]. Therefore, we hypothesized that PMK-1 is required for VL-004 activity. Although VL-004 increased the resistance of animals bearing the pmk-1(km25) deletion allele (), the magnitude of the VL-004 effect was significantly lower than the VL-004-treated wild types. Notably, pmk-1(km25) mutants are more sensitive to PQ than wild-type worms (); however, the difference in resistance is much smaller than the difference between VL-004-treated wild-type vs. pmk-1 mutants. Thus, PMK-1 is important to VL-004 activity to some extent.
Collectively, the above set of experiments shows that VL-004 acts through canonical aging genes.
VL-004 extends C. elegans lifespan and healthspan
We explored the effect of VL-004 on C. elegans lifespan at two concentrations that provided significant protection against PQ, i.e., 0.25 mM and 4 mM (). For comparison, we used Spd at these concentrations. Both concentrations of VL-004 significantly extended the life of the worms, while Spd had no effect (), reconfirming VL-004 superiority vis a vis Spd.
Figure 5. VL-004 promotes longevity and healthspan. (A) Survival curves of worms in the presence of VL-004, Spd, or vehicle. The summary of lifespan data is presented in Table S3.(B) Rhodamin-phalloidin staining of body-wall muscles of vehicle and VL-004 treated worms (in day 3 and day 11 post L1). Scale bar: 100 μm. (C) Quantification of rhodamin-phalloidin staining (head region, tip on the nose to the posterior bulb of the pharynx). N ≥ 35/per treatment, normalized to worm size by dividing fluorescence by head width (under the posterior bulb of the pharynx). n = 3. Speed (D) and thrashing (E) measurements. The speed/thrashing of worms was measured in the absence of bacteria. Speed (n = 6, N ≥ 48 worms/per treatment). Thrashing (n = 4, N ≥ 24/per treatment). (F) Food intake. (n ≥ 6, N ≥ 24 worms/per treatment). (G) Oxygen consumption rate (OCR) of wild-type worms treated with either vehicle or 4 mM VL-004 at day 3 and 11 (post L1). (n = 6, N ≥ 120 worms/per treatment). (H) OCR Quantification. Basal, maximal (FCCP), and non-mitochondrial (azide) are presented. (I) ATP levels in wild-type animals treated for 3 and 11 days as in (G). N = 3 (day 11) or N = 6 (day 3). (J) TMRE staining of 11 days SJ4103 worms grown with 4 mM VL-004 or vehicle. TMRE staining was normalized to mitochondrial GFP intensity. n = 3, N ≥ 48 worms/per treatment. (K) Mitochondrial to nuclear DNA ratio (mtDNA:nDNA). n = 9. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS = non-significant.
To test whether this increase in lifespan is mitophagy dependent, we investigated the effect of 4 mM VL-004 on dct-1(tm376) mutants’ lifespan. VL-004 did not extend the lifespan of dct-1 mutants (Figure S6A), suggesting that its function in lifespan extension is mitophagy-dependent.
Mitochondrial superoxide signal extends C. elegans lifespan [Citation54]. Therefore, we asked whether VL-004 lengthen lifespan by increasing mitochondrial superoxide. To that end, we exposed wild-type worms to 4 mM VL-004 for 48 h (L1 to young adults) and measured mitochondrial superoxide level using MitoSOX dye [Citation54,Citation55]. In addition, we included a positive control (10 μM FCCP). Importantly, we co-stained the worms with MitoTracker Green dye [Citation56,Citation57] to normalize the MitoSOX signals vs. mitochondrial mass. As expected, FCCP increased mitochondrial superoxide production (Figure S6B-C), while vehicle and VL-004 treated worms had similar superoxide levels, suggesting that VL-004 effect on lifespan is not mediated by mitochondrial superoxide production.
Outstandingly, worms treated with VL-004 were faster and looked healthier than control worms throughout the lifespan experiment. To quantify it, we measured the effect of VL-004 on worms’ muscle mass, speed on solid medium, swimming (thrashing), and food intake (pharyngeal pumping). We used phalloidin, a high-affinity F-actin probe, to estimate muscle mass in worms treated with 4 mM VL-004. We performed these measurements in young (three days post L1, day 3) and older worms (eleven days post L1, day 11). 11-days vehicle-treated worms displayed a significant decrease in muscle mass, but it did not occur in VL-004 treated worms (). Correspondingly, the speed of 11-days VL-004 treated worms was similar to that of young ones (), while the speed of 11-days vehicle worms was significantly decreased. The thrashing and food intake activities of the worms treated with VL-004 and vehicle were similar on day 3 (, respectively). In contrast, on day 11, these activities were significantly higher in VL-004 treated compared with day 11 controls.
To examine whether these VL-004 effects on muscle mass and activity are mitophagy dependent, we repeated the above experiments using 11 days old dct-1(tm376) mutants. VL-004 did not preserve muscle mass and activity in dct-1 mutants (Figure S7A-E), suggesting that its muscle-preservation capacity is mitophagy dependent.
VL-004 retains muscle strength in older worms, alongside of being a potent mitophagy inducer. Since muscle functionality depends on optimal mitochondrial activity, we hypothesized that VL-004 maintains aerobic respiration in aged worms. Therefore, we measured oxygen consumption rate (OCR) and ATP level in worms treated with vehicle or 4 mM VL-004 on days 3 and 11.
In the OCR experiments, basal respiration is first measured in resting worms. Then maximal OCR is measured upon the addition of the uncoupler FCCP. Finally, non-mitochondrial OCR is recorded after adding azide, which blocks the activities of complexes IV and V [Citation58]. On day 3, VL-004 slightly, though significantly, decreased the basal and maximal OCRs (). On the other hand, on day 11 the maximal OCR was considerably higher in VL-004 treated worms, whereas the basal OCR was similar to the vehicle controls.
To complement it, we performed ATP measurements on day 3 and day 11. Intriguingly, VL-004 increased the ATP/per protein content in day 3 worms (). A possible explanation is that mitophagy activation by VL-004 creates a selective pressure that preserves the most efficient mitochondria so that ATP level per protein content increases. If true, these mitochondria might produce more ATP without increasing the basal OCR ().
On day 11, ATP levels per protein content were similar between VL-004 and vehicle-treated worms. It is important to note that the basal OCR was similar between treatments on day 11 (). Only when we pushed the worm via FCCP to maximal OCR did we observe the difference in mitochondrial potential reflecting the activity of VL-004. Therefore, the similar ATP to protein content we observed in day 11-resting worms is consistent with OCR measurements.
To explore whether the effects of VL-004 on OCR and ATP on day 11 and day 3 are mitophagy dependent, we repeated these experiments with dct-1(tm376) mutants. VL-004 did not increase ATP and OCR levels in dct-1 mutants (Figure S7F-H), suggesting that these VL-004 functions are mitophagy dependent.
To further explore the effect of VL-004 on mitochondrial activity on day 11, we measured mitochondrial membrane potential using the TMRE dye [Citation59]. VL-004 increased the TMRE signals in day 11 worms compared with controls (, and Figure S8A), suggesting these mitochondria are polarized and healthy. Furthermore, VL-004 did not increase the mitochondrial membrane potential of dct-1(tm376) worms (Figure S7I), suggesting that this VL-004 function is mitophagy dependent.
We also observed an increase in the mtDNA:nDNA ratio (), indicating that VL-004-induced-mitophagy is accompanied by mitochondrial biogenesis. These results are consistent with VL-004 health-promoting activity as decreased mitochondrial membrane potential typifies damaged mitochondria and aging cells [Citation60,Citation61]. At the same time, increased mtDNA copy number is associated with cellular health and protection against oxidative stress [Citation62]. Notably, a previous study shows that pink-1 RNAi decreases TMRE staining in C. elegans worms [Citation59]. In accordance with this observation, we have found that VL-004 did not increase the mtDNA:nDNA of 11-day pink-1(tm1779) mutants (Figure S7J), further supporting the importance of mitophagy for VL-004 function. Together, our findings demonstrate that VL-004 extends the lifespan and healthspan of worms in a mitophagy-dependent manner. Therefore, it fulfills the third criterion we set for a powerful MAC.
VL-004 is not a mitochondrial uncoupler
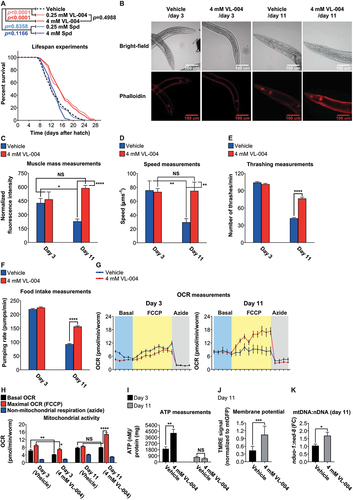
Sub-toxic levels of mitochondrial uncouplers were shown to extend the lifespan in several model organisms, including yeast [Citation63], C. elegans [Citation64], and mice [Citation65]. Our data suggest that VL-004 is not a mitochondrial uncoupler. First, day 11 worms show an increased TMRE signal (). Second, day 3 and day 11 VL-004-treated worms show increased or no change in ATP levels, respectively (). However, we examine the effect of VL-004 in these experiments after several days of treatment. To determine the immediate effect of VL-004 on mitochondrial activity, we measured the OCR’s of worms upon treatment with the following stimulus train: VL-004 (at 0.25 mM, 1 mM, and 4 mM), followed by 10 μM FCCP (a positive control presenting a potent mitochondrial uncoupler), and 40 mM azide (to terminate mitochondrial activity). In contrast to FCCP, VL-004 did not affect the worms’ OCR at any of the tested concentrations (), suggesting that it is not a mitochondrial uncoupler. Interestingly, 0.25 mM slightly, however significantly, increased the OCR in the presence of FCCP.
Figure 6. VL-004 does not change worms’ oxygen consumption rate. OCR (oxygen consumption rate) of wild-type worms (day 3 post L1) treated with the following stimulus train: VL-004 (at 0.25 mM, 1 mM, and 4 mM), 10 μM FCCP, and 40 mM azide. The experiments were performed in the absence of bacteria (A) or presence of heat-killed bacteria (C). n = 3, N ≥ 120 worms/per treatment). (B and D) OCR Quantification. Basal, vehicle/VL-004 treatments, maximal (FCCP), and non-mitochondrial (azide) are presented. Asterisks indicate significance compared with the vehicle treatment (maximal OCR comparison). *p < 0.05.
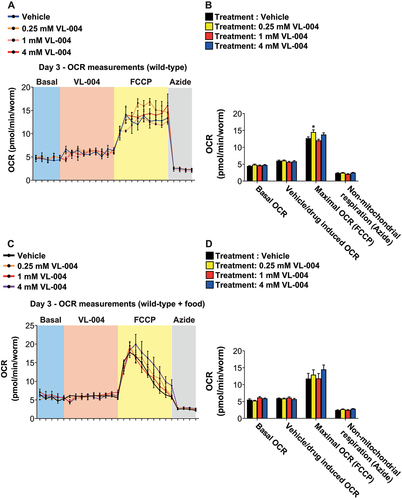
The above Seahorse experiments were performed in the absence of food. Because the lack of food inhibits pharyngeal pumping, and food stimulates it [Citation66], a genuine question is whether VL-004 is accessible to the worm during the experiment. To address it, we repeated the experiments in the presence of heat-killed bacteria. Notably, we calibrated the amount of food to ensure consistent pharyngeal pumping for 6 h; a significantly longer duration than the above experiment. Furthermore, VL-004 did not affect the worm’s OCR also in the presence of food (). Together, this set of experiments further supports our conclusion that VL-004 is not a mitochondrial uncoupler.
Is there a “time window” for VL-004 activity?
So far, we have described experiments in which the VL-004 treatment was started before adulthood (in either the L1 or L4 stage). To explore whether post-adulthood VL-004 treatment is also efficient, we exposed adult worms (6 days post L1. i.e., day 6) to 0.25 mM VL-004 for 48 h and examined their survival in PQ. We also included controls in which we treated L1 worms with VL-004 for 48 h. i.e., until the worms reached adulthood (i.e., day 3). The survival of day 3 and day 6 VL-004 treated worms was identical (), suggesting that the protective activity of VL-004 is independent of a particular developmental stage.
Figure 7. VL-004 kinetics and activity in worm models of neurodegenerative diseases. (A) Stacked bar graph comparing the survival of 1- and 6-days worms (in 200 mM PQ, after 3 h and 6 h) treated with 0.25 mM VL-004 for 48 h. (B-D) Bar graphs showing worms’ survival in 200 mM PQ. The worms (3 days post L1) were treated with 0.25 mM VL-004 for a fixed period of time, and their survival was measured after 3 hours in PQ. 3 and 6 h (B), 9 and 12 h (C), and 16 and 24 h (D). n = 6. Total number of worms: (A) ≥ 133; (B) ≥ 130; (C) ≥ 142; (D) ≥ 138. (E) Kaplan–Meier curves comparing the paralysis rate of AM141 worms grown on 0.25 mM or 4 mM of Spd or VL-004, n = 3, the total number of worms N ≥ 174, Day 0 = L4 stage of development. (F) Paralysis rates of AM141 worms bearing the dct-1(tm376) and pink-1(tm1779) mutations, compared to AM141 controls. n = 3, total number of worms N ≥ 92. (G) Thrashing measurements, AM725 transgenic worms. n ≥ 6. N ≥ 17 worms/per treatment. Asterisks represent significance compared to vehicle control within the comparison group, i.e., wild-type of mutated SOD-1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS = non-significant.
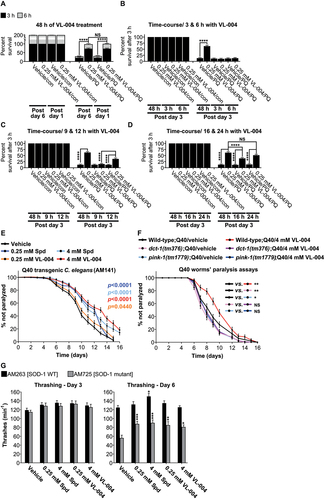
To further characterize the kinetics of the VL-004 effect, we performed time-course experiments. We pretreated adult worms (day 3) with 0.25 mM VL-004 for increasing periods of time (3, 6, 9, 12, 16, and 24 h) and then tested their survival in PQ. The 3, 6, and 9 h VL-004 treatment did not protect against PQ. However, 12, 16, and 24 h pretreatment provide significant progressive protection, compared with vehicle controls (). Notably, 6 h treatment with either 4 mM VL-004, or Spd induced robust mitophagy in EVG1421 worms (Figure S8B, C), suggesting that mitophagy enhancement by VL-004 is essential but not sufficient for protection against oxidative injury.
This set of experiments suggests that the processes mediating the activity of VL-004 advance gradually, over hours, and reach maximal activity after around 24 h.
VL-004 ameliorates protein toxicity in several C. elegans models of neurodegenerative diseases
Impaired mitophagy appears to play a crucial role in the onset and/or progression of several neurodegenerative diseases, including Huntington disease and amyotrophic lateral sclerosis [Citation67]. Therefore, we tested the effect of VL-004 and Spd in C. elegans models for these diseases.
First, we performed experiments with a C. elegans model for Huntington disease, i.e., the AM141 transgenic strain. These worms overexpress a 40 polyglutamine (Q40) stretch in the body wall muscles and show progressive paralysis with age [Citation68,Citation69]. We tested the effect of VL-004 and Spd at 0.25 and 4 mM concentrations compared to the vehicle. All concentrations of VL-004 and Spd significantly ameliorated Q40 induced paralysis (). Moreover, the 4 mM worked best for both compounds and to a similar degree.
To explore whether the VL-004 protective effect is mitophagy dependent, we introduced the Q40 extrachromosomal array into dct-1(tm376) and pink-1(tm1779) mutants, and repeated the paralysis assay using 4 mM VL-004 and vehicle as a control. VL-004 did not ameliorate the paralysis phenotype of dct-1 and pink-1 mutants (), highlighting that mitophagy is required for VL-004 protective activity. Intriguingly, the paralysis phenotype was significantly harsher in vehicle-treated dct-1 and pink-1 mutants than in vehicle-treated worms with wild-type genetic background (), emphasizing the importance of steady-state mitophagy for combating proteotoxicity.
Second, we explored the functions of VL-004 and Spd in a C. elegans model for familial amyotrophic lateral sclerosis (ALS) on days 3 and 6, using the AM725 transgenic strain [Citation70] that expresses the human ALS-associated insertion/frameshift mutation G127X, which truncates the last 21 amino from the C terminus of SOD-1 and destabilizes the protein [Citation71]. For comparison, we used the AM263 strain that expresses the human wild-type SOD-1 [Citation70]. On day 3 the thrashing of the two strains was similar, and there was no difference between vehicle and compounds treated worms. However, on day 6, the thrashing rate of vehicle-treated AM725 worms was significantly lower than AM263 worms (<0.0001), suggesting that truncated SOD-1 toxicity is already manifested. Both 0.25 mM and 4 mM VL-004 and Spd treatment significantly ameliorate the mutated SOD-1 toxicity to a similar extent ().
In sum, we demonstrated that VL-004 and Spd alleviate the symptoms associated with proteotoxicity models in C. elegans.
VL-004 induces mitophagy and protects against stress in human cells
As mitophagy is conserved from yeast to mammals [Citation72], we tested whether VL-004 induces mitophagy also in human cells and protects them from oxidative stress. To that end, we treated HaCaT cells (a human epithelial cell line [Citation73]) with VL-004 for 24 h and measured the colocalization of mitochondria and lysosomes using Cytopainter (ab139487). In addition, we tested the effect of Spd and FCCP (due to its toxicity, FCCP was given for 2 h only). We used VL-004 and Spd at 100 µM because this Spd dose induces autophagy in human cells [Citation74]. VL-004 induced robust mitophagy in HaCaT cells, similar to FCCP and Spd (), suggesting that it is a potent MAC also in mammalian cells.
Figure 8. VL-004 activity in mammalian cells. (A) Colocalization of mitochondria and lysosome. Arrowheads indicate representative colocalization. The inset represents a nine-fold enlargement – scale bar: 50 μm. (B) Colocalization quantification. n ≥ 467 cells/per treatment.
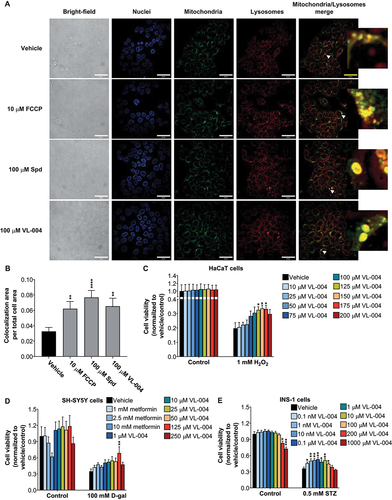
We then examined whether VL-004 would protect HaCaT cells from hydrogen peroxide (H2O2) injury. Cells were treated with increasing concentrations of VL-004 (10–200 μM) for 24 h, exposed then to 1 mM H2O2 for 3 h, and assessed for survival using methylene blue [Citation75]. VL-004 did not show any toxicity in control cells while providing significant protection against H2O2-induced oxidative injury at 125, 150, and 175 μM concentrations (), indicating that its antioxidant activity is preserved.
D-Galactose (D-gal) accelerates cellular senescence and aging phenotypes [Citation76,Citation77]. Previous studies show that D-gal induces oxidative stress in the human SH-SY5Y neuroblastoma cells [Citation78]. Moreover, mitophagy induction protects these cells from oxidative damage [Citation79]. Thus, we treated cells with increasing concentrations of VL-004 (1–250 μM) for 24 h, exposed them to 100 mM D-gal for 19 h, and measured cell survival using MTT [Citation80]. In addition, we tested the effect of metformin, at various doses, because it was shown to protect against D-gal toxicity [Citation81]. 125 μM VL-004, but not metformin, significantly protected against D-gal (), suggesting VL-004 is superior to metformin in this assay system.
The diabetogenic antibiotic streptozotocin (STZ) (Wu and Huan, 2008) appears to mediate its toxic effect, at least in part, by ROS [Citation82,Citation83], as transgenic mice with increased pancreatic islets expression of SOD2/MnSOD (superoxide dismutase 2, mitochondrial) and catalase are less vulnerable to damage by this agent [Citation84]. Rat insulinoma cells (INS-1), which are widely used as a model for islet beta-cell function [Citation85], were, therefore, exposed to increasing concentrations of VL-004 and tested for resistance to 0.5 mM STZ. Intriguingly, VL-004 was already effective at the low nanomolar range (), while at concentrations above 100 μM it compromised control cells viability and probably therefore was not effective in protection against STZ.
Is VL-004 protective activity in cells mitophagy/autophagy-dependent? To answer this, we performed two experiments: First, we repeated the D-gal senescence assays with SH-SY5Y cells treated with PINK1 specific siRNA (TriFECTa kit, IDT) or scrambled siRNA as a negative control (i.e., universal’ negative control duplex [Citation86]). Indeed, PINK1 silencing inhibited the protective effect of VL-004 (Figure S9A), suggesting that mitophagy is vital for VL-004 activity in human cells. Second, we inhibited autophagy using bafilomycin A1 (BafA1, 70 nM) [Citation87] and repeated the D-gal senescence experiment. BafA1 also suppressed the protective effect of VL-004 (Figure S9B), suggesting that autophagosome-lysosome fusion is essential for VL-004 function.
Finally, to confirm that VL-004 induces mitophagy in SH-SY5Y cells, we used the cox8-mCherry-EGFP reporter [Citation88]. Similar to the mito-Rosella sensor, this mitochondria reporter shows red and green fluorescence under a neutral pH environment. However, its EGFP fluorescence is quenched in the acidic environment of the lysosome. VL-004 induced significant mitophagy (Figure S9C) in a similar magnitude to the FCCP positive control.
Together, this set of experiments demonstrates that VL-004 is also a potent mitophagy inducer in mammalian cells and confers protection against oxidative injury and senescence.
MACs structure-activity relationships: preliminary characterization
Our lesson from the initial structural characterization of MACs is that eliminating the internal amine of Spd improves the mitophagic properties of the molecule (). Thus, a symmetric structure of two positively charged residues, flanking an uninterrupted linear carbon chain, constitutes a prototypic MAC. To further explore it, we examined the effect of the carbon chain length on its bioactivity. Shortening the eight-carbon chain of VL-004 to a diamine with six carbons (VL-006, ) abrogated its bioactivity while extending it by two carbons (VL-007, ) improved its activity against oxidative stress. VL-006 did not protect against PQ at all concentrations tested (Figure S9D, E). By contrast, VL-007 showed superiority vs. VL-004 at 0.0625 mM (). It showed similar protection at 0.25 mM, while at 4 mM VL-007 did not protect from PQ toxicity (Figure S9E), suggesting that this compound’s safety range is narrower.
Figure 9. Carbon chain length and substituents affect diamine activity. (A and B) Stacked bar graphs showing worms’ Survival in 200 mM PQ after 3 h and 6 h. Graphs represent the average of six independent experiments: (A) ≥ 113; asterisks indicate significance compared to vehicle control; (B) ≥ 168. (C) Survival curves of worms in the presence of VL-850 or vehicle. The summary of lifespan data is presented in Table S3. (D) Colocalization of mitochondria and lysosome. Arrowheads indicate representative colocalization. The inset represents an eight-fold enlargement – scale bar: 50 μm. (E) Colocalization quantification. N = 3, ≥338 cells per treatment.(F) HaCat cells were incubated with VL-850 at the indicated concentrations for 24 h, then treated with 1 mM CoCl2 for 24 h. Cell viability was measured after 48 h by the methylene blue assay. Right panel, representative assay plates, top row – vehicle-treated, low – 100 μM VL-850. N ≥ 3.
Next, we examined the effect of VL-004 amines’ derivatization on its bioactivity: i.e., N, N-dimethyl-1-8-diaminooctane (VL-008), identical to VL-004, but with monomethylated amines. VL-008 was significantly less potent than VL-004 at lower concentrations and equally effective at 4 mM, suggesting that the methyl groups may interfere with target binding (Figure S9F). The replacement of two internal carbons in VL-004 by oxygen, generating 2,2’-(ethylenedioxy)bis(ethylamine) (VL-009), led to a complete loss of the protective activity against PQ (Figure S9G). These findings further refine our characterization of a useful MAC. Namely, an uninterrupted linear aliphatic chain of at least eight carbons in length, flanked by primary amines at both ends.
In light of the above, we designed and tested the activity of an uninterrupted eight-carbon chain flanked by aminooxy residues at both ends (VL-850, ). First, we examined VL-850 activity in C. elegans, using the PQ-protection assay, at two low concentrations. i.e., 0.01562 and 0.0625 mM, compared with VL-004. VL-850 provided significant protection against PQ at both concentrations and to a similar extent. In contrast, VL-004 provided protection only at the higher concentration of 0.0625 mM, which was still significantly lower than VL-850 even at 0.01562 mM. These VL-850 concentrations significantly extended C. elegans lifespan, and in this experimental setting, the 0.0625 mM concentration was superior to that of 0.01562 mM (). Thus, VL-850 improved activity points to possible directions for MACs’ structural optimization.
Next, we examined whether VL-850 induces mitophagy in human cells. For this, we treated HaCat cells with 100 μM VL-850 or 10 μM FCCP (as a positive control) and measured the colocalization of mitochondria and lysosomes using Cytopainter. VL-850 induced significant mitochondria & lysosome colocalization, to a similar extent as FCCP ().
Consequently, we tested whether VL-850 protects against oxidative stress: HaCat cells were treated with 10 μM or 100 μM VL-850 for 24 h and exposed to oxidative stress by 1 mM cobalt chloride (CoCl2); an ROS-producing agent [Citation89,Citation90]. After 24 h, we replaced the CoCl2-containing medium with a fresh one and measured cell survival after 48 h, using methylene blue. Notably, the viability of VL-850 treated cells was similar to control cells that were not treated with CoCl2 (), demonstrating VL-850 capacity to protect human cells from oxidative injury, already at 10 μM.
Thus, the novel MAC, VL-850, is a potent mitophagy inducer protecting against oxidative damage in C. elegans and human cells.
Discussion
From Spd to synthetic MACs
Spermidine (Spd) is a known mitophagy enhancer [Citation15,Citation17,Citation91,Citation92], and this study aimed to develop improved Mitophagy Activating Compounds (MACs), based on this initial finding. VL-004, a simplified version of Spd structure, fulfills the criteria we set: it induces mitophagy, protects from oxidative damage, and promotes healthspan in older animals. Moreover, VL-004 extends lifespan and protects the organism from toxic protein aggregates in several disease models in C. elegans.
The prolongation of both lifespan () and healthspan (e.g., muscle mass and motility, ) by the same agent is not obvious; on the contrary, relative health duration and longevity were found to be mutually exclusive [Citation93]. We further characterized the structural requirements essential for effective MAC activity as an uninterrupted linear aliphatic chain of at least eight carbons in length, flanked by primary amines on both ends (, and S9D-G). Our structure-activity relationships study culminated in the design of a novel, significantly more potent MAC, VL-850 ().
Mitophagy-induction and oxidative-damage protection
Both VL-004 and Spd induce mitophagy in C. elegans and human cells (). However, VL-004 was more potent than Spd at 1 mM concentration in C. elegans (). Notably, VL-004 induces the expression of key autophagy genes (), including the LC3 and GABARAP related genes lgg-1 and lgg-2, and the autophagy receptor sqst-1. Moreover, it appears to recruit LGG-1 to autophagosome structures (). Therefore, we suggest that VL-004 is a potent inducer of macroautophagy. In addition, VL-004 protected against oxidative injury in a superior manner compared to Spd and several Spd derivatives (). Thus, although both compounds induce mitophagy, the extent of their downstream beneficial effect is incomparable.
Performance related to lifespan and toxic protein aggregation models in C. elegans
VL-004 extends C. elegans lifespan at both 0.25 mM and 4 mM concentrations and to a similar extent (). By contrast, Spd did not extend lifespan at these concentrations. Although Spd was reported to extend the lifespan of C. elegans at 0.2 mM [Citation17], this discrepancy might relate to the use of UV-killed bacteria in that study. In contrast, we used live OP50 bacteria that can metabolize Spd. In this respect, one of the limitations of Spd as an anti-aging drug is its metabolic degradation, including some harmful products [Citation19,Citation20]. The fact that the range of concentrations at which VL-004 prolongs life is broad and its function against oxidative injury is independent of bacterial viability (Figure S2D) suggests that it is safe and stable.
We then compared the efficacy of both in two disease models in C. elegans (). Interestingly, Spd is superior to VL-004 in the polyglutamine disease model at 0.25 mM concentration (p = 0.0344) and has similar potency at 4 mM (). Moreover, Spd and VL-004 have comparable potency in the SOD1 ALS model (). In conclusion, VL-004 is more effective than Spd in protecting against oxidative stress and prolonging lifespan. However, it has slightly lesser or similar potency in toxic polyglutamine and ALS disease models. Respectively.
Anti-aging/stress response transcription factors mediate the VL-004 activity
FOXO3/FOXO3a and TFEB are key transcription factors controlling the mitophagy transcriptome [Citation94]. FOXO3 regulates key autophagy/mitophagy genes, including PINK1, LC3B, ATG9, and BNIP3L/NIX [Citation94–96]. This function appears to be evolutionary conserved, and studies in flies and mammals show that FOXO controls the activity of PINK in neurons and immune cells [Citation97,Citation98]. TFEB regulates the expression of genes crucial to mitophagy, and this activity is modulated by a complex lysosomal-signaling platform [Citation94]. Also, TFEB is a major regulator of lysosomal biogenesis, thus controlling lysosome number and activity [Citation99]. Thereby, TFEB regulates mitophagy at multiple stages, from the encapsulation of mitochondria in autophagosomes to degradation in the autolysosome. Thus, TFEB appears to be a promising target for treating age-related diseases. Our studies show that the FOXO3 and TFEB orthologs, DAF-16 and HLH-30, respectively, are essential for VL-004 activity (3A, D), suggesting that VL-004 acts through conserved signaling pathways. Fascinatingly, DAF-16 and HLH-30 appear to act as a complex regulating various stress responses in a context-dependent manner [Citation100]. Indeed, they co-occupy similar promoter regions and thus regulate similar target genes and act together to promote longevity and resilience against oxidative injury [Citation100].
VL-004 activity is PINK-1-dependent, but PDR-1-independent
An unexpected outcome of our studies is that the PRKN ortholog, PDR-1, is not required for VL-004 activity (, and Figure S4 D-F). Importantly, in agreement with previous studies [Citation2], PDR-1 was required for mitophagy induction by mitochondrial uncoupler (Figure S4F). Therefore, it appears that healthy mitophagy induced by VL-004 is PINK-1-dependent yet, PDR-1-independent.
Examples for PINK1-dependent PRKN-independent mitophagy were reported in recent studies [Citation101]. Baehrecke and colleagues show that ubiquitin-binding protein Vps13D regulates mitophagy in a Pink1-dependent park-independent manner in Drosophila. Intriguingly, the C. elegans C25H3.11 protein is a close homolog of Vps13D. We will explore whether C25H3.11 is necessary for VL-004 activity.
Moreover, a study by Kanki and colleagues demonstrate that the anticancer agent gemcitabine stabilizes PINK1, thus induce mitophagy, in a PRKN-independent way [Citation102]. Also, they show that the E3 ubiquitin ligase MUL1 is important for gemcitabine-induced mitophagy. Although there is no close homolog of MUL1 in C. elegans, another E3 ubiquitin ligase (s) may act together with PINK-1 to facilitate healthy mitophagy by VL-004.
Materials and methods
Reagents
1, 8-Diaminooctane (Sigma, D22401-25 G), 2-mercaptoethanol (Sigma, M6250), acetone (Sigma, 179,973), adenosine 5’-triphosphate disodium salt hydrate (Sigma, A7699-1 G), ampicillin (Thermofisher, BP1760-25), BactoTM agar (Gibco, 214,010), BactoTM peptone (Gibco, 211,677), BactoTM tryptone (Gibco, 211,705), BactoTM yeast extract (Gibco, 212,750), BSA fraction V (MP biomedicals, 1,600,069), calcium chloride (Sigma, C4901), carbenicillin (Sigma, C1389), carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma, C2759), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (Sigma, C2920), cobalt chloride (Sigma, 60,820), D-galactose (Next Advance, ZrOB05), dimethyl sulfoxide (Sigma, D8418), DMEM high glucose (Biological Industries, 01–055-1A), DMEM low glucose (Sigma, D5921), FBS heat inactivated (Biological Industries, 04 − 127–1A), Fluorescent Mounting Medium (GBI Labs, E18-100/E18-18), Gibson Assembly® Master Mix (NEB, E2611L), glutaraldehyde (Sigma, G5882), glycerol (Gadot, 7846), HEPES (Biological Industries, 03–025-1B), hydrochloric acid (Sigma, 320,331), hydrogen peroxide (Sigma, 216,763), IPTG (Bio-Lab, 16,242,352), isopropanol (Thermo Fisher, 67–63-0), KpnI (NEB, R3142S), L-glutamine (Biological Industries, 03–020-1B), luminescent ATP detection assay kit (Abcam, ab113849), magnesium sulfate (Sigma, 208,094), methyl viologen dichloride hydrate (Sigma, 856,177–1 G), methylene blue (Alfa Aesar- Thermo Scientific, 42,771.AE), Nonidet P-40 (Sigma, 74,385), paraformaldehyde 16% (Electron Microscopy Sciences, 15,720), penicillin-streptomycin-neomycin (Biological Industries, 03–032-1B), phenoxyisopropanol (Sigma, 484,423), phosphate-buffered saline (Biological Industries, 02–023-1A), Pierce BCA Protein Assay Kit (Thermo Fisher, 23,225), Pluronic F-127 (Sigma, P2443), Poloxamer 188 (Sigma, P5556), Polybead® Microspheres 0.10 μm (Polysciences, 00876–15), potassium phosphate dibasic (Sigma, P3786), potassium phosphate monobasic (Millipore, 1.04873.1000), proteinase K (Sigma, 70,663), rhodamine-phalloidin (Thermo Fisher, R415), RPMI (Biological Industries, 01–100-1A), SalI (NEB, R0138S), SDS (Sigma, L3771), SeaKem LE Agarose (Lonza, 50,004), sodium chloride (Bio-Lab, 1,903,059,100), sodium hydroxide (Gadot, 1310–73-2), sodium phosphate dibasic (Sigma, 4273), sodium pyruvate (Alfa Aesar, J61840), spermidine (Sigma, S2626), streptozotocin (Sigma, S0130), tetracycline hydrochloride (Sigma, 87,128–25 G), tetramethylrhodamine ethyl ester (TMRE; Thermo Fisher, T669), thiazolyl blue tetrazolium bromide (MTT; Alfa Aesar by Thermo Scientific, L11939.03), tris hydrochloride (Sigma, 10,812,846,001), Trizma base (Sigma, T1503-1 KG), trypsin-EDTA (Biological Industries, 03–0521A), Tween-20 (Sigma, P1379).
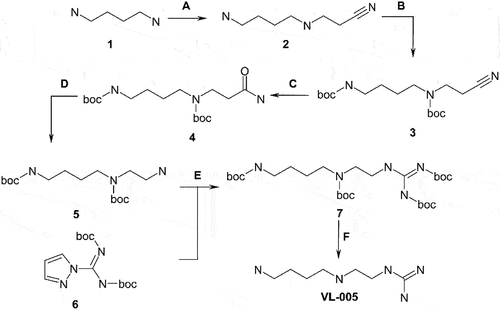
The synthetic procedure for MetSpd, VL-002, VL-003, VL-005, and VL-850
Compounds synthesis is described in detail below.
Compound stock solutions
Unless otherwise mentioned, the compounds were solubilized in either M9 buffer (C. elegans experiments; 22 mM KH2PO4 [Merck, 104,873] 42 mM Na2HPO4 [Sigma, 04273], 86 mM NaCl [Bio-Lab, 1,903,059,100), and 1 mM MgSO4 [Sigma, M7506]) or PBS (mammalian cell experiments) to a final concentration of 200 mM, and titrated (if needed) to pH 7.4. Notably, VL-850 was solubilized in DMSO to a final concentration of 100 mM, and then diluted 20-fold in HEPES buffer to a pH 7.4. The compounds were aliquoted and stored at −20°C until used.
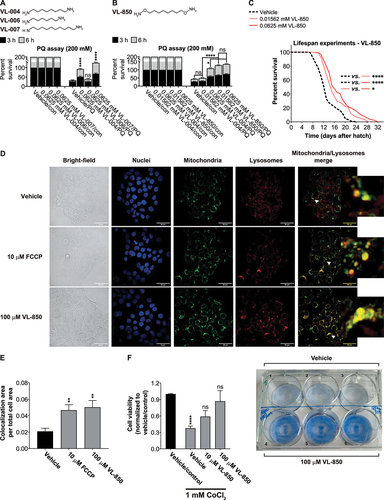
Synthetic procedure for MetSpd, VL-002, VL-003, VL-005, and VL-850
MetSpd
Synthesis was carried out following the scheme given below:
Step A: To a solution of compound 1 (30.0 g, 337 mmol) in anhydrous dichloromethane (500 mL) a solution of Na2CO3 (67.8 g, 640 mmol) in water (900 mL) was added followed by the addition of benzyl chloroformate (48 mL, 338 mmol). The reaction mass was stirred overnight at room temperature and then layers were separated. The aqueous layer was extracted with dichloromethane. The combined organics were washed with 1 M hydrochloric acid and aqueous NaHCO3, dried over Na2SO4, and evaporated under reduced pressure to obtain 63.2 g (283 mmol, 84%) of compound 2.
Step B: To a solution of compound 2 (63.1 g, 283 mmol) and triethylamine (118 mL, 847 mmol) in anhydrous DMSO (450 mL) sulfur trioxide pyridine complex (135 g, 848 mmol) was added, and the resulting mixture was stirred overnight at room temperature. Then it was poured onto crushed ice and extracted with dichloromethane. The combined organic layer was washed with 30% aqueous citric acid and aqueous NaHCO3, dried over Na2SO4, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to give 23.0 g (104 mmol, 37%) of compound 3.
Step C: To a solution of compound 4 (11.9 g, 104 mmol) in anhydrous methanol (120 mL) molecular sieves (20 g) were added. Then a solution of compound 3 (21.9 g, 99.0 mmol) and acetic acid (11.3 mL, 198 mmol) in methanol (300 mL) was added, followed by NaBH3CN (9.33 g, 148 mmol). The reaction mass was stirred overnight at room temperature and filtered. The filtrate was evaporated under reduced pressure, and the residue was mixed with an aqueous K2CO3 solution. The product was extracted with ethyl acetate. The combined organic extracts were dried over Na2SO4 and evaporated under reduced pressure. The crude material was purified by silica gel column chromatography to give 13.6 g (42.6 mmol, 43%) of compound 5.
Step D: A mixture of compound 5 (13.6 g, 42.6 mmol), Pd/C (10.0 g), and Pd(OAc)2 (10.0 g) in methanol (300 mL) was stirred under atmosphere of hydrogen at room temperature for 21 days. The solid was filtered off, and the filtrate was evaporated under reduced pressure. The residue was mixed with 6 M hydrochloric acid and evaporated under reduced pressure. The crude material was recrystallized from methanol to obtain 2.30 g (8.57 mmol, 20%) of 1-methyl-spermidine (MetSpd) as trihydrochloride salt.
VL-002 and VL-003
Synthesis was carried out following the scheme given below:

Step A: A mixture of potassium phthalimide (1) (100 g, 540 mmol), tetrabutylammonium chloride (8.00 g, 28.8 mmol), and 4-bromo-1-butene (66 mL, 650 mmol) in acetonitrile (1000 mL) was refluxed overnight, cooled to room temperature, and diluted with water. The precipitated solid was collected by filtration, washed with water (3 × 150 mL), and dried to obtain 81.0 g (403 mmol, 75%) of compound 7.
Step B: To a cooled to 0°C solution of compound 2 (81.0 g, 403 mmol) in anhydrous chloroform (900 mL) m-CPBA (104 g, 603 mmol) was added, and the resulting mixture was stirred overnight at room temperature. The precipitated solid was filtered off and rinsed with chloroform. The filtrate and rinses were washed with aqueous Na2S2O3 solution and NaHCO3 solution, dried over Na2SO4, and evaporated under reduced pressure to obtain 85.1 g (392 mmol, 97%) of compound 3
Step C: To a solution of compound 3 (85.1 g, 392 mmol) in anhydrous ethanol (1000 mL) dibenzylamine (85.0 g, 431 mmol) was added, and the reaction mass was refluxed overnight. The resulting mixture was concentrated to ½ of the initial volume and filtered. The obtained solid was washed with hexane and dried to obtain 120 g (289 mmol, 74%) of compound 4.
Step D: To a cooled to 0°C solution of compound 4 (75.0 g, 181 mmol) in anhydrous dichloromethane (750 mL) a solution of Morph-DAST (33 mL, 271 mmol) in dichloromethane (300 mL) was added and the reaction was stirred for 2 h. Then it was poured onto crushed ice and layers were separated. The aqueous layer was extracted with dichloromethane. The combined organic layers were washed with NaHCO3 solution, dried over Na2SO4, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to give 40.0 g (96.0 mmol, 53%) of compound 5.
Step E: To a solution of compound 5 (39.5 g, 94.8 mmol) in ethanol (1500 mL) hydrazine hydrate (36.5 mL, 750 mmol) was added, and the reaction mass was refluxed for 2 h. After the mixture cooled down to room temperature, it was filtered, and the filter cake was rinsed with ethanol. The filtrate and rinses were evaporated under reduced pressure, and the residue was dissolved in NaOH solution. The product was extracted with dichloromethane. The combined organic layer was dried over Na2SO4, and evaporated under reduced pressure to obtain 21.5 g (75.1 mmol, 80%) of compound 6.
Step F: To a solution of compound 6 (8.70 g, 30.4 mmol) in anhydrous methanol (50 mL) molecular sieves (10 g) were added. Then a solution of compound 3 (6.40 g, 28.9 mmol) and acetic acid (3.3 mL, 57.7 mmol) in methanol (100 mL) was added, followed by NaBH3CN (2.70 g, 43.0 mmol). The reaction mass was stirred overnight at room temperature and filtered. The filtrate was evaporated under reduced pressure, and the residue was mixed with an aqueous K2CO3 solution. The product was extracted with ethyl acetate. The combined organic extracts were dried over Na2SO4 and evaporated under reduced pressure. The crude material was purified by silica gel column chromatography to give 9.20 g (24.8 mmol, 63%) of compound 7.
Step G: A mixture of compound 7 (6.61 g, 17.8 mmol), Pd/C (1.10 g, 5% wt.), and methanol (200 mL) was degassed and then stirred under pressure of hydrogen (3 atm.) at 50°C for 8 h. The catalyst was removed by filtration, and the filtrate was evaporated in vacuo to obtain 3.00 g (16.9 mmol, 95%) of target compound VL-003.
Step H: To a solution of compound 6 (11.9 g, 41.0 mmol) in THF (150 mL) 1 M solution of NaHCO3 (6.95 g, 82.7 mmol) was added, followed by Boc2O (10 mL, 43.5 mmol). The reaction mass was stirred overnight at room temperature, and layers were separated. The organic layer was evaporated under reduced pressure. The aqueous layer was extracted with ethyl acetate. The residue, obtained after evaporation of THF, was mixed with the ethyl acetate solution, dried over Na2SO4, and evaporated under reduced pressure to obtain 15.5 g (40.1 mmol, 98%) of compound 8.
Step I: A mixture of compound 8 (15.4 g, 39.8 mmol), Pd/C (2.00 g, 10% wt.), and methanol (200 mL) was degassed and then stirred under pressure of hydrogen (10 atm.) at 40°C for 12 h. The catalyst was removed by filtration, and the filtrate was evaporated in vacuo to obtain 7.30 g (35.4 mmol, 89%) of compound 9, pure enough for the next step.
Step J: To a solution of compound 9 (6.96 g, 33.7 mmol) in anhydrous methanol (100 mL) molecular sieves (10 g) were added. Then a solution of compound 3 (7.11 g, 32.1 mmol) and acetic acid (3.7 mL, 64.7 mmol) in methanol (100 mL) was added followed by NaBH3CN (3.03 g, 48.2 mmol). The reaction mass was stirred overnight at room temperature and filtered. The filtrate was evaporated under reduced pressure and the residue was mixed with aqueous K2CO3 solution. The product was extracted with ethyl acetate. The combined organic extracts were dried over Na2SO4 and evaporated under reduced pressure. The crude material was purified by silica gel column chromatography to give 6.30 g (15.3 mmol, 48%) of compound 10.
Step K: To a solution of compound 10 (6.25 g, 15.2 mmol) in anhydrous dichloromethane (100 mL) trifluoroacetic acid (5 mL, 65.3 mmol) was added dropwise, and the resulting mixture was stirred overnight at room temperature. The volatiles were evaporated, and the residue was dissolved in ethyl acetate. The solution was washed with aqueous K2CO3, and the aqueous layer was back-extracted with ethyl acetate. The combined organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was hydrogenated (1 atm. of H2) in methanol (200 mL) in the presence of Pd/C (2.00 g) and Pd(OAc)2 (2.00 g) for 72 h. The catalysts were filtered off, and the filtrate was evaporated under reduced pressure to obtain 2.00 g (11.3 mmol, 80%) of target compound VL-002.
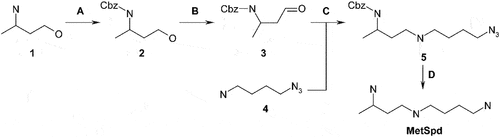
VL-005
Synthesis was carried out following the scheme given below:
Step A: To a solution of compound 1 (100 g, 1134 mmol) in anhydrous methanol (500 mL) acrylonitrile (37.0 mL, 565 mmol) was added, and the resulting mixture was stirred overnight at room temperature (RT). The volatiles were evaporated, and the crude product was purified by vacuum distillation (b.p. 135°C at 2 mmHg) to obtain 34.2 g (242 mmol, 21%) of compound 2.
Step B: To a solution of compound 2 (34.2 g, 242 mmol) in anhydrous dichloromethane (240 mL) a solution of Boc2O (122 mL, 531 mmol) in anhydrous dichloromethane (80 mL) was added dropwise and the resulting mixture was stirred overnight at RT. The obtained solution was washed with 1 M hydrochloric acid and aqueous K2CO3, dried over Na2SO4, and evaporated under reduced pressure to obtain 96.8 g of crude compound 3. The material was used as is in the next step.
Step C: To a solution of compound 3, obtained in the previous step, (96.8 g) in methanol (700 mL) 25% aqueous ammonia (2000 mL) was added, and the mixture was cooled to 0°C. 50% H2O2 was added, and the reaction mass was left to stir for 72 h. The product was extracted with dichloromethane (3 × 500 mL). The combined organic extract was washed with aqueous Na2S2O3, dried over Na2SO4, and evaporated under reduced pressure. The crude product was recrystallized from hexane/2-propanol 10:1 mixture to obtain 47.1 g (131 mmol, 54% over 2 steps) of compound 4.
Step D: To a cooled to 0°C mixture of compound 4 (47.1 g, 131 mmol), DMF (125 mL), and water (125 mL) PIDA (63.3 g, 197 mmol) was added. After the mixture was stirred for 15 min it was treated with anhydrous pyridine (5.30 mL, 65.8 mmol) and left to stir for 48 h at RT. The volatiles were evaporated; the residue was mixed with water, and the obtained solution was extracted with ethyl acetate (3 × 250 mL). The organic extract was washed with brine (3 × 300 mL), dried over Na2SO4, and evaporated under reduced pressure. The obtained solid was mixed with 1 M KHSO4 solution, washed with ethyl acetate (3 × 100 mL), and basified with 4 M aqueous NaOH. The product was extracted with dichloromethane (3 × 200 mL). The combined organic extract was dried over Na2SO4, and evaporated under reduced pressure to obtain 8.00 g (24.1 mmol, 20%) of compound 5.
Step E: A mixture of compound 5 (8.00 g, 24.1 mmol), compound 6 (9.00 g, 29.0 mmol), and anhydrous THF (85 mL) was stirred overnight at RT. and then evaporated under reduced pressure. The residue was dissolved in ethyl acetate; the solution was washed with water, dried over Na2SO4, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to give 9.00 g (15.7 mmol, 66%) of pure compound 7.
Step F: To a cooled to 0°C solution of compound 7 (9.00 g, 15.7 mmol) in dioxane (25 mL) 4 M dioxane/HCl (75 mL) was added dropwise, and the resulting mixture was stirred overnight at RT. The volatiles were evaporated, and the residue was recrystallized from methanol to obtain 2.50 g (8.84 mmol, 52%) of target compound Z2901777223 as trihydrochloride salt.
VL-850

Step A: To a mixture of compound 1 (18.0 g, 110 mmol) and triethylamine (14.0 g, 138 mmol) in DMF (150 mL) a solution of 1,8-dibromooctane (15.0 g, 55.0 mmol) in DMF (20 mL) was added, and the reaction mass was stirred for 72 h at RT. The obtained solution was poured into water (500 mL). The precipitated solid was collected by filtration, washed with water (3 × 100 mL) and MTBE (2 × 50 mL), and air-dried to obtain 14.0 g (58% yield) of compound 2.
Step B: To a cooled to 10°C stirring solution of compound 1 (14.0 g, 85.8 mmol) in THF (200 mL) 40% aqueous methylhydrazine (11 mL) was added, and the reaction mass was stirred for 12 h at RT. The precipitated solid was collected by filtration and rinsed with THF (2 × 50 mL). The filtrate and rinses were evaporated under reduced pressure and the residue was mixed with hexane (100 mL). The insoluble solid was filtered off and the filtrate was evaporated in vacuo. The residue was purified by vacuum distillation to obtain 3.80 g (67% yield) of target compound VL-850.
Worm strains and bacteria
Worm strains and bacteria are listed in Table S1.
Oligonucleotides
All oligonucleotides used to amplify specific sequences from the genome and coding DNA, or to determine mRNA levels by qRT-PCR are listed in Table S2.
C. elegans and cell culture – Maintenance and handling
C. elegans
Culture preparation
- In general, unless otherwise noted, C. elegans strains were grown under standard laboratory conditions (Brenner, 1974) in 35-mm Petri dishes containing 4 ml of nematode growth medium (NGM; 3 g sodium chloride (NaCl) 20 g Bacto agar and 2.5 g of Bacto peptone, as described below))-agar seeded with 250 μl OP50 bacteria (CGC) at RT (21°C). The strains we used in this study are listed above. These strains were either generated in this study using our laboratory N2 strain or outcrossed with it at least three times unless indicated otherwise.
Experimental NGM plates were prepared as described in [Citation103] with the following modifications. To make 1 L of NGM-agar, we dissolved 3 g sodium chloride (NaCl) in 900 ml DDW, added 20 g Bacto agar, 2.5 g of Bacto peptone, and DDW to a total volume of 1 L. Then, we autoclaved the suspension, let it cool to ~55°C in a heating bath, and added 1 ml of 0.1 M CaCl2, 1 ml 0.1 M MgSO4, 25 ml of 1 M potassium phosphate buffer pH 6, and 2 ml of 5 mg/ml cholesterol (Fisher Scientific, AFF-J13580-06; the cholesterol is dissolved in ethanol). Finally, we added MACs/vehicles at the concentrations indicated in the article. The poured plates were allowed to dry, at RT, for 24 h, seeded with 100 µl of OP50 bacteria (OD600 = 0.6), and kept at RT for additional 24 h. The experimental plates were stored at 4°C, and used within a week.
Worms synchronization
- We used two methods to generate synchronized worms: (1) Alkaline-hypochlorite (Brenner, 1974) – in most cases; (2) Egg-laying (in ).
Alkaline hypochlorite method: Gravid hermaphrodites were collected into 15-ml tubes by washing the NGM plates three times with M9 buffer (22 mM KH2PO4, 42 mM Na2HPO4, pH 7, 86 mM NaCl, and 1 mM MgSO4). Tubes were centrifuged for 1 min (1690 x g, 1 min), and the supernatant was removed until 1.5 ml of volume remained. Then, we added 1.5 ml of hypochlorite solution (0.5 N NaOH, 1.25% NaOCl) to each tube and inverted it five times. We used a syringe with a 21-gauge needle to assist the release of embryos and aspirated the worm suspensions back and forth several times. After 5 min, the embryos were sedimented by centrifugation (1690 x g for 1 min), and most of the hypochlorite solution was removed. Each tube was washed 3 times with 5 ml of M9 buffer. Next, we removed most of the M9 buffer (without disturbing the embryo pellet) and added 1 ml of fresh M9 buffer to each tube. The tubes were rotated for 16 h at RT (9 rpm). The hatched L1 larvae were collected by centrifugation (1690 x g for 1 min), counted, transferred to seeded NGM plates, and grown until the desired developmental stage.
Egg-laying method
We transferred ten gravid hermaphrodites into a seeded NGM plate and let them lay eggs for 6 h. Afterward, we removed these worms from the plates. The hatched L1s were grown until the desired developmental stage.
Mammalian cell culture
The cell lines used in this study are HaCaT [Citation73], SH-SY5Y (Abcam, ab275475), and INS-1 [Citation104]; the HaCaT and INS-1 cell lines were generously provided to us by Prof. Ron Kohen (Institute for Drug Research, School of Pharmacy, Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem), and Prof. Gil Leibowitz (The Diabetes Unit and the Endocrine Service, Hadassah-Hebrew University Medical Center, Jerusalem, Israel), respectively. The HaCaT and SH-SY5Y cells were maintained in DMEM medium supplemented with 10% FBS (heat-inactivated), 2% L-glutamine. INS-1 cells were maintained in RPMI media supplemented with 2% L-glutamine, 1% penicillin-streptomycin-neomycin, 10% FBS (heat-inactivated), 1% 1 M HEPES, pH 7.2, 1% sodium pyruvate, and 50 μM β-mercaptoethanol. In general, we split the cells into a 1:4 or 1:6 cells /fresh- medium ratios when they reached 70–80% confluency.
Cell Counting – Trypsinized cells were counted using CellDrop, an automated cell counter (DenNovix) in 5x replicates per cell line.
Method details
C. elegans
Paraquat (PQ) resistance assay
Paraquat assays were conducted as described in [Citation105,Citation106] with some modifications. Synchronized ~200 L1 larvae were grown in 35 mm seeded NGM plates supplemented with indicated drug concentrations or with a vehicle as a control until the desired developmental stage. The worms were collected from the plate with M9 buffer, washed twice with the same buffer, and transferred into a 96-well plate (~12 worms per well, 100 µl). PQ (200 mM, final concentration) or M9 (control) were added to each well, and the plate was placed on an orbital shaker at 350 rpm at 21°C. We used an eyelash pick to score worms’ survival (we touched each worms several times over the course of a few seconds) when PQ was added (time 0) and 3, 6, and 24 h afterward. We performed six biological repeats for experiments, with a minimum of 120 worms per condition.
For the heat-killed bacteria experiment (Fig. S1D), liquid OP50 E. coli culture was grown to an OD600 of 0.6 and concentrated 10-fold by centrifugation at 3000 × g for 5 min at 4°C. 5 ml of concentrated bacteria was washed three times with M9 and incubated for 45 min at 65°C. Then, 100 µl of heat-killed bacteria were added to each control/experimental plate and let dry for ~16 h. Then, the course of the experiment was as described above.
RNAi assays
RNAi experiments were conducted as described previously [Citation107].
HT115 bacteria expressing RNAi against pha-4 (Vidal library) or empty vector (L4440; Addgene, 1654; deposited by Andrew Fire) were grown in 2XYT (16 g tryptone, 10 g yeast extract, 5 g NaCl in 1 L of sterile water, pH 7.0) containing 100 μg/ml ampicillin and 12.5 μg/ml tetracycline at 37°C to an OD600 ~ 0.7. We put 100 μl from these bacteria into 3.5 cm NGM-agar plates containing 25 μg/ml carbenicillin and 1 mM IPTG pre-dried for 48 h at 21°C. Synchronized L1 larvae (prepared as described above) were put on the RNAi plates (~20 worms per plate) and grown until the young adult stage.
Phalloidin staining
We followed the protocol described in [Citation108] with some modifications. In brief, day 3 and day 11 worms were washed off the plates with M9 buffer, collected into a 1.7 ml microcentrifuge tube, and washed twice with the same buffer. After the last wash, the excess buffer was removed until ~10 μl remained, the worms were put in liquid nitrogen, and thawed on ice. Then, the worms were fixed by 5 min incubation in 100 μl methanol (pre-cooled to −20°C), brief centrifugation to remove the methanol (800 x g for 30 sec), and 5 min in 100 µL acetone (pre-cooled to −20°C) on ice. Then, 1 ml of blocking solution (PBS, 3% BSA, 1% Tween-20) was added, and the worms were sedimented to remove the acetone. Afterward, the worms were incubated blocking solution for 20 min. The worms were centrifuged, and the blocking solution was replaced with 100 μl rhodamine-phalloidin solution (750 μM in blocking solution). The worms were stained for 1 h, RT, in the dark under agitation. Afterward, the worms were washed three times with blocking solution, put on a 2% agarose pad (excess of liquid was absorbed with a Kimwipe), and cover with 10 µL antifade solution (E18-100/E18-18). Worms were imaged using an Olympus IX71S1F-3-5 inverted microscope equipped with UPlanFLN20X and Q-Imaging Rolera EM-C2™ camera, and neutral density filters (32ND50/32ND12/32ND6). Image analysis was performed with ImageJ [Citation109]. At least 30 animals were imaged for each condition, three biological repeats.
Off-food speed assay
Speed experiments were conducted as described previously [Citation106].
Low-peptone imaging plates
We prepared imaging plates (55 mm) contained 8 ml of low peptone NGM (0.3% NaCl, 2% Bacto agar, 0.013% Bacto Peptone, 0.01 mM CaCl2, 0.01 mM MgSO4, 25 mM Potassium Phosphate Buffer pH 6.0, 0.005 mg/ml cholesterol) and let them dry for ~18 h at RT. Prior to the experiment, we placed a copper ring (diameter 17 mm) at the center of the plate – we heated the ring with fire for a few seconds before inserting it so that the agar melted at the ring circumference. Thus, a seal was formed that prevented the worms from escaping.
Speed assay
Eight to ten day 3 and day 11 worms were picked into a 100 µl M9 buffer placed on a low-peptone plate and left for 30 min to digest remaining bacteria in their gut. During this time, the M9 buffer was replaced twice by removing most of the liquid with a pipette tip and adding the same volume of M9 buffer back. Then, the washed worms were transferred into a low-peptone imaging plate into the center of the copper. Excess of liquid was gently removed with a Kimwipe, and the worms were left for 5 min to acclimate. Then, worms’ movement was recorded for 10 min at a frame rate of 0.5 frame/sec using a Q-Imaging MicroPublisher 5.0 RTV Microscope Camera mounted onto an Olympus SZ61 stereomicroscope. Speed analysis was performed using custom-written MATLAB software [Citation110]. We analyzed the speed of at least 48 animals per treatment in six independent experiments.
Thrashing experiments
Day 5 and day 10 worms were washed off the plates with M9 buffer, collected into a 1.7-ml microcentrifuge tube, and washed twice with the same buffer. Then, we transferred a single worm into a 96-plate well containing 50 μl of M9 buffer, allowed to acclimatize for 1 min, and counted the number of body bends (thrashing) for 1 min. We analyzed the speed of at least 24 animals per treatment in four independent experiments.
Food intake measurements
Day 3 and day 11, worms were washed off the plates as described above and transferred into an NGM plate seeded with 10 µL OP50 bacteria. After 1 min of acclimatization, we counted the contractions of the posterior bulb of the pharynx over the course of 1 min. At least 30 worms were examined from each group in six independent experiments.
Paralysis assays
Paralysis assays were conducted as described in [Citation111] with some modifications. Synchronized worms (by egg-laying at 21°C) were put on plates containing compounds at different concentrations or vehicles (15 worms per plate). Worms were transferred to fresh plates every day until the post-fertile stage and then every 3–4 days until the end of the experiment. Worms that move their head in response to prodding with a wormpick, but did not show a full body wave propagation were scored as paralyzed. Worms that displayed internal progeny hatching (worm bagging), ruptured, burrowed in the agar, or crawled off the plates were censored.
Lifespan analysis
We performed lifespan experiments as described in [Citation106]. Synchronized L1 larvae (sodium hypochlorite method) were put on plates containing compounds at different concentrations or vehicles (20–25 worms per plate). Worms were transferred to fresh plates every day until the post-fertile stage and then every 2–3 days until the end of the experiment. Worms that did not display muscle contraction in response to prodding with a wormpick were scored as dead, while worms that showed bagging, ruptured, burrowed in the agar, or crawled off the plates were censored. However, they were included in the lifespan data analysis as censored animals (as described previously [Citation112]). All life span studies were performed at 21°C.
Oxygen consumption rate experiments
For the seahorse experiments described in , we followed the protocol described in [Citation58]. Notably, since SeaHorse XFe96 does not have a cooling system, the instrument room was temperature-controlled to 10°C using air conditioning. In this way, the plate temperature was ~21°C, and therefore suitable for C. elegans experiments.
For the acute OCR experiments (), we followed the procedure described in [Citation58] with the following changes:
OCR in the absence of food (): We subjected the worms to the following stimulus train: Basal OCR – 5 cycles of Mixing phase (5 min) – Resting phase (3 min) – Measuring phase (2 min). Then, we employ 9 cycles of the same mixing-resting-measuring protocol for VL-004/vehicle and FCCP treatments. Finally, we employ 4 cycles of the above same mixing-resting-measuring protocol for the azide treatment.
OCR in the presence of food (): We subjected the worms to the above stimulus train. However, these experiments were done in the presence of heat-killed OP50 bacteria (prepared as described in the Paraquat (PQ) resistance assay above); we added 25 μl bacteria into 175 μl M9 buffer (one 96 well volume).
ATP measurements
We followed the protocol described previously [Citation113] with modifications. Day 3 and day 11 worms were washed off the plates with M9 buffer, and collected into a 1.7 ml microcentrifuge tube containing 500 μl of ice-cold M9 buffer. Then, the worms were washed 3 times with an ice-cold M9 buffer (2 min, 900 x g). After the last wash, excess of buffer was removed until 500 μl remained. Then, the tube was transferred to a 95°C thermoblock for 15 min to inactivate ATPases and immediately frozen at – 80°C until use. Then, the worms were thawed on ice and crushed as described in [Citation105] using a Bullet Blender Homogenizer (Next Advance, Inc) (5 min at speed 9). Then, worms were further lysed using an ultrasonic bath (8 min, maximum magnitude) and centrifuged at 13,300 x g for 15 min. We then transferred the supernatant (S1) to a new tube and kept the pellet for total protein estimation. In addition, we kept 50 μl aside of S1 for protein estimation and used 100 μl of S1 to determine ATP content with a CellTiter-Glo® Luminescent kit (Promega, G7570) to measure ATP according to manufacturer instruction. Total protein estimation: We normalized the ATP content to worms’ protein content. Since many membrane/cuticle proteins cannot be extracted using M9 buffer, we used a strong detergent buffer to elute the remaining proteins from the worm-bead pellet. For this, we added 100 μl of SDS buffer (60 mM Tris pH6.8, 5% glycerol, 2% SDS) to the pellet and incubated it at 65°C for 10 min. Next, we punched a single hole in the bottom of the tube, inserted it into a new tube, and wrapped the connection point with parafilm. We centrifuged the tubes at 500 x g for 1 min and collected the flow (P1). We mixed 50 μl of each S1 and P1 and then estimated protein concentration using the BCA kit. We calculated ATP content per μg of protein.
Mitophagy imaging (C. elegans)
Colocalization
Worms treated with vehicle or compounds were washed off the plate with M9 buffer into a 1.7 ml microcentrifuge tube, washed twice more with the same buffer, and excess of buffer was removed to maximum without disturbing the worms. Then, we incubated the worms in 200 µl M9 buffer containing 0.1% Poloxamer 188, 0.1% Pluronic F127, 2 µl Lysosome/Mitochondria/Nuclear Staining Cytopainter Reagent (Abcam, ab139487) for 1 hr at RT with rotation (9 rpm). The staining solution was washed off three times and worms were placed on NGM plates supplemented with corresponding treatments (e.g., vehicle-treated worms were put in a vehicle containing plate, and VL-004 treated worms were put in a VL-004-plate, etc.) for 45 min at RT to reduce background staining. Worms were washed off the plates, cleaned with M9 buffer as described above, and fixed with 1% paraformaldehyde (PFA) on ice for 30 min. PFA was removed by three more washes with M9 buffer. Worms were mounted on 5% agarose pads and imaged with Nicon Yokogawa W1 Spinning Disk microscope fitted with 2 SCMOS ZYLA cameras. We used a 60X CFI Plan-Apochromat Lambda air objective to acquire the images.
Image analysis
We measured the colocalization of mitochondria and lysosome signals using the ImageJ Colocalization plugin (as described in [Citation114]) using the following parameters: Ratio = 75%, Threshold red channel = 80.0, Threshold green channel = 50.0. Output is a binary image containing colocalized puncta. We focused only on puncta in the head body wall muscle. If an image also had pharyngeal or nose stain, these regions were removed manually. Area of all puncta larger than 0.25 pixels was summed up.
Mito-Rosella
Worms expressing Mito-Rosella constructs were treated with vehicles and compounds as described in the Results section. To image the worms, we prepared 10% agarose pad in M9 and placed a 2 µl drop of Polybeads at its center. Up to 10 worms were transferred into the drop and covered with a coverslip. The worms were imaged using an Olympus IX71S1F-3-5 inverted microscope equipped with UAPON 60X (Olympus, Tokyo, Japan). Image analysis: Images were analyzed using ImageJ software.
(1) Neuronal mito-Rosella: Mitochondrial fluorescence was measured in the nerve cord, using the vulva as a reference point. To determine fluorescence intensity, we subtracted the background in both red and green channels; Process -> Subtract Background (we chose the radius of the rolling ball to be 50 pixels). Then, we used the auto threshold function to identify mitochondrial fluorescence in the red channel; Image -> Adjust -> Threshold (auto). Regions of mitochondrial-fluorescence were recorded using the region of interest manager (ROI); Analyze -> Analyze Particles (regions > 4 square pixels). ROI integrated densities, red and green channels, was measured.
(2) Body-wall-muscle mito-Rosella: We quantified the red/green signals only in the head BWM. Background subtraction, and thresholding were performed as described for the neuronal mito-Rosella. Then, we used the polygon selection tool to select mitochondria only in the head BWM, and generated a binary image mask; Edit – > Selection – > Create Mask. Then we looked for intersection of the Mask and tresholded red channel image by Process – > Image Calculator (“AND” operation). The resulted image was used to select mitochondria located in head muscle and add them to ROI manager; Analyze -> Analyze Particles (regions > 4 square pixels). The integrated densities of ROI in the red and the green channels were measured.
Autophagy imaging
Worms expressing intestinal mCherry-LGG-1 (the EVG414 strain [Citation37]) were treated with vehicle, or 4 mM VL-004 (as described above). As a positive control for robust mitophagy, we starved the worms. For this, we collected the worms as described for the mitochondria/lysosome “colocalization” experiments above. Then, we incubated the worms in 500 μl M9 buffer in 1.7 ml microcentrifuge tube on a rotator for 4 h. To measure autophagy, we transferred the worms (young adult stage) onto a 5% agarose pads supplemented with 0.7% phenoxyisopropanol, and imaged them with an Olympus IX71S1F-3-5 inverted microscope equipped with UAPON 40X (Olympus, Tokyo, Japan). Images were analyzed using ImageJ software. We measured the total area of puncta and normalized it to the area of the worm. To highlight puncta only, we first removed the background using Process-> Subtract Background. The rolling ball radius was set to 300 pixels. We thresholded the image until only the puncta area appeared on a resulting binary image. We measured the area of the puncta using Analyze -> Analyze particles. The resulting area was divided by the area of the worm being measured.
MitoSOX staining
Worms were prepared as described for the mitochondria/lysosome “colocalization” experiments above. Then, they were incubated in 200 µl of M9 supplemented with 0.1% Poloxamer 188, 0.1% Pluronic F127, 10 µM Mitochondrial Superoxide Indicator (Abcam, ab228567), 1 µM MitoTracker Green (ThermoFisher SCIENTIFIC, M7514) for 90 min at RT with rotation (9 rpm). The staining solution was washed off three times, and worms were placed on NGM plates supplemented with corresponding treatments for 45 min at RT. Worms were transferred onto a 5% agarose pads supplemented with 0.7% phenoxyisopropanol, and imaged with an Olympus IX71S1F-3-5 inverted microscope equipped with UAPON 20X (Olympus, Tokyo, Japan). Images were analyzed using ImageJ software. We subtracted the background signal of each channel and then calculated mean intensity in the head region only. MitoSOX signal was normalized to MitoTracker Green signal.
TMRE staining
Worms were prepared as described for the mitochondria/lysosome “colocalization” experiments above. Notably, in these experiments, we used SJ4103 worms that expressed mitochondrial-targeted GFP in their body wall muscles. Worms were incubated in 200 µl M9 buffer supplemented with 4 μM tetramethylrhodamine, ethyl ester (TMRE), 0.1% Poloxamer 188, 0.1% Pluronic F127, for 60 min at RT with rotation (9 rpm). Then the worms were washed with M9 three times and transferred to NGM plates supplemented with corresponding treatments for 45 min at 21°C. Worms were imaged as described in MitoSOX protocol above. Images were analyzed using ImageJ software. We subtracted the background signal of each channel and then calculated mean intensity in the head region only. MitoSOX signal was normalized to MitoTracker Green signal.
Molecular biology
Transgenes
In brief, we used modified polycistronic mCherry pPD95.75 expression vector [Citation115] to express mito-Rosella in neurons and BWM. Microinjections were performed as described previously [Citation116].
Prgef-1::hsp-6::mito-Rosella
mtRosella plasmid was generated by Gibson assembly cloning [Citation117]. rgef-1 promoter was amplified from with: 5’ CAAGAATGATCGTGCGGAAAGCATTTTTCGTCGTCGTCGTCGATGC 3’ and 5’ CCACGATGCCTGTAGCAATGGCAAC 3’. The Rosella sequence was amplified from pAS1NB m Rosella I (Plasmid #71247) with 5’ TAATAATAAAAAGAAGGATGATCAATCAACTGTTCCACGTGCTCGAGATCCACCTGTTGCTACAATGGCCTCCTCCGAGGAC 3’ and 5’ GGCGCTCAGTTGCAATTCTACTCATTATTTGTATAGTTCATCCATGC 3’. The DNA sequence encoding HSP-6 was amplified with 5’ CGGCATCGACGACGACGACGAAAAATGCTTTCCGCACGATCATTC 3’ and 5’ GACGTCCTCGGAGGAGGCCATTGTAGCAACAGGTGGATCTCGAGCACGTGGAACAGTTGATTGGTTTTGCTCCTTCTTTGGCTC 3’. Equal amount of all fragments (100 ng) were incubated for 4 h at 50°C with Gibson Assembly Mix (NEB). The construct was verified by sequencing using the primers listed in Table S2. The resulting plasmid was injected into N2 worms with rol-6(SU1006) co-injection marker at 2.5 ng/µl and 50 ng/µl, respectively.
Pmyo-3::Tom70::mito-Rosella transgene
The plasmid was generated by Gibson assembly cloning, as described above. Pmyo-3 promoter was amplified using 5’ GTC GAT CTC CGG AAG TGA TTA TAG TCT CTG TTT TCG TTA ATT TTG AAT TTT GC 3’ and 5’ GTCTTGTTCCTTGTAATGAAGCTCTTCATTTTTTTCTAGATGGATCTAGTGGTCGTGG 3’. The DNA sequence encoding the first 30 amino acids of the yeast Tom70 was amplified with 5’ CCACGACCACTAGATCCATCTAGAAAAAAATGAAGAGCTTCATTACAAGGAACAAGAC 3’ and 5’ ATGACGTCCTCGGAGGAGGCTGGTTGTAATAATAGTAGGCACCGATG 3’. The Rosella sequence was amplified from pAS1NB m Rosella I (Plasmid #71247) with 5’ GCCATCGGTGCCTACTATTATTACAACGCCTCCTCCGAGGACGTCAT 3’ and 5’ GGCGCTCAGTTGCAATTCTACTCATTATTTGTATAGTTCATCCATGC 3’. The pPD95.75 expression backbone fragments were amplified using primers dividing the ampicillin resistance region into two parts: 5’ CCACGATGCCTGTAGCAATGGCAAC 3’ and 5’ GTTGCCATTGCTACAGGCATCGTGG 3’. Gibson reactions were performed as described above. The construct was verified by sequencing. The resulting plasmid was injected into N2 worms with rol-6(SU1006) co-injection marker at 2.5 ng/µl and 25 ng/µl, respectively.
Pha-4 3’ UTR RNAi plasmid
To target the 3’ untranslated region of pha-4, we inserted the 3’ UTR pha-4 DNA sequence into the L4440 RNAi plasmid. The 3’ UTR region was amplified with 5’ GTG GGT TGG TAC CAT CTC CAA TTC ACA CGT TCT CCT TTT ATT GG 3’ and 5’ GTA CGA AGT CGA CCA AAT TCA ATA AAA TAT GAA AAA CTC AAA ACA AAA TAC C 3’ oligonucleotides, and inserted using KpnI and SalI. The resulted sequence was:
5’ ATC TCC AAT TCA CAC GTT CTC CTT TTA TTG GAA CAG GGC ATA GGA TAC TGC TGA TGC TAA TAC CGT GAA AAA AAT GTT CTA TGG ATG GAA AAT GAT TAT TTT TTG CGA TCC TCC TGC CAT TGG TTT CAA CCG GCG TGC TTG TGT CAC CTC ATC GTT GTT GTA AAT ACT GTT GAG AAT TTA TAT TCA TTC TTC GTC CCA TGT TCT CTT TTT TTA AAC TGA ATT TGA ATT TAT ACT GGT TCC CCT CTA CCA GAT GCC TTA ACT TTT ACC AGA AAT TGA TTT TTA TTC CTC CAT GTG TAA AAT CAA AAC TTT ACC GGT TCC GAA TTT AAT TCT GAA TGT TTT CTG CAA TAA GTA TAC ACC TAT CAA TAT CAA CAT AAT ATT CCC CAA TGT TTT TGA TGA ATT TAT TTT TTC CAC AAT AGA TAT GTT CCA TTT TTA TCC GCA GGG ACT TGA CTC CTC TTT TCC ATG TTT TCC AGG CTC CTT CTA CTT TAT GAC TTC CTA AAT GTC TTT GTA TCT AAC ACT ATT CAC TTC TTT CCA AAT TCT CTC CAT TTT CTC ATG GTA TTT TGT TTT GAG TTT TTC ATA TTT TAT TGA ATT TGT ATT TTC TTT ATT ATC GTT TTA AAT AAA AAA TT 3’.
The pha-4 (3’ UTR) RNAi vector was transformed HT115 (DE3) bacteria, and used a described above.
RNA purification and quantitative reverse transcription-quantitative PCR
RNA purification and RT-qPCR experiments were conducted as described in [Citation105] with some modifications. Worms (~2000 young adults per biological repeat per condition) grown on either 4 mM VL-004 or vehicle were washed off the plates with M9 buffer and collected into a 1.7 ml microcentrifuge as described above. After the final centrifugation, the M9 buffer was replaced with 200 μl of GeneZol (GeneAid, GZX200), 0.1 g of 0.5 mm zirconium oxide beads (Next Advance, ZROB05) was added into the tube and the tube was put at −80°C for ~16 h. Then, worms were lysed with Bullet Blender (NEXT ADVANCE; 2 min, speed 8, 3 min, speed 9). Total RNA was immediately isolated using TriRNA Pure Kit (GeneAid, GZXD200) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA with qScript™ cDNA Synthesis Kit (QuantaBio, 95,047). qPCR was performed using the iTaq™ Universal SYBR Green Supermix (Bio-Rad Laboratories Ltd, 1,725,124), CFX Connect TM Real-Time System (Bio-Rad), with the following program: Initial 3 min at 95°C followed by 39 cycles of 10s at 95°C and 30s at 60°C, and finally 10s at 95°C. At the end of each qPCR experiment, we analyzed the melting curve (carried out between 60°C to 95°C) to confirm the specificity of amplicons. Three technical replicates were performed for each gene. Gene expression was normalized to ned-8 and ubq-2, and analyzed using Bio-Rad CFX Manager 3.1 software. The primer sequences of the normalizing and experimental genes are shown in Table S2.
Mammalian cell culture
Cell viability assays
Methylene blue
HaCat cells were seeded in 96-well plates at a density of 10,000 cells per well, in 100 µl DMEM high glucose media with phenol red, 2% L-glutamine, 1% tetracycline, and 10% heat-inactivated FBS. The cells were incubated at 37°C, 5% CO2. After 24 h, when cells showed 50–60% confluence, we added the drugs and vehicle and incubated the cells under the same conditions for additional 24 h. We added H2O2 to a final concentration of 1 mM to induce oxidative injury and incubated the cells for 3 h under the same conditions. Then, the cells were washed three times with 100 µl PBS and supplemented with 100 µl of fresh medium. Importantly, this medium did not contain drug or vehicle and was identical to the medium we used to seed the cells. After 24-h incubation, the cells were fixed with 50 µl of 2.5% glutaraldehyde in PBS for 10–15 min at RT. Fixed cells were washed three times with DDW, and let dry for 24 h at RT. Then, the cells were incubated with 100 µl 1% methylene blue (dissolved in boric acid) for 1 h at RT. Cells were rinsed three times with water and dried overnight. Notably, it is essential to ensure that the cells are dried to minimize moisture variation among the samples. We added 100 µl 0.1 N HCl to each well, wrapped the plate with parafilm, and put it at 37°C for 1 hr to solubilize the methylene blue. We read the absorbance at 650 nm using a Tecan plate reader. Each measurement was performed in triplicate, with four biological repeats.
Cell recovery
These experiments were performed as described above with the following differences: (1) We used 6-wells plates. Therefore, the number of seeded HaCat cells was
3,00,000; (2) We used 1 mM CoCl2 to induce oxidative damage, for 16 h; (3) We determined cell density 48 h post CoCl2 treatment. In this period, the medium was replaced after 24 h post CoCl2.
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
We used the MTT assay to assess cell viability.
INS-1 cells
were seeded in 96-well plates (Falcon 96 well clear bottom) at a density of 50,000 cells per well in 100 µl RPMI media with phenol red, 2% L-glutamine, 1% P/N/S, 10% heat-inactivated FBS, 1% 1 M HEPES, 1% sodium pyruvate, and 50 μM 2-mercaptoethanol. The cells were incubated at 37°C, 5% CO2. After 24 h, when cells showed 50–60% confluence, we added the drugs and vehicle and incubated the cells under the same conditions for additional 24 h. Then, we added streptozotocin (0.5 mM final concentration) and incubated the cells under the same conditions for 19 h. Next, 20 μl of MTT (5 mg/ml in PBS) was added to each well, and the plate was incubated at 37°C, 5% CO2 for 3.5 h. To solubilize MTT crystals, the medium was carefully removed and replaced with 150 μl 4 mM HCl, 0.1% Nonidet P-40 in isopropanol. Plates were agitated on orbital shaker for 15 min at RT while protected from light. Absorbance was measured at 590 nm and 620 nm (as a reference). Each measurement was performed in triplicate, with seven biological repeats.
SH-SY5Y cells
MTT assays were performed as described above, except for the following differences: (1) We seeded 50,000 cells per well in DMEM high glucose media with phenol red, 2% L-glutamine, 1% tetracycline, and 10% heat-inactivated FBS for 48 h (2) To induce cellular senescence and oxidative stress, cells were incubated in 100 mM D-galactose (in DDW) for 19 h.
Mitophagy imaging (cells)
Colocalization
These experiments were performed as described for the C. elegans, with the following differences: (1) HaCat cells were seeded in 8 well ibidi µslide at a density of 34,000 cells per well, in 300 µl DMEM high glucose media without phenol red, 2% L-glutamine, 1% tetracycline, and 10% heat-inactivated FBS. The cells were incubated at 37°C, 5% CO2. After 24 h, when cells showed 50–60% confluence, we added the drugs and vehicle and incubated the cells under the same conditions for additional 6 h; notably, we used FCCP as a positive control (10 μM for 2 h). Then, we replaced the medium with 200 μl medium containing 0.4 μl Cytopainter Reagent (Abcam, ab139487) and incubated the cells for 30 min at 37°C, 5% CO2. The cells were washed three times with PBS, fixed with 3.7% of PFA for 15 min at RT, and washed again with PBS (three times). Cells were kept at 4°C and imaged, as described in the “Mitophagy imaging (C. elegans)” colocalization section above, within 24 h.
Cox8-EGFP-mCherry imaging
SHSY-5Y cells were plated in a 6-well tissue culture plate at a density of 1,000,000 cells per well in complete growth media (DMEM high glucose media, 2% L-glutamine, 10% FBS and 1% tetracycline) and incubated at 37°C for 24 h to 70% confluence. Then, the cells were transiently transfected with the cox8-mCherry-EGFP reporter [Citation88] (Addgene, 78,520; deposited by David Chan) according to the jetOPTIMUS (Polyplus, 117–07) instructions. After 48 h, the transfected cells were seeded into 8-well ibidi µslide (ibidi, 80,826; 50,000 cells per well). After 24 h, we added 125 µM VL-004, 10 μM FCCP, or vehicle as described above for the HaCat cell. Cells ere imaged them with an Olympus IX71S1F-3-5 inverted microscope equipped with UAPON 40X (Olympus, Tokyo, Japan) and neutral density filters (32ND50/32ND12/32ND6). At least 320 cells were imaged for each condition, with three biological repeats. Images were analyzed using ImageJ software.
siRNA transfection
SHSY-5Y cells were plated in a 6-well tissue culture plate at a density of 500,000 cells per well in complete growth media (DMEM high glucose media, 2% L-glutamine, 10% FBS and 1% tetracycline) and incubated at 37°C for 48 h to 40% confluence. PINK1 was depleted with a TriFECTa Dicer-Substrate RNAi kit (Integrated DNA Technologies, RNA-10025-PR) employing three PINK-1-specific Dicer-Substrate siRNA duplexes, described below.
Duplex 1–5′-AGUCACUUACAGAAAAUCCAAGAGA −3′ and 5′-UCUCUUGGAUUUUCUGUAAGUGACUGC −3′;
Duplex 2–5′- GUUCUGGACCAGCUACUGAAUUATT −3′ and 5′-AAUAAUUCAGUAGCUGGUCCAGAACGC −3′;
Duplex 3–5′-UAUCUGAUAGGGCAGUCCAUUGGTA −3′ and 5′-UACCAAUGGACUGCCCUAUCAGAUACU −3′.
In addition, a “universal” siRNA that targets a site absent from human, mouse, and rat genomes was used as a negative control [Citation86]. siRNA transfection was performed using INTERFERin (Polyplus, 101,000,028) following the manufacturers’ protocols. In brief, SH-SY5Y cells were incubated for 24 h with the siRNA transfection complexes (total concentration of 25 nM, i.e., 8.33 nM of each diRNA duplex). PINK1 depletion was confirmed by qPCR. Six independent experiments, each in triplicates were performed.
MTT assays were performed as described above (for the SH SY-5Y cells), except for the following differences: (1) We seeded 120,000 of siRNA transfected SHSY-5Y cells per 96 well; (2) Senescence was induced with 300 mM D-galactose.
Autophagy inhibition by bafilomycin A1
The autophagy-inhibition experiments in SH SY-5Y cells were performed as described above, with the following change. Bafilomycin A1 (70 nM; Cell Signaling Technology, 54645S) was added to the experimental wells 2 h prior to the VL-004 (or vehicle) treatment.
Statistical analysis
For comparison between two groups, we used the Mann-Whitney test. For comparison between more than two groups, when one parameter was explored (e.g., the effect of VL-004), we used an unpaired one-way ANOVA (Brown-Forsythe and Welch ANOVA tests with Welch’s correction). For comparison between more than two groups, when two parameters are explored (e.g., the effect of VL-004 in young and aged worms), we used Two-way ANOVA with Bonferroni/ Dunnett’s posttest. For lifespan and paralysis assays, we used the log-rank (Mantel-Cox) test. Data are presented as mean ± SEM (standard error of the mean) or as a Kaplan-Meier curve. All statistical analysis was performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com.
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Einav Gross ([email protected]).
Materials availability
There are restrictions to the availability of VL-001, VL-002, VL-003, VL-005, and VL-850 due to patent/licensing and production limitations. Any inquiries may be mailed to Einav Gross ([email protected]).
Supplemental Material
Download Zip (33.4 MB)Acknowledgments
We thank members of the Gross laboratory for critical reading of the manuscript, comments and advice. We thank the Japan National Bioresource Project for the Nematode C. elegans and the CGC, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440), for providing some of the strains. We thank N. Tavernarakis and E. Cohen for C. elegans and bacteria strains. This research was supported by a grant from Vitalunga Ltd, the Cleveland Clinic Center for Transformative Nanomedicine, and the ISRAEL SCIENCE FOUNDATION (grant No. 989/19). Graphical Abstract was generated with BioRender.com.
Disclosure statement
The following patent application (No. PCT/IL2019/050278) is related to this work.
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2078069.
Additional information
Funding
Related Research Data
References
- Kowald A, Kirkwood TBL. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J Theor Biol. 2000;202(2):145–160.
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521(7553):525–528. PubMed PMID: 25896323
- Palikaras K, Lionaki E, Tavernarakis N. Coupling mitogenesis and mitophagy for longevity.Autophagy.2015;11(8):1428–1430; Epub 2015/06/18. PubMed PMID: 26083448; PubMed Central PMCID: PMCPMC4590656.
- Onishi M, Yamano K, Sato M, et al.Molecular mechanisms and physiological functions of mitophagy.EMBO J.2021;40(3):e104705; Epub 2021/01/14. PubMed PMID: 33438778; PubMed Central PMCID: PMCPMC7849173.
- Yamashita S-I, Kanki T. How autophagy eats large mitochondria: autophagosome formation coupled with mitochondrial fragmentation. Autophagy. 2017;13(5):980–981.
- Diot A, Morten K, Poulton J. Mitophagy plays a central role in mitochondrial ageing.Mamm Genome.2016;27(7–8):381–395; Epub 2016/06/29. PubMed PMID: 27352213; PubMed Central PMCID: PMCPMC4935730.
- Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401–412. Epub 2019/02/12 PubMed PMID: 30742114.
- Liu J, Liu W, Li R, et al.Mitophagy in Parkinson’s disease: from pathogenesis to treatment.Cells.2019;8(7):712; Epub 2019/07/25. PubMed PMID: 31336937; PubMed Central PMCID: PMCPMC6678174.
- Clark EH, Vazquez de la Torre A, Hoshikawa T, et al. Targeting mitophagy in Parkinson’s disease. J Biol Chem. 2020;296:100209. Epub 2020/12/30. PubMed PMID: 33372898; PubMed Central PMCID: PMCPMC7948953.
- Drummond MJ, Addison O, Brunker L, et al. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J Gerontol A Biol Sci Med Sci. 2014;69(8):1040–1048. Epub 2014/02/15 PubMed PMID: 24526667; PubMed Central PMCID: PMCPMC4111292.
- Bellanti F, Lo Buglio A, Vendemiale G. Mitochondrial impairment in Sarcopenia. Biology (Basel). 2021;10(1). DOI:10.3390/biology10010031 Epub 2021/01/10 PubMed PMID: 33418869; PubMed Central PMCID: PMCPMC7825073.
- Qi Y, Qiu Q, Gu X, et al. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci Rep. 2016;6(1):24700.
- Yang X, Zhang M, Dai Y, et al. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging (Albany NY). 2020;12(17):16852–16866. Epub 2020/09/10 PubMed PMID: 32902411; PubMed Central PMCID: PMCPMC7521492.
- Palikaras K, Princz A, Tavernarakis N. Mitophagy modulators. In: Rattan SIS, editor. Encyclopedia of biomedical Gerontology. Oxford: Academic Press; 2020. p. 433–446.
- Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22(12):1428–1438.
- Schroeder S, Hofer SJ, Zimmermann A, et al. Dietary spermidine improves cognitive function. Cell Rep. 2021;35(2):108985.
- Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11(11):1305–1314.
- Minois N, Carmona-Gutierrez D, Bauer MA, et al. Spermidine promotes stress resistance in Drosophila melanogaster through autophagy-dependent and -independent pathways. Cell Death Dis. 2012;3(10):e401. Epub 2012/10/13 PubMed PMID: 23059820; PubMed Central PMCID: PMCPMC3481127.
- Igarashi K, Ueda S, Yoshida K, et al.Polyamines in renal failure.Amino Acids.2006;31(4):477–483; Epub 2006/03/24. PubMed PMID: 16554974.
- Pegg AE. Toxicity of polyamines and their metabolic products.Chem Res Toxicol.2013;26(12):1782–1800; Epub 2013/11/15. PubMed PMID: 24224555.
- Cochemé HM, Murphy MP. Complex I is the major site of mitochondrial superoxide production by Paraquat*. J Biol Chem. 2008;283(4):1786–1798.
- Yoneda T, Benedetti C, Urano F, et al.Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones.J Cell Sci.2004;117(Pt 18):4055–4066; Epub 2004/07/29. PubMed PMID: 15280428.
- Fang EF, Palikaras K, Sun N, et al. In vitro and in vivo detection of mitophagy in human cells, C. Elegans, and mice. J Vis Exp. 2017; 129. Epub 2017/12/30. 10.3791/56301. PubMed PMID: 29286376; PubMed Central PMCID: PMCPMC5755444.
- Palikaras K, Tavernarakis N. In vivo mitophagy monitoring in Caenorhabditis elegans to determine mitochondrial homeostasis. Biol Protoc. 2017;7(7). DOI:10.21769/BioProtoc.2215 Epub 2017/04/14 PubMed PMID: 28405595; PubMed Central PMCID: PMCPMC5386141.
- Koentjoro B, Park JS, Sue CM. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease.Sci Rep.2017;7(1):44373; Epub 2017/03/11. PubMed PMID: 28281653; PubMed Central PMCID: PMCPMC5345073.
- Nahapetyan H, Moulis M, Grousset E, et al. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death Dis. 2019;10(2):119.
- Lee SH, Du J, Stitham J, et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol Med. 2016;8(7):779–795. Epub 2016/05/26 PubMed PMID: 27221050; PubMed Central PMCID: PMCPMC4931291.
- Schaar CE, Dues DJ, Spielbauer KK, et al. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015;11(2):e1004972. Epub 2015/02/12 PubMed PMID: 25671321; PubMed Central PMCID: PMCPMC4335496.
- Head BP, Zulaika M, Ryazantsev S, et al.A novel mitochondrial outer membrane protein, MOMA-1, that affects cristae morphology in Caenorhabditis elegans.Mol Biol Cell.2011;22(6):831–841; Epub 2011/01/21. PubMed PMID: 21248201; PubMed Central PMCID: PMCPMC3057707.
- Cabreiro F, Au C, Leung KY, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153(1):228–239. Epub 2013/04/02 PubMed PMID: 23540700; PubMed Central PMCID: PMCPMC3898468
- Palmisano NJ, Meléndez A. Autophagy in C. elegans development. Dev Biol. 2019;447(1):103–125.
- Manil-Ségalen M, Lefebvre C, Jenzer C, et al. The C. elegans LC3 acts downstream of GABARAP to Degrade autophagosomes by interacting with the HOPS Subunit VPS39. Dev Cell. 2014;28(1):43–55.
- Kumsta C, Chang JT, Lee R, et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat Commun. 2019;10(1):5648.
- Konstantinidis G, Tavernarakis N. Molecular basis of neuronal autophagy in ageing: insights from Caenorhabditis elegans.Cells.2021;10(3):694; Epub 2021/04/04. PubMed PMID: 33800981; PubMed Central PMCID: PMCPMC8004021.
- Zhou X, Zhao X, Zhou W, et al. Impaired placental mitophagy and oxidative stress are associated with dysregulated BNIP3 in preeclampsia. Sci Rep. 2021;11(1):20469.
- Lizama-Manibusan B, McLaughlin B. Redox modification of proteins as essential mediators of CNS autophagy and mitophagy.FEBS Lett.2013;587(15):2291–2298; Epub 2013/06/19. PubMed PMID: 23773928; PubMed Central PMCID: PMCPMC5125300.
- Miedel MT, Graf NJ, Stephen KE, et al. A Pro-Cathepsin L mutant is a luminal substrate for endoplasmic-reticulum-associated degradation in C. elegans. PloS one. 2012;7(7):e40145.
- Lin K, Dorman JB, Rodan A, et al.daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans.Science.1997;278(5341):1319–1322; Epub 1997/11/21. PubMed PMID: 9360933.
- Lin K, Hsin H, Libina N, et al.Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling.Nat Genet.2001;28(2):139–145; Epub 2001/05/31. PubMed PMID: 11381260.
- Xu X, Kim SK, Copenhaver GP. The GATA transcription factor egl-27 delays aging by promoting stress resistance in Caenorhabditis elegans. PLoS Genet. 2012;8(12):e1003108.
- Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction.Annu Rev Biochem.2008;77(1):727–754; Epub 2008/04/01. PubMed PMID: 18373439.
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans.Proc Natl Acad Sci U S A.1998;95(22):13091–13096; Epub 1998/10/28. PubMed PMID: 9789046; PubMed Central PMCID: PMCPMC23719.
- Houthoofd K, Braeckman BP, Lenaerts I, et al. No reduction of metabolic rate in food restricted Caenorhabditis elegans. Exp Gerontol. 2002;37(12):1359–1369.
- Bar DZ, Charar C, Dorfman J, et al. Cell size and fat content of dietary-restricted Caenorhabditis elegans are regulated by ATX-2, an mTOR repressor. Proc Natl Acad Sci U S A. 2016;113(32):E4620–9. Epub 2016/07/28 PubMed PMID: 27457958; PubMed Central PMCID: PMCPMC4987808.
- Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. Epub 2008/02/19 PubMed PMID: 18281291; PubMed Central PMCID: PMCPMC2447655.
- Zhang Y, Shao Z, Zhai Z, et al. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009;4(7):e6348. PubMed PMID: 19633713; PubMed Central PMCID: PMC2711329.
- Jiang H, Guo R, Powell-Coffman JA. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia.Proc Natl Acad Sci U S A.2001;98(14):7916–7921; Epub 2001/06/28. PubMed PMID: 11427734; PubMed Central PMCID: PMCPMC35443.
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013;4(1):2267.
- Hansen M, Chandra A, Mitic LL, et al.A role for autophagy in the extension of lifespan by dietary restriction in C. elegans.PLoS Genet.2008;4(2):e24; Epub 2008/02/20. PubMed PMID: 18282106; PubMed Central PMCID: PMCPMC2242811.
- Nhan JD, Turner CD, Anderson SM, et al. Redirection of SKN-1 abates the negative metabolic outcomes of a perceived pathogen infection. Proc Natl Acad Sci U S A. 2019;116(44):22322–22330. Epub 2019/10/16 PubMed PMID: 31611372; PubMed Central PMCID: PMCPMC6825279.
- Tang L, Dodd W, Choe K. Isolation of a hypomorphic skn-1 Allele that does not require a balancer for maintenance.G3 (Bethesda).2015;6(3):551–558; Epub 2015/12/31. PubMed PMID: 26715089; PubMed Central PMCID: PMCPMC4777118.
- Inoue H, Hisamoto N, An JH, et al. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 2005;19(19):2278–2283. PubMed PMID: 16166371; PubMed Central PMCID: PMCPMC1240035.
- Chikka Madhusudana R, Anbalagan C, Dvorak K, et al. The mitochondria-regulated immune pathway activated in the C. elegans intestine is neuroprotective. Cell Rep. 2016;16(9):2399–2414.
- Yang W, Hekimi S, Tissenbaum HA. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8(12):e1000556. PubMed PMID: 21151885; PubMed Central PMCID: PMC2998438.
- Robinson KM, Janes MS, Pehar M, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103(41):15038–15043. PubMed PMID: WOS:000241247300012.
- Kwon YJ, Guha S, Tuluc F, et al. High-throughput BioSorter quantification of relative mitochondrial content and membrane potential in living Caenorhabditis elegans. Mitochondrion. 2018;40:42–50. Epub 2017/10/08. PubMed PMID: 28986305; PubMed Central PMCID: PMCPMC5858966.
- Presley AD, Fuller KM, Arriaga EA. MitoTracker Green labeling of mitochondrial proteins and their subsequent analysis by capillary electrophoresis with laser-induced fluorescence detection.J Chromatogr B Analyt Technol Biomed Life Sci.2003;793(1):141–150; Epub 2003/07/26. PubMed PMID: 12880861.
- Koopman M, Michels H, Dancy BM, et al. A screening-based platform for the assessment of cellular respiration in Caenorhabditis elegans. Nat Protoc. 2016;11(10):1798–1816.
- Kim H, Perentis RJ, Caldwell GA, et al. Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson’s models. Cell Death Dis. 2018;9(5):555.
- Panel M, Ghaleh B, Morin D. Mitochondria and aging: a role for the mitochondrial transition pore?Aging Cell.2018;17(4):e12793; Epub 2018/06/12. PubMed PMID: 29888494; PubMed Central PMCID: PMCPMC6052406.
- Velarde MC. Mitochondrial and sex steroid hormone crosstalk during aging.Longev Healthspan.2014;3(1):2; Epub 2014/02/06. PubMed PMID: 24495597; PubMed Central PMCID: PMCPMC3922316.
- Ikeda M, Ide T, Fujino T, et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PloS one. 2015;10(3):e0119687. Epub 2015/03/31 PubMed PMID: 25822152; PubMed Central PMCID: PMCPMC4379048.
- Barros MH, Bandy B, Tahara EB, et al.Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae.J Biol Chem.2004;279(48):49883–49888; Epub 2004/09/24. PubMed PMID: 15383542.
- Morcos M, Du X, Pfisterer F, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008;7(2):260–269. Epub 2008/01/29 PubMed PMID: 18221415.
- Caldeira da Silva CC, Cerqueira FM, Barbosa LF, et al. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7(4):552–560.
- Avery L, Horvitz HR. Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans.J Exp Zool.1990;253(3):263–270; Epub 1990/03/01. PubMed PMID: 2181052.
- Rodolfo C, Campello S, Cecconi F. Mitophagy in neurodegenerative diseases. Neurochem Int. 2018;117:156–166.
- Morley JF, Brignull HR, Weyers JJ, et al.The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans.Proc Natl Acad Sci U S A.2002;99(16):10417–10422; Epub 2002/07/18. PubMed PMID: 12122205; PubMed Central PMCID: PMCPMC124929.
- Machiela E, Dues DJ, Senchuk MM, et al. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol Dis. 2016;96:1–11.
- Gidalevitz T, Krupinski T, Garcia S, et al.Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity.PLoS Genet.2009;5(3):e1000399; Epub 2009/03/07. PubMed PMID: 19266020; PubMed Central PMCID: PMCPMC2642731.
- Jonsson PA, Ernhill K, Andersen PM, et al. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127(Pt 1):73–88. Epub 2003/10/10 PubMed PMID: 14534160.
- Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297. Epub 2012/12/26. PubMed PMID: 23267366; PubMed Central PMCID: PMCPMC3525948.
- Boukamp P, Petrussevska RT, Breitkreutz D, et al.Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line.J Cell Biol.1988;106(3):761–771; Epub 1988/03/01. PubMed PMID: 2450098.
- Pietrocola F, Lachkar S, Enot DP, et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015;22(3):509–516. Epub 2014/12/20 PubMed PMID: 25526088; PubMed Central PMCID: PMCPMC4326581.
- Karni R, Jove R, Levitzki A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene. 1999;18(33):4654–4662.
- Cui X, Zuo P, Zhang Q, et al. Chronic systemic D-galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: protective effects of R-alpha-lipoic acid. J Neurosci Res. 2006;84(3):647–654. Epub 2006/05/20 PubMed PMID: 16710848.
- Delwing-de Lima D, Hennrich SB, Delwing-Dal Magro D, et al. The effect of d-galactose induced oxidative stress on in vitro redox homeostasis in rat plasma and erythrocytes. Biomed Pharmacother. 2017;86:686–693.
- Liu YY, Nagpure BV, Wong PT, et al.Hydrogen sulfide protects SH-SY5Y neuronal cells against d-galactose induced cell injury by suppression of advanced glycation end products formation and oxidative stress.Neurochem Int.2013;62(5):603–609; Epub 2013/01/01. PubMed PMID: 23274001.
- Di Rita A, D’Acunzo P, Simula L, et al. AMBRA1-mediated mitophagy counteracts oxidative stress and apoptosis induced by neurotoxicity in human neuroblastoma SH-SY5Y cells. Front Cell Neurosci. 2018;12(92). DOI:10.3389/fncel.2018.00092
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays.J Immunol Methods.1983;65(1–2):55–63; Epub 1983/12/16. PubMed PMID: 6606682.
- Rahimi VB, Askari VR, Mousavi SH. Ellagic acid reveals promising anti-aging effects against d-galactose-induced aging on human neuroblastoma cell line, SH-SY5Y: a mechanistic study. Biomed Pharmacother. 2018;108:1712–1724. Epub 2018/10/31. PubMed PMID: 30372874.
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Fifth edition. ed. Oxford: Oxford University Press; 2015. p. xxxviii, 905 pages, 16 unnumbered pages of plates p.
- Nahdi AMTA, John A, Raza H. Elucidation of molecular mechanisms of streptozotocin-induced oxidative stress, apoptosis, and mitochondrial dysfunction in Rin-5F Pancreatic β-Cells. Oxid Med Cell Longev. 2017;2017:7054272.
- Chen H, Li X, Epstein PN. MnSOD and Catalase Transgenes Demonstrate That Protection of Islets From Oxidative Stress Does Not Alter Cytokine Toxicity. Diabetes. 2005;54(5):1437–1446.
- Hohmeier HE, Mulder H, Chen G, et al.Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion.Diabetes.2000;49(3):424–430; Epub 2000/06/27. PubMed PMID: 10868964.
- Rose SD, Collingwood MA, Behlke MA. Optimizing knockdown of gene expression using the TriFECTa™ Dicer-substrate RNAi reagent system. Nat Methods. 2006;3(9):V–VII.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy. 2021;17(1):1–382. Epub 2021/02/27 PubMed PMID: 33634751; PubMed Central PMCID: PMCPMC7996087.
- Rojansky R, Cha M-Y, Chan DC. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife. 2016;5:e17896.
- Yuan Y, Hilliard G, Ferguson T, et al.Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha.J Biol Chem.2003;278(18):15911–15916; Epub 2003/02/28. PubMed PMID: 12606543.
- Cervellati F, Cervellati C, Romani A, et al. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free Radic Res. 2014;48(3):303–312.
- Rider JE, Hacker A, Mackintosh CA, et al.Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide.Amino Acids.2007;33(2):231–240; Epub 2007/03/31. PubMed PMID: 17396215.
- Madeo F, Eisenberg T, Pietrocola F, et al. Spermidine in health and disease. Science. 2018;359(6374). DOI:10.1126/science.aan2788 Epub 2018/01/27 PubMed PMID: 29371440
- Zhang William B, Sinha Drew B, Pittman William E, et al. Extended Twilight among Isogenic C. elegans Causes a Disproportionate Scaling between Lifespan and Health. Cell Syst. 2016;3(4):333–45.e4.
- Tan S, Wong E. Mitophagy Transcriptome: mechanistic Insights into Polyphenol-Mediated Mitophagy. Oxid Med Cell Longev. 2017;2017:9028435.
- Papa L, Germain D. SirT3 regulates the mitochondrial unfolded protein response.Mol Cell Biol.2014;34(4):699–710; Epub 2013/12/11. PubMed PMID: 24324009; PubMed Central PMCID: PMCPMC3911493.
- Audesse AJ, Dhakal S, Hassell LA, et al.FOXO3 directly regulates an autophagy network to functionally regulate proteostasis in adult neural stem cells.PLoS Genet.2019;15(4):e1008097; Epub 2019/04/12. PubMed PMID: 30973875; PubMed Central PMCID: PMCPMC6478346.
- Koh H, Kim H, Kim MJ, et al.Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN-induced kinase 1 (PINK1) null mutant.J Biol Chem.2012;287(16):12750–12758; Epub 2012/03/02. PubMed PMID: 22378780; PubMed Central PMCID: PMCPMC3339960.
- Mei Y, Zhang Y, Yamamoto K, et al.FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation.Proc Natl Acad Sci U S A.2009;106(13):5153–5158; Epub 2009/03/12. PubMed PMID: 19276113; PubMed Central PMCID: PMCPMC2654023.
- Wang S, Chen Y, Li X, et al. Emerging role of transcription factor EB in mitochondrial quality control. Biomed Pharmacother. 2020;128:110272.
- Lin -X-X, Sen I, Janssens GE, et al. DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat Commun. 2018;9(1):4400.
- Shen JL, Fortier TM, Wang R, et al. Vps13D functions in a Pink1-dependent and Parkin-independent mitophagy pathway. J Cell Biol. 2021;220(11). DOI:10.1083/jcb.202104073 Epub 2021/08/31 PubMed PMID: 34459871; PubMed Central PMCID: PMCPMC8406608.
- Igarashi R, Yamashita S-I, Yamashita T, et al. Gemcitabine induces Parkin-independent mitophagy through mitochondrial-resident E3 ligase MUL1-mediated stabilization of PINK1. Sci Rep. 2020;10(1):1465.
- Livshits L, Gross E. A method for measuring sulfide toxicity in the nematode Caenorhabditis elegans. MethodsX. 2017;4:250–255.
- Asfari M, Janjic D, Meda P, et al. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130(1):167–178.
- Romero-Afrima L, Zelmanovich V, Abergel Z, et al. Ferritin is regulated by a neuro-intestinal axis in the nematode Caenorhabditis elegans. UNSP 101359. 10.1016/j.redox.2019.101359. PubMed PMID: WOS:000501490700032 Redox Biol. 2020;28.
- Abergel R, Livshits L, Shaked M, et al.Synergism between soluble guanylate cyclase signaling and neuropeptides extends lifespan in the nematode Caenorhabditis elegans.Aging Cell.2017;16(2):401–413; Epub 2017/01/06. PubMed PMID: 28054425; PubMed Central PMCID: PMCPMC5334569.
- Livshits L, Chatterjee AK, Karbian N, et al. Mechanisms of defense against products of cysteine catabolism in the nematode Caenorhabditis elegans. Free Radic Biol Med. 2017;104:346–359. Epub 2017/02/10. PubMed PMID: 28179109.
- Mattout A, Pike Brietta L, Towbin Benjamin D, et al. An EDMD Mutation in C. elegans Lamin Blocks Muscle-Specific Gene Relocation and Compromises Muscle Integrity. Curr Biol. 2011;21(19):1603–1614.
- Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics Int. 2004;11(7):36–42.
- Abergel Z, Chatterjee AK, Zuckerman B, et al. Regulation of Neuronal Oxygen Responses in C. elegans Is Mediated through Interactions between Globin 5 and the H-NOX Domains of Soluble Guanylate Cyclases. J Neurosci. 2016;36(3):963–978. PubMed PMID: 26791224; PubMed Central PMCID: PMC4719025.
- Cohen E, Bieschke J, Perciavalle RM, et al.Opposing activities protect against age-onset proteotoxicity.Science.2006;313(5793):1604–1610; Epub 2006/08/12. PubMed PMID: 16902091.
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20(23):2131–2136. PubMed PMID: 21093262; PubMed Central PMCID: PMC3058811.
- Palikaras K, Tavernarakis N. Intracellular Assessment of ATP Levels in Caenorhabditis elegans. Biol Protoc. 2016;6(23). DOI:10.21769/BioProtoc.2048 Epub 2017/02/15 PubMed PMID: 28194429; PubMed Central PMCID: PMCPMC5303341
- Runwal G, Stamatakou E, Siddiqi FH, et al.LC3-positive structures are prominent in autophagy-deficient cells.Sci Rep.2019;9(1):10147; Epub 2019/07/14. PubMed PMID: 31300716; PubMed Central PMCID: PMCPMC6625982.
- Gross E, Soltesz Z, Oda S, et al. GLOBIN-5-Dependent O2 Responses Are Regulated by PDL-1/PrBP That Targets Prenylated Soluble Guanylate Cyclases to Dendritic Endings. J Neurosci. 2014;34(50):16726–16738. PubMed PMID: 25505325; PubMed Central PMCID: PMC4261097.
- Mello CC, Kramer JM, Stinchcomb D, et al. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10(12):3959–3970. PubMed PMID: 1935914; PubMed Central PMCID: PMC453137.
- Gibson DG, Young L, Chuang R-Y, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345.