
ABSTRACT
Newly emerging transformed epithelial cells are recognized and apically removed by surrounding normal cells through a biological event termed “cell competition”. However, little is known about the mechanisms underlying this process. In a recent study, we describe that RASG12V/RasV12-transformed cells surrounded by normal cells exhibit decreased lysosomal activity accompanied with accumulation of autophagosomes. Restoration of low lysosomal activity or inhibition of autophagosome formation significantly antagonizes apical extrusion of RASG12V cells, suggesting that non-degradable autophagosomes are required for cell competition. Notably, analysis of a cell competition mouse model demonstrates that macroautophagy/autophagy-ablated RASG12V cells are less readily eliminated by cell competition, and remaining transformed cells destroy ductal integrity, leading to chronic pancreatitis. Thus, our findings illuminate a critical role for non-degradable autophagosomes in cell competition and reveal a homeostasis-preserving role of autophagy upon emergence of transformed cells.
Cell competition is a process wherein cell populations with an identical lineage, but different properties, compete with each other; one population is eliminated as a loser and the other survives as a winner. When transformed cells carrying genetic insults, including the active RAS mutant, elevated expression of SRC or ERBB2, or activation of YAP1 (Yes1 associated transcriptional regulator) emerge within an epithelial layer, they compete with surrounding normal epithelial cells and are apically eliminated via cell competition. To date, it has been revealed that elaborate molecular mechanisms such as cytoskeletal reorganization, membrane trafficking and repulsive reactions are involved in the process of apical elimination of transformed cells.
The role for autophagy in cancer is complex and may differ depending on tumor stages. During the mid-to-late stages of cancer development, autophagy has been implicated to promote tumor progression by exerting a cytoprotective effect in response to cellular stress or increased metabolic demands imposed by rapid proliferation of cancer cells. Conversely, it is conceived that autophagy countervails oncogenesis based on a series of studies using autophagy-deficient mice, yet the tumor-suppressive mechanism through which autophagy acts remains largely puzzling.
In a recent study, we reported that accumulated autophagic vacuoles in RASG12V-transformed cells play a crucial role for cell competition [Citation1]. To evaluate autophagic activity in transformed cells surrounded by normal epithelial cells, we established Madin-Darby canine kidney (MDCK) or MDCK-pTRE3G MYC-RASG12V cell lines that stably express GFP-LC3. When RASG12V cells are co-cultured with MDCK cells at a ratio of 1:50, the number of GFP-LC3-positive puncta in RASG12V cells is profoundly higher than that in RASG12V cells cultured alone. Given that bafilomycin A1 treatment does not alter the value, autophagic flux is prevented in RASG12V cells non-cell autonomously. This result was supported by experiments using an autophagic flux probe, GFP-LC3-RFP-LC3ΔG, demonstrating that interaction with neighboring normal cells reduces autophagic flux in RASG12V cells. We then explored the fate of undegradable autophagic structures by electron microscopy observations and found that both autophagosomes and autolysosomes accumulate in RASG12V cells co-cultured with normal cells. To explore underlying mechanism of hindered autophagic flux, lysosomal activity of transformed loser cells was carefully evaluated. Incorporation of LysoTracker, which labels acidic, functional lysosomes is profoundly diminished in RASG12V cells confronted with normal cells. The comparable non-cell autonomous lysosomal dysfunction is also observed when another indicator of lysosomal activity, Magic Red, is exploited. Reduced lysosomal activity is substantially alleviated by addition of forskolin which acidifies lysosomes by elevating intracellular cAMP levels, and forskolin treatment inhibits apical extrusion of RASG12V cells in a dose-dependent manner. These results indicated that by perturbing autophagy, lysosomal dysfunction positively regulates cell competition.
Previous studies suggest that normal epithelial cells squeeze adjacent transformed cells by locating the cytoskeletal protein FLN (filamin) at the boundary. This brought us to examine involvement of FLN-mediated mechanical cues in the altered lysosomal-autophagic activity. FLN-knockdown in normal cells does not cause either decreased lysosomal activity or accumulation of autophagosomes in RASG12V cells, suggesting that mechanical input from surrounding normal cells induces autophagic perturbation caused by reduced lysosomal activity. We also demonstrated that knockdown of ATG13, a key molecule of the autophagy initiation complex, in RASG12V-transforemd cells profoundly suppresses apical extrusion of RASG12V cells, signifying that autophagosome formation per se is required for transformed cells to be apically expelled. Furthermore, FLN accumulation in normal cells is substantially abolished when co-cultured with ATG13-knockdown RASG12V cells. Collectively, these findings indicate that there exist mutual regulatory mechanisms between accumulated autophagosomes in RASG12V-transformed cells and FLN accumulation in neighboring normal cells, which collaboratively regulate apical elimination of transformed cells ().
Figure 1. A schematic diagram depicts a model where autophagy plays a tumor-suppressive role via cell competition during the initial stage of carcinogenesis.
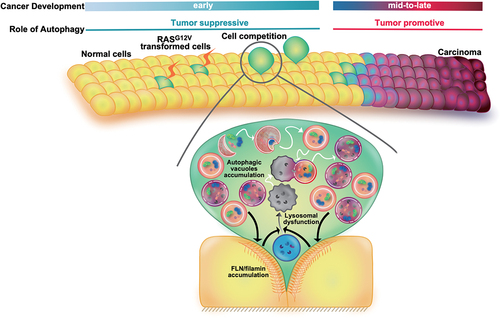
We finally explored autophagic activity in vivo using a cell competition mouse model wherein bicistronic expression of HRASG12V and eGFP is under the control of a floxed STOP transcriptional cassette. Injection of low-dose tamoxifen induces expression of transgenes in a mosaic fashion, and we found that the number of LC3-positive puncta in RASG12V cells confronted with normal cells substantially increases compared to that in those surrounded by one another. In addition, lysosomal activity is found to be depressed non-cell autonomously, demonstrating that attenuated degradation of autophagosomes caused by lysosomal dysfunction occurs in vivo. Further, we showed that ablation of autophagosome formation by Atg5-knockout in RASG12V cells interferes with apical extrusion in pancreas. The remaining Atg5-knockout RASG12V cells in pancreatic ducts form an irregular, stratified epithelium with ductal papillary architecture, and eventually cause chronic pancreatitis.
Collectively, our study demonstrated that autophagy countervails oncogenesis by actively removing transformed cells via cell competition (). More precise elucidation of autophagy-mediated cell competition will guide a novel anti-cancer therapy in the future.
Disclosure statement
All authors have no conflicts of interest to declare.
Additional information
Funding
Reference
- Akter E, Tasaki Y, Mori Y, et al. Non-degradable autophagic vacuoles are indispensable for cell competition. Cell Rep. 2022 Aug 30;40(9):111292.