
ABSTRACT
Mitophagy is a key intracellular process that selectively removes damaged mitochondria to prevent their accumulation that can cause neuronal degeneration. During mitophagy, PINK1 (PTEN induced kinase 1), a serine/threonine kinase, works with PRKN/parkin, an E3 ubiquitin ligase, to target damaged mitochondria to the lysosome for degradation. Mutations in the PINK1 and PRKN genes cause early-onset Parkinson disease that is also associated with mitochondrial dysfunction. There are a large number of reports indicating the critical role of PINK1 in mitophagy. However, most of these findings were obtained from in vitro experiments with exogenous PINK1 expression and acute damage of mitochondria by toxins. Recent studies using novel animal models suggest that PINK1-PRKN can also function independent of mitochondria. In this review, we highlight the major differences between in vitro and in vivo models for investigating PINK1 and discuss the potential mechanisms underlying these differences with the aim of understanding how PINK1 functions under different circumstances.Abbreviations: AAV: adeno-associated viruses;AD: Alzheimer disease; CCCP: carbonyl cyanidem-chlorophenyl hydrazone; HD: Huntington disease; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MTS: mitochondrial targeting sequence; PD: Parkinson diseases; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; ROS: reactive oxygen species; UIM, ubiquitin interacting motif.
Introduction
Mitochondria perform a diverse range of critical cellular functions, including producing energy, buffering cytosolic calcium flux, synthesizing lipids, and participating in apoptosis-programmed cell death [Citation1]. Since mitochondria are involved in energy production through oxidative phosphorylation, cellular redox balances, and apoptosis [Citation1], mitochondrial dysfunction can result in increased oxidative stress, reduced ATP production, and decreased cell survival, therefore playing a vital role in many neurodegenerative disorders. In addition, mitochondria are prone to damage, and this damage results in the release of toxic levels of reactive oxygen species (ROS) to cause cell death. To combat this, there are a number of quality control systems to repair damaged mitochondria and to protect the overall integrity of the mitochondria network. However, when mitochondria damage is too severe to repair, mitochondria can be selectively degraded via the autophagy pathway, a process referred to as mitophagy [Citation2].
Mitochondria dysfunction, including impaired fission/fusion, altered transport and morphology, and abnormal mitochondrial protein levels and activity, is a common feature of many neurodegenerative disorders such as Parkinson (PD), Alzheimer (AD) and Huntington (HD) diseases [Citation3–6]. Mutations in two PD related genes have been reported to play key roles in mitochondria quality control: PINK1, encoding a serine/threonine kinase; and PRKN/parkin, encoding a cytosolic ubiquitin ligase. A number of in vitro studies have demonstrated that PINK1 works together with PRKN to remove unhealthy mitochondria by lysosomes through the mitophagy process. These studies have expanded the roles of mitochondrial dysfunction and mitophagy in PD and provided a wealth of information about how PINK1-PRKN pathway regulates mitochondria homeostasis.
Although there is wide acceptance for the role of PINK1-PRKN-mediated mitophagy, it is notable that PINK1-PRKN independent mitophagy also plays significant physiological roles and involves various receptors such as cardiolipin, a phospholipid that is protective against neural apoptosis [Citation7,Citation8]. It is also important to recognize the limitations of the studies that support the current prevailing model for PINK1-PRKN-mediated mitophagy. In particular, much of the work that supports this model relied on the use of mitochondria toxins, overexpressed PINK1 or PRKN, and immortalized cell lines that do not exactly mimic the physiological or pathological conditions in vivo. There is still lack of in vivo evidence for PINK1-PRKN-mediated mitophagy in animal models. Furthermore, non-mitochondrial dependent functions of PINK1 and PRKN have also been reported, but whether the mitochondria-independent function of these two proteins is related to PD pathogenesis or other pathological conditions remains elusive. Thus, in this review, we will discuss the differences in PINK1-PRKN-mediated mitophagy between in vitro and in vivo models, aiming at elucidating how PINK1 and PRKN function in vivo and how their dysfunction is involved in PD and other physiological or pathological conditions.
Association of PD with mitochondrial dysfunction
Parkinson disease (PD) is the second most common neurodegenerative disorder that affects more than 1% people over age 60 [Citation9]. Clinical signs of PD include tremor, bradykinesia, postural instability, and rigidity. Pathologically, PD is characterized by age-dependent and progressive loss of dopamine neurons in the substantia nigra and accumulation of α-synuclein positive inclusions (Lewy bodies) [Citation10]. The idea that mitochondrial dysfunction may be involved in the selective neurodegeneration of PD was first suggested by the discovery of toxic effects of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). Accidental exposure to MPTP was found to cause parkinsonism and DA neurodegeneration [Citation11]. Follow-up studies revealed that MPTP is metabolized to MPP+, which is a mitochondrial complex I inhibitor and is selectively taken up into DA neurons [Citation12–14]. In addition to MPTP, other mitochondrial toxins (such as pesticide rotenone and Paraquat) have been shown to cause or correlate with increased risk of PD [Citation15–17]. Importantly, postmortem PD patient brain tissues display well-documented evidence of mitochondrial dysfunction including complex I deficiency [Citation18,Citation19], bioenergetics defects, age-dependent accumulation of mtDNA deletions [Citation20,Citation21], and dysregulation in the expression of various mitochondrial proteins [Citation22]. The strong link between mitochondrial dysfunction and PD indicates that DA neurons is sensitive to mitochondrial dysfunction and has stimulated interest in studying the association of mitochondrial dysfunction with genetic mutations that also cause PD.
Although the majority of PD is sporadic, specific genetic mutations have been reported in rare familial cases. Among the dozens of genes responsible for familial PD, PINK1 and PRKN are two most extensively characterized genes whose mutations were found to result in early-onset (before the age of 45) and autosomal recessive PD that is also featured by neurodegeneration in association with mitochondria dysfunction [Citation23–25]. PINK1 is a mitochondrial kinase encoding a 581-aa protein with an N-terminal mitochondrial targeting sequence (MTS) followed by a putative transmembrane domain and a serine/threonine kinase domain. On the other hand, PRKN is a 465-aa E3 ubiquitin ligase that contains an N-terminal ubiquitin-like (UBL) domain and C-terminal double Ring finger motifs connected by an IBR domain [Citation26]. Drosophila genetic studies have revealed that PINK1 and PRKN function in the same biological pathway and that PRKN acts downstream of PINK1 [Citation27–29]. Further biochemical analysis of PINK1-PRKN uncovers their function in detecting mitochondrial damage and recruiting ubiquitin machinery to remove dysfunctional mitochondria via lysosomes, establishing the prevalent theory that PINK1-PRKN work together in the same pathway to protect mitochondria.
In vitro study of the PINK1-PRKN pathway and mitophagy
Mitophagy is the selective degradation of damaged mitochondria by targeting them to the lysosomes, a critical intracellular process for maintaining mitochondria homeostasis and preventing neuronal death. Mounting evidence from biochemical and in vitro studies demonstrated that PINK1 and PRKN work together to induce mitophagy, as both proteins target damaged mitochondria to the lysosomes for clearance of the unhealthy mitochondria [Citation24]. Full-length PINK1 is a 581-aa Ser/Thr kinase consisting of a N-terminal mitochondria/matrix-targeting signal (1–34 aa), a putative transmembrane (TM) domain (85–110 aa), a ser/ther kinase domain (156–511 aa) and a C-terminal extension (512–581 aa, highly conserved) [Citation24]. In vitro studies of cell lines revealed that endogenous PINK1 is synthesized constitutively in the cytosol as a full-length precursor (~63-68 kDa). In healthy cells, the full-length 63-kDa PINK1 is transferred across the outer mitochondrial membrane and then cleaved at A103 and F104 to a 52-kDa isoform by inner mitochondrial membrane-bound proteases. The N-terminal cleaved 52-kDa isoform of PINK1 is then released into cytosol and degraded via ubiquitin-proteasome system [Citation24,Citation30,Citation31]. As such, endogenous PINK1 is undetectable in cells with physiologically polarized mitochondria. However, when mitochondria are damaged, the full-length PINK1 is accumulated and stabilized on the mitochondrial membrane to phosphorylate PRKN (an E3 ubiquitin-ligase) and ubiquitin, leading to activation of mitochondrial quality control pathways [Citation32]. During this process, PINK1 acts as a key sensor of mitochondrial damage.
PRKN, a cytosolic E3 Ub ligase, acts downstream of PINK1 when its S65 residue is phosphorylated by PINK1 [Citation33]. The UBL domain of PRKN also plays a special role in PRKN activation, which involves substrate recognition, recruiting and binding to SH3 or ubiquitin interacting motif (UIM) domains, proteasome association, and regulation of cellular PRKN levels and activity [Citation34]. Structural and biochemical analysis of PRKN showed that PRKN exists in a compact and autoinhibited conformation in the basal, unstimulated state, where the UBL domain exerts an inhibitory effect on both E2 binding and transthiolation [Citation35–39]. Upon being phosphorylated and activated by PINK1 at Ser65, PRKN modifies various outer mitochondrial membrane proteins, predominantly with K11- and K63-linked UB chains [Citation40], and thus increases the pool of phospho-ubiquitin to recruit autophagy receptors such as SQSTM1/p62, OPTN, and MAP1LC3/LC3 [Citation41–44] to damaged mitochondria, resulting in the removal of damaged mitochondria via mitophagy. It has been extensively shown that PRKN ubiquitinates a wide variety of cytosolic and outer mitochondrial membrane proteins upon mitochondrial depolarization [Citation45,Citation46].
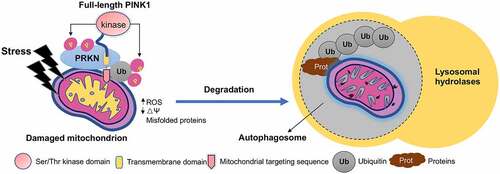
In numerous cell lines and mouse embryonic fibroblasts, exposure to mitochondrial toxins such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP), antimycin A, and rotenone, triggers the accumulation of PINK1 on mitochondria and then signals PRKN and ubiquitin by phosphorylation to ubiquitinate the damaged mitochondrial proteins for removing the damaged mitochondria by the lysosomes [Citation24,Citation43,Citation47–50]. Analysis of the crystal structure of insect PINK1 bound to ubiquitin further provides a structural base for the interactions of PINK1 with PRKN and ubiquitin [Citation51,Citation52]. All together, these findings strongly indicate that PINK1 confers protection against mitochondrial dysfunction-dependent cell death induced by both intrinsic stress and environmental insults [Citation53–56] ().
Figure 1. In vitro studies of PINK1-PRKN-mediated mitophagy. In cultured cells stressed by mitochondrial toxin CCCP, full-length PINK1 is targeted to damaged mitochondria. PINK1 then phosphorylates PRKN and ubiquitin to form a complex that add polyubiquitination chains to mitochondrial proteins, followed by degradation by lysosomes. The ubiquitinated proteins could also undergo degradation by the proteasome.
Despite convincing in vitro studies indicating the critical role of PINK1 in mitophagy, it seems that the regulation of mitophagy by PINK1 is also cell type dependent. The study of cultured mouse cortical neurons with PRKN overexpression revealed that PRKN recruitment and translocation to depolarized mitochondria occurred only after 12 h of treatment with mitochondrial toxins, suggesting that PINK1-PRKN-mediated mitophagy in neurons is a quite slower process than that in non-neuronal cells [Citation57]. Different types of neurons show variability in the intracellular environment and mitochondria stresses, which may complicate the study of PINK1-PRKN-mediated mitophagy in neurons. Immortalized cells typically rely on glycolysis and could potentially tolerate substantial loss of mitochondria, whereas neurons rely primarily on oxidative phosphorylation for energy production [Citation58–60], implying that additional mechanisms regulate mitophagy in neurons. Beyond mitophagy, overexpression of PINK1 or PRKN in rat hippocampal axons decreases mitochondrial movement, suggesting that PINK1-PRKN also mediate mitochondrial motility in cultured neurons [Citation54].
In vivo investigation of PINK1-PRKN-associated mitochondrial function
Although current evidence from cultured cells does provide strong support for PINK1-PRKN-mediated mitophagy, it is also important to recognize the limitations of the in vitro work that relies on PRKN or PINK1 overexpression and acute mitochondrial injury. In particular, there is a pressing need for obtaining in vivo evidence to support the role of PINK1-PRKN-mediated mitophagy. To this end, a variety of animal models have been established to investigate the in vivo function of PINK1 and PRKN (). The first in vivo evidence indicating that PINK1 and PRKN could work together to regulate mitochondrial quality control came from genetic complementation studies in Drosophila. In 2003, Drosophila genetic studies revealed that prkn null mutant flies exhibit locomotor defects, muscle degeneration, and mitochondrial dysfunction, thereby providing in vivo fact for involvement of PRKN in mitochondrial quality control [Citation27]. Then in 2006, other genetic studies further revealed that Pink1 null mutant flies also showed apoptotic muscle degeneration, mitochondrial defects, and male sterility as seen in park mutant flies [Citation28,Citation29]. Notably, park overexpression rescues male sterility and mitochondrial defects in Pink1 mutant flies, but overexpression of Pink1 does not rescue the phenotypes of park mutants, suggesting that Pink1 and park function in the same pathway and that park acts downstream of Pink1. Furthermore, overexpression of Drp1 (Dynamin related protein 1) or inhibition of Marf/MFN, which is involved in mitochondrial fusion-fission dynamics, also recovered mitochondrial defects and muscle degeneration in the park and Pink1 mutant flies [Citation61–63]. These findings indicated that in Drosophila Pink1-park maintain mitochondrial integrity when dealing with energetically demanding tissues.
Table 1. PINK1 or PRKN knockout animal models.
The in vitro studies and fly model investigation have made incredible progress in understanding the roles of PINK1-PRKN and mitophagy, but a major challenge remains in the field is to accurately and faithfully detect PINK1-PRKN-mediated mitophagy in the mammalian brain. A number of pink1 prkn knockout mouse models have been established for investigating the effects of PINK1-PRKN on mitochondria and neuronal survival. However, these pink1 prkn knockout mouse models fail to replicate selective and overt neurodegeneration seen in PD [Citation66]. Even 2-year-old triple knockout mice that lack Pink1, Prkn and Park7/DJ-1, which are three main genes causative of early onset PD, do not show obvious neurodegeneration [Citation67]. Absence of neuronal loss in these knockout mice could be due to the existence of compensatory mechanisms that are able to cope with PINK1-PRKN-PARK7/DJ-1 deficiency in mouse models or the life span of mouse that is too short to reach a threshold of damage sufficient to induce an overt phenotype.
To address the above possibilities, conditional prkn knockout mice, which were established via AAV-mediated Cre-loxp induction, showed DA neuronal loss in aged mice [Citation68]. However, a recent study reported that AAV-mediated Cre expression would induce a massive decrease in neuronal populations of substantia nigra [Citation69]. It seems that additional stress and insults need to be added to induce neurodegeneration in pink1-prkn knockout mice. For example, crossing the POLG (mtDNA mutator) mouse with prkn knockouts produced offspring with selective degeneration of dopaminergic neurons and locomotor deficits, though loss of PRKN did not affect overall levels of mtDNA somatic mutations [Citation70]. Furthermore, dopaminergic neuronal loss was observed in pink1 knockout mice in the presence of α-synuclein or MPTP induced toxicity [Citation71,Citation72]. Intestinal bacterial pathogens were also recently found to promote neurodegeneration in pink1 knockout mice [Citation73]. Pink1 KO rats were reported to have age-dependent loss of TH (tyrosine hydroxylase)-positive neurons [Citation74], but this mild degeneration could not be confirmed by other studies [Citation75]. These findings indicate that the rodent brains are more resistant to pink1 mutations when compared to the human brain since a patient carrying homozygous point mutations in either the PINK1 or PRKN gene would suffer neurodegeneration.
The pink1 or prkn KO mouse models are also unable to show striking defects in mitophagy. Two reporter systems, mito-QC and mt-Keima, have contributed to our understanding of physiological mitophagy. Recent studies using Mito-QC mouse and Drosophila models demonstrated that basal mitophagy activity is not affected by the loss of PINK1 [Citation76,Citation77]. Although mt-Keima is more sensitive than mito-QC to detect PINK1-PRKN-mediated mitophagy in mice after exhaustive exercise, the basal level of mitophagy is independent of PINK1 in the absence of exhaustive exercise [Citation78]. Furthermore, using both Keima- and tandem mCherry-GFP-based reporters in zebrafish revealed that disruption of pink1 or prkn had no effect on mitophagy [Citation79]. In Drosophila, however, PINK1 was found to be essential for the induction of mitophagy in response to hypoxic exposure and rotenone treatment [Citation80] and to be required for an age-dependent increase in mitophagy [Citation81]. It is possible that PINK1-PRKN-mediated mitophagy is species dependent and occurs under pathological conditions. A knockin mouse model, in which PRKN cannot be phosphorylated by PINK1, also showed no clear neurodegeneration or nigrostriatal mitophagy impairment [Citation82]. In addition, mass spectrometry analysis of pink1 knockout rodents did not show significant alterations in the expression levels of mitochondrial proteins [Citation83], and inconsistent or mild alterations of mitochondrial function were found among the PINK1 KO animal models [Citation84,Citation85]. All these raise an important issue of whether PINK1 acts differentially in vitro and in vivo to regulate mitochondrial function.
Lack of neuronal loss and obvious alterations in mitophagy in pink1 knockout mice has motivated researchers to utilize large animals to create PD models. Recent development of CRISPR-Cas9 allowed for disrupting the genes of PINK1-PRKN in large animals for investigating the consequences of loss of function. Interestingly, CRISPR/Cas9-mediated PINK1-PRKN deletion in pigs did not produce any obvious neurodegeneration and severe phenotypes [Citation86,Citation87], like pink1 or prkn KO mouse models [Citation53,Citation67,Citation88]. These findings suggest that PINK1-PRKN deficiency may induce species-dependent neuropathology and underscore the importance in using non-human primates to explore the function of PINK1 and PRKN [Citation89].
Recently, a few groups have used CRISPR-Cas9 to target the monkey PINK1 gene to generate monkey models of PD [Citation90–93]. When the monkey PINK1 exon 2 and exon 4 were targeted by two gRNAs in embryos, a large PINK1 DNA fragment was deleted, resulting in almost complete elimination of PINK1 expression in the monkey brain [Citation90]. The newborn monkeys showed severe neurodegeneration or died postnatally, demonstrating for the first time that PINK1 is essential for neuronal survive in the developing primate brain [Citation90]. Furthermore, disruption of PINK1 gene via AAV-mediated CRISPR/Cas9 in adult monkey brain also led to severe neurodegeneration and motor deficits of monkeys [Citation93]. It is worth noting that the homozygous deletion of a large region of PINK1 gene has not been found in humans, perhaps because such deletion is embryonic lethal in humans [Citation94]. Interestingly, using the paired gRNA-Cas9-D10A nickases to disrupt the monkey PINK1 gene in the fertilized monkey oocytes did not model PD phenotypes in the live young monkeys [Citation91]. On the other hand, co-editing PINK1 and PARK7/DJ-1 genes via AAV-delivered CRISPR-Cas9 system in adult monkey substantia nigra could mediate severe nigra dopaminergic cell loss and motor function deficits [Citation92]. It seems likely that the phenotypes of PINK1-targeted monkeys rely on the extent to which PINK1 is depleted.
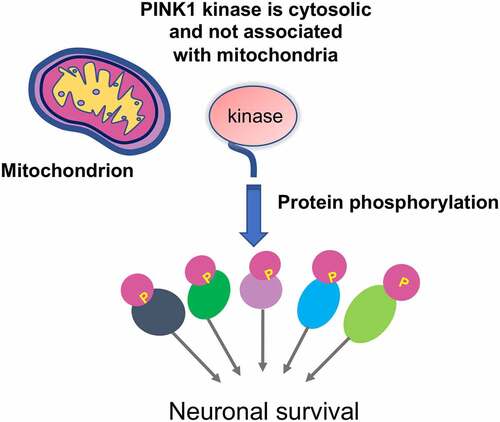
The severe neuronal loss in PINK1 mutant monkey brain is in clear contrast to the absence of neurodegeneration in mouse models that have completely deleted the Pink1 gene, suggesting that PINK1ʹs function is at least species-dependent. The PINK1 mutant monkey model also allowed for studies of the effect of PINK1 on mitochondria. Interestingly, although PINK1 deficiency could cause severe neuronal loss, there was no obvious differences in mitochondrial protein expression, morphology, and metabolic activities between wild type and PINK1-targed monkey brains [Citation93]. Instead, loss of PINK1 caused marked reduction of phosphorylation of proteins that are important for neuronal survival, which is also supported by the selective expression of PINK1 kinase form (55-kDa) that lacks N-terminal mitochondria targeting domain in the monkey brain [Citation93]. In support of the notion that PINK1 exists as a cytoplasmic protein in the primate brain, fractionation analysis indicated that the majority of the monkey PINK1 is cytosolic [Citation93]. Consistently, most of substrates phosphorylated by PINK1 are also cytoplasmic proteins [Citation93]. Thus, PINK1 deficiency triggers neurodegeneration in the developing monkey brain by affecting protein phosphorylation without impacting mitochondrial homeostasis ().
Figure 2. In vivo studies of PINK1. In the primate brain, PINK1 kinase (55-kDa) is stably expressed in the cytoplasm and is not associated with mitochondria. It phosphorylates a large number of proteins to maintain neuronal survival.
PINK1 expression levels account for different results from in vitro and in vivo models
Understanding why PINK1 acts differentially in vitro and in vivo, especially on mitophagy, is important for uncovering how PINK1 engages in important cellular processes and PD pathogenesis. A number of cell lines-based studies have revealed that PINK1-PRKN axis regulates mitochondria clearance via mitophagy upon acute mitochondrial toxin treatment that results in the dissipation of mitochondrial membrane potential. An important issue is whether acute and excessive stress on mitochondria can also occur in vivo. PD is characterized by age-dependent neurodegeneration, a process that is likely involved in chronic and progressive damage on mitochondria. It remains to be investigated whether the effect of chronic and cumulative stress on mitochondria can trigger PINK1-PRKN-mediated mitophagy.
Also, regulation of mitochondrial metabolic function and mitophagy is not identical in different types of cells. For example, immortalized cells typically rely on glycolysis due to their origin as cancer cells and could potentially tolerate substantial loss of mitochondria, whereas neurons rely primarily on oxidative phosphorylation for energy production such that loss of entire mitochondrial pool would be fatal [Citation58–60]. CCCP-induced mitochondrial depolarization results in mitochondria translocation of PRKN in cultured cell lines, which, however, was difficult to be seen in cultured primary neurons [Citation95,Citation96].
A more critical phenomenon is different expression levels of PINK1 in various types of cells and different species. Most of the in vitro studies used overexpression of PINK1-PRKN to investigate their translocation on mitochondria and regulation on mitophagy because endogenous PINK1 is hardly detected. The same scenario also applies to small mammals. It has been well recognized that endogenous PINK1 in rodents is at the undetectable level [Citation30,Citation93,Citation97,Citation98]. Endogenous PINK1 is thought to be synthesized constitutively in the cytosol as a full-length precursor (~63-68 kDa). Upon import into mitochondria, PINK1 is proteolytically cleaved to produce its mature form (~52-55 kDa) that is subsequently re-translocated to the cytosol, resulting in a rapid turnover and low steady state levels [Citation30]. Thus, overexpression of PINK1 in vitro and endogenous level of PINK1 in vivo apparently account for differential effects seen in vitro and in vivo.
Establishment of PINK1 knockout monkey models demonstrated for the first time that loss of PINK1 can mediate striking neuronal loss in the mammalian brains and also provided important insight into the selective neurodegeneration in PD. Being able to detect endogenous PINK1 expression at the protein level has been a very challenging and difficult issue in the field. Using five different antibodies against three epitopes in human PINK1, Yang et al. uncovered the expression of PINK1 that is selectively abundant in the primates [Citation93]. PINK1 is abundantly expressed in the primate brain, but not in the peripheral tissues, and exists as a kinase form to phosphorylate a large number of neuronal proteins [Citation90,Citation93]. These findings strongly support the idea that PINK1 functions as a cytosolic kinase to maintain neuronal viability. In line of this concept, a number of previous in vitro studies have indicated that cytosolic PINK1 plays vital roles in various intracellular function and neuronal survival [Citation99–103]. They also explain why loss of Pink1 does not induce neurodegeneration in the mouse brain because the undetectable endogenous PINK1 is dispensable for the development and maturation of rodent neuronal cells. However, the expression of PINK1 is critical for PRKN activation, as PRKN remains inactive and becomes activated when it is phosphorylated by PINK1 [Citation33,Citation104,Citation105]. Thus, the intrinsically low level of PINK1 in the rodent brain would confer a minor impact of prkn knockout in the mouse brain.
The differences between in vitro and in vivo models for studying PINK1-PRKN function raise crucial issues that need to be well addressed. First, why is PINK1 undetectable in mice but abundant in the primates? The regulation of PINK1 expression is more likely mediated at translational and/or protein stability level, as PINK1 mRNA is ubiquitously and abundantly expressed across different species [Citation106]. Thus, rigorous investigation of mechanisms underlying PINK1 protein expression and cleavage is required. The undetectable expression of PINK1 in the rodent models makes it difficult to use mice to perform this important investigation. Use of large mammals such as monkeys for investigation could elucidate the mechanism for species-dependent expression of PINK1 and may also explain the absence of neuropathological phenotypes of pig models that have deleted the PINK1 gene [Citation86,Citation87]. For example, the primate-specific CASP4 (caspase 4), which is not expressed in the mouse brain, was able to cleave TARDBP/TDP-43 to cause truncated TARDBP to redistribute in the cytoplasm, resulting in the cytoplasmic toxicity in the monkey brain [Citation107]. The cytoplasmic truncated TARDBP can selectively reduce SQSTM1 expression in the monkey brain by inhibiting SQSTM1 mRNA stability via its binding to the unique 3ʹUTR sequence (GU/UG) of the primate SQSTM1 transcript [Citation108]. The findings from TARDBP and PINK1 mutant monkey models indicate the same disease-causing gene may have different gene expression patterns or regulatory mechanisms, which thus contribute to different phenotypes and pathological changes in different species. Second, to what extent dose PINK1-PRKN-mediated mitophagy play a role in vivo, especially under pathological conditions? There is no doubt that PINK1-PRKN mediate mitophagy in cultured cells under stress conditions and may also function to protect mitochondrial integrity in vivo under particular conditions. Does full-length PINK1, which is able to localize to mitochondria, or cytosolic PINK1 kinase play a more important role for cell survival and mitochondrial function, and how do these different forms of PINK1 coordinate their functions in vivo? Although loss of PINK1 in the monkey model causes neuronal loss without significant impact on mitochondrial morphology and protein levels, it remains to be investigated whether PINK1 in the primate brain is targeted to mitochondria when chemical-induced mitochondrial damage occurs. In addition, since female pink1 knockout rats were found to display early onset Parkinsonian behaviors [Citation109], it would be interesting to examine whether sex also influences the severity of phenotypes in the PINK1 mutant monkey models.
Concluding remarks
The use of cell culture models and mitochondrial toxins has elucidated the molecular mechanism by which PINK1 and PRKN act together to degrade damaged mitochondria. However, more studies under the in vivo physiological or pathological conditions need to be conducted to validate PINK1-PRKN-mediated mitophagy and its relevance to neuropathology when both PINK1 and PRKN are expressed at the endogenous level. For example, the current non-human primate studies were focused on the aspect of neurodegeneration caused by loss of PINK1. It remains to be explored whether the established methods to measure the basal mitophagy activities can be applied to monkeys via transgenic expression of the Mito-QC reporter. There has been wealth of information from in vitro studies regarding PINK1ʹs proteolytical process, PRKN recruitment, phosphorylation of ubiquitin and mitochondria proteins. Whether these important in vitro events also occur in the non-human primates under physiological conditions remains to be rigorously investigated. Despite the limitations in using the non-human primate models due to their time-consuming and high-cost experiments, it is clear that the non-human primate model would be very valuable to address these outstanding issues. In addition, PINK1 expression levels appear to account for differences seen in the in vitro and in vivo models and are also found to be altered in tumor cells, and its function is not restricted to the brain and is beyond mitochondria [Citation32]. Thus, combination of in vitro and in vivo experiments to investigate PINK1 would continuously delineate the functions of this important and multifaceted protein. Understanding how PINK1 functions differentially under divergent conditions would also help develop the therapeutics for diseases that are related to dysfunction of PINK1.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20(7):745–754.
- Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. 2016;26(10):733–744.
- Keeney PM, Xie J, Capaldi RA, et al. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26(19):5256–5264.
- Wang X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci Ther. 2009;15(4):345–357.
- Panov AV, Gutekunst C-A, Leavitt BR, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5(8):731–736.
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795.
- Chao H, Lin C, Zuo Q, et al. Cardiolipin-dependent mitophagy guides outcome after traumatic brain injury. J Neurosci. 2019;39(10):1930–1943.
- Teresak P, Lapao A, Subic N, et al. Regulation of PRKN-independent mitophagy. Autophagy. 2022;18(1):24–39.
- Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373(9680):2055–2066.
- Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–2303.
- Langston JW. Epidemiology versus genetics in Parkinson’s disease: progress in resolving an age-old debate. Ann Neurol. 1998;44(3 Suppl 1):S45–52.
- Javitch JA, D’Amato RJ, Strittmatter SM, et al. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A. 1985;82(7):2173–2177.
- Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36(26):2503–2508.
- Ramsay RR, Singer TP. Energy-dependent uptake of N-methyl-4-phenylpyridinium, the neurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria. J Biol Chem. 1986;261(17):7585–7587.
- Liou HH, Tsai MC, Chen CJ, et al. Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology. 1997;48(6):1583–1588.
- Tanner CM, Kamel F, Ross GW, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119(6):866–872.
- Betarbet R, Sherer TB, MacKenzie G, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–1306.
- Schapira AH, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1(8649):1269.
- Schapira AH, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54(3):823–827.
- Giannoccaro MP, La Morgia C, Rizzo G, et al. Mitochondrial DNA and primary mitochondrial dysfunction in P arkinson’s disease. Mov Disord. 2017;32(3):346–363.
- Dolle C, Flønes I, Nido GS, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun. 2016;7(1):13548.
- Toulorge D, Schapira AH, Hajj R. Molecular changes in the postmortem parkinsonian brain. J Neurochem. 2016;139 Suppl 1:27–58.
- McInerney-Leo A, Hadley DW, Gwinn-Hardy K, et al. Genetic testing in Parkinson’s disease. Mov Disord. 2005;20(1):1–10.
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273.
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160.
- Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608.
- Greene JC, Whitworth AJ, Kuo I, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100(7):4078–4083.
- Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–1166.
- Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157–1161.
- Liu Y, Guardia-Laguarta C, Yin J, et al. The ubiquitination of PINK1 is restricted to its mature 52-kDa form. Cell Rep. 2017;20(1):30–39.
- Rub C, Wilkening A, Voos W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017;367(1):111–123.
- Arena G, Valente EM. PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease. J Pathol. 2017;241(2):251–263.
- Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–153.
- Seirafi M, Kozlov G, Gehring K. Parkin structure and function. FEBS J. 2015;282(11):2076–2088.
- Chaugule VK, Burchell L, Barber KR, et al. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011;30(14):2853–2867.
- Riley BE, Lougheed JC, Callaway K, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013;4(1):1982.
- Spratt DE, Julio Martinez-Torres R, Noh YJ, et al. A molecular explanation for the recessive nature of parkin-linked Parkinson’s disease. Nat Commun. 2013;4(1):1983.
- Trempe JF, Sauvé V, Grenier K, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340(6139):1451–1455.
- Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32(15):2099–2112.
- Ordureau A, Sarraf S, Duda D, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56(3):360–375.
- Narendra D, Kane LA, Hauser DN, et al. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6(8):1090–1106.
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–48.
- Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314.
- Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–376.
- Chan NC, Chan DC. Parkin uses the UPS to ship off dysfunctional mitochondria. Autophagy. 2011;7(7):771–772.
- Vives-Bauza C, Przedborski S. PINK1 points Parkin to mitochondria. Autophagy. 2010;6(5):674–675.
- Narendra D, Tanaka A, Suen D-F, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803.
- Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221.
- Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14(10):1929–1938.
- Schubert AF, Gladkova C, Pardon E, et al. Structure of PINK1 in complex with its substrate ubiquitin. Nature. 2017;552(7683):51–56.
- Gan ZY, Callegari S, Cobbold SA, et al. Activation mechanism of PINK1. Nature. 2022;602(7896):328–335.
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105(32):11364–11369.
- Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906.
- Huang Z, Ren S, Jiang Y, et al. PINK1 and Parkin cooperatively protect neurons against constitutively active TRP channel-induced retinal degeneration in Drosophila. Cell Death Dis. 2016;7(4):e2179.
- Huang E, Qu D, Huang T, et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun. 2017;8(1):1399.
- Cai Q, Zakaria H, Simone A, et al. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22(6):545–552.
- Grenier K, Kontogiannea M, Fon EA. Short mitochondrial ARF triggers Parkin/PINK1-dependent mitophagy. J Biol Chem. 2014;289(43):29519–29530.
- Amadoro G, Corsetti V, Florenzano F, et al. Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: the pink-parkin pathway. Front Aging Neurosci. 2014;6:18.
- Kann O, Kovács R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292(2):C641–57.
- Deng H, Dodson MW, Huang H, et al. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105(38):14503–14508.
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378(3):518–523.
- Yang Y, Ouyang Y, Yang L, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105(19):7070–7075.
- Gispert S, Ricciardi F, Kurz A, et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One. 2009;4(6):e5777.
- Goldberg MS, Fleming SM, Palacino JJ, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278(44):43628–43635.
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66(5):646–661.
- Kitada T, Tong Y, Gautier CA, et al. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem. 2009;111(3):696–702.
- Shin JH, Ko H, Kang H, et al. Paris (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144(5):689–702.
- Rezai Amin S, Gruszczynski C, Guiard BP, et al. Viral vector-mediated Cre recombinase expression in substantia nigra induces lesions of the nigrostriatal pathway associated with perturbations of dopamine-related behaviors and hallmarks of programmed cell death. J Neurochem. 2019;150(3):330–340.
- Pickrell AM, Huang C-H, Kennedy S, et al. Endogenous parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron. 2015;87(2):371–381.
- Haque ME, Thomas KJ, D’Souza C, et al. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A. 2008;105(5):1716–1721.
- Oliveras-Salva M, Tong Y, Gautier CA, et al. Alpha-synuclein-induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol Aging. 2014;35(11):2625–2636.
- Matheoud D, Cannon T, Voisin A, et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature. 2019;571(7766):565–569.
- Dave KD, De Silva S, Sheth NP, et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis. 2014;70:190–203.
- de Haas R, Heltzel LCMW, Tax D, et al. To be or not to be pink(1): contradictory findings in an animal model for Parkinson’s disease. Brain Commun. 2019;1(1):fcz016.
- McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(2):439–449 e5.
- Lee JJ, Sanchez-Martinez A, Martinez Zarate A, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018;217(5):1613–1622.
- Liu YT, Sliter DA, Shammas MK, et al. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy. 2021;17(11):3753–3762.
- Wrighton PJ, Shwartz A, Heo J-M, et al. Quantitative intravital imaging in zebrafish reveals in vivo dynamics of physiological-stress-induced mitophagy. J Cell Sci. 2021;134(4). DOI:10.1242/jcs.256255.
- Kim YY, Um JH, Yoon JH, et al. Assessment of mitophagy in mt-Keima Drosophila revealed an essential role of the PINK1-Parkin pathway in mitophagy induction in vivo. FASEB J. 2019;33(9):9742–9751.
- Cornelissen T, Vilain S, Vints K, et al. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in drosophila. Elife. 2018;7:e35878.
- McWilliams TG, Barini E, Pohjolan-Pirhonen R, et al. Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol. 2018;8(11):180108.
- Stauch KL, Villeneuve LM, Purnell PR, et al. SWATH-MS proteome profiling data comparison of DJ-1, Parkin, and PINK1 knockout rat striatal mitochondria. Data Brief. 2016;9:589–593.
- Zhuang N, Li L, Chen S, et al. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016;7(12):e2501.
- Yamada T, Dawson TM, Yanagawa T, et al. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy. 2019;15(11):2012–2018.
- Wang X, Cao C, Huang J, et al. One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci Rep. 2016;6(1):20620.
- Zhou X, Xin J, Fan N, et al. Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell Mol Life Sci. 2015;72(6):1175–1184.
- Zhou H, Falkenburger BH, Schulz JB, et al. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3(4):242–250.
- Yin P, Li S, Li X-J, et al. New pathogenic insights from large animal models of neurodegenerative diseases. Protein Cell. 2022;13(10):707–720.
- Yang W, Liu Y, Tu Z, et al. CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res. 2019;29(4):334–336.
- Chen ZZ, Wang JY, Kang Y, et al. PINK1 gene mutation by pair truncated sgRNA/Cas9-D10A in cynomolgus monkeys. Zool Res. 2021;42(4):469–477.
- Li H, Wu S, Ma X, et al. Co-editing PINK1 and DJ-1 genes via adeno-associated virus-delivered CRISPR/Cas9 system in adult monkey brain elicits classical parkinsonian phenotype. Neurosci Bull. 2021;37(9):1271–1288.
- Yang W, Guo X, Tu Z, et al. PINK1 kinase dysfunction triggers neurodegeneration in the primate brain without impacting mitochondrial homeostasis. Protein Cell. 2022;13(1):26–46.
- Yang W, Li S, Li XJ. A CRISPR monkey model unravels a unique function of PINK1 in primate brains. Mol Neurodegener. 2019;14(1):17.
- Van Laar VS, Arnold B, Cassady SJ, et al. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20(5):927–940.
- Rakovic A, Shurkewitsch K, Seibler P, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem. 2013;288(4):2223–2237.
- Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9(11):1758–1769.
- Zhou C, Huang Y, Shao Y, et al. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105(33):12022–12027.
- Fedorowicz MA, Vries‐Schneider RLA, Rüb C, et al. Cytosolic cleaved PINK 1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2014;15(1):86–93.
- Qin S, Ye L, Zheng Y, et al. Cytosolic PINK1 orchestrates protein translation during proteasomal stress by phosphorylating the translation elongation factor eEF1A1. FEBS Lett. 2021;595(4):507–520.
- Lee Y, Stevens DA, Kang S-U, et al. PINK1 primes parkin-mediated ubiquitination of paris in dopaminergic neuronal survival. Cell Rep. 2017;18(4):918–932.
- Wang KZQ, Steer E, Otero PA, et al. PINK1 interacts with VCP/p97 and activates PKA to promote NSFL1C/p47 phosphorylation and dendritic arborization in neurons. eNeuro. 2018;5(6):ENEURO.0466–18.2018.
- Soman SK, Tingle D, Dagda RY, et al. Cleaved PINK1 induces neuronal plasticity through PKA-mediated BDNF functional regulation. J Neurosci Res. 2021;99(9):2134–2155.
- Gladkova C, Maslen SL, Skehel JM, et al. Mechanism of parkin activation by PINK1. Nature. 2018;559(7714):410–414.
- Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080.
- Blackinton JG, Anvret A, Beilina A, et al. Expression of PINK1 mRNA in human and rodent brain and in Parkinson’s disease. Brain Res. 2007;1184:10–16.
- Yin P, Guo X, Yang W, et al. Caspase-4 mediates cytoplasmic accumulation of TDP-43 in the primate brains. Acta Neuropathol. 2019;137(6):919–937.
- Yin P, Bai D, Deng F, et al. SQSTM1-mediated clearance of cytoplasmic mutant TARDBP/TDP-43 in the monkey brain. Autophagy. 2021;18(8):1955–1968.
- Marquis JM, Lettenberger SE, Kelm-Nelson CA. Early-onset Parkinsonian behaviors in female Pink1-/- rats. Behav Brain Res. 2020;377:112175.