
ABSTRACT
Macroautophagy/autophagy is a highly conserved catabolic process pivotal to cellular homeostasis and support of tumorigenesis. Being a potential therapeutic target for cancer, we have worked to understand the implications of autophagy inhibition both systemically, and tumor-specifically. We utilized inducible expression of Atg5 shRNA to temporally control autophagy levels in a reversible manner to study the effects of tumor-intrinsic and systemic autophagic loss and restoration on established KrasG12D/+;trp53−/− (KP) lung tumor growth. We reported that transient systemic ATG5 loss significantly reduces KP lung tumor growth. Through in vivo isotope tracing and metabolic flux analyses, we noted that systemic ATG5 knockdown significantly reduces the uptake of glucose and lactate in lung tumors, leading to impaired TCA cycle metabolism and biosynthesis. Additionally, we observed an increased tumor T cell infiltration in the absence of systemic ATG5, which is essential for T cell-mediated tumor killing. Moreover, the impaired tumor metabolism and increased T cell infiltration are sustained when autophagy is restored in a short term. Finally, we found that intermittent systemic ATG5 knockdown, a mock therapy situation, significantly prolongs the lifespan of mice bearing KP lung tumors. Our findings lay the proof of concept for inhibition of autophagy as a valid approach to cancer therapy.
The search for an effective therapeutic strategy in treating non-small cell lung cancer (NSCLC) never ends. Therapies targeting oncogenic RAS, or its downstream effectors have not proven effective in treating KRAS-derived NSCLCs with the exception of KRASG12C. Additionally, though immunotherapy with pembrolizumab and nivolumab is an approved treatment, only a fraction of NSCLC patients treated show a positive response. These facts underscore the importance in uncovering new targets for novel therapies.
Our group and others have shown that both tumor-intrinsic and host autophagy supports the tumorigenesis of different cancers using genetically engineered mouse models (GEMMs). It does this through several processes: the inhibition of TRP53/p53, maintenance of redox homeostasis, inhibiting antigen presentation and maintaining levels of essential amino acids. Autophagy at the host level helps maintain the levels of arginine, alanine, and other amino acids within the tumor microenvironment (TME) while also suppressing anti-tumor T cell responses. Utilizing a KrasG12D/+;trp53−/− (KP) atg7 GEMM (KrasLSL-G12D/+;trp53Flox/Flox;atg7Flox/Flox) we found that autophagy supports KP lung tumorigenesis. However, this loss of autophagy is irreversible, and the deletion occurs simultaneously with the activation of oncogenic KRAS and TRP53. To understand the effect of loss of autophagy on established tumors, a conditional whole-body atg7 knockout GEMM was generated and revealed that host autophagic loss impairs established tumor growth. Though tumor regression occurs within 4–5 weeks, permanent loss of Atg7 leads to a significantly reduced lifespan due to neurodegeneration. This finding underscores the importance of a new GEMM, where the inhibition of autophagy is controllable both temporally and in permanence.
The new GEMMs were generated by crossing KP mice with mice expressing an inducible Tet-regulated Atg5 shRNA detectable by green fluorescence protein (GFP) either systemically (rtTA3;shRNA-Atg5;KP) or tumor-specifically (LSL-rtTA3;shRNA-Atg5;KP) [Citation1]. In contrast to our previous discovery, tumor-specific Atg5 knockdown shows no notable change in established KP lung tumor growth. We suggest that the inhibition of Atg5 through shRNA could be a less potent inhibition of autophagy than the complete gene knockout in the atg7;KP model. However, systemic Atg5 knockdown presents with a significant reduction in established KP lung tumor growth. Furthermore, when autophagy is restored in a short term, deleterious lung tumor growth is irreversible, whereas autophagy ablation-mediated tissue damage is recovered. These findings suggest a therapeutic window for autophagy inhibition in the treatment of lung cancer.
Knowing that autophagy provides key metabolic fuel for major energy and biosynthesis pathways from our prior work, we sought to investigate the impact of systemic autophagic inhibition on tumor metabolism. We performed in vivo [U13C6]-glucose labeling on mice bearing KP lung tumors after acute Atg5 knockdown systemically or tumor-specifically followed by tumor metabolomics using liquid chromatography-mass spectrometry (LC-MS). In the systemic knockdown model, we found that glucose uptake in KP lung tumors is significantly lower than in Atg5 wild-type (WT) mice. Additionally, this loss is paired with a reduced glucose carbon flux to tumor TCA cycle metabolites but not to liver TCA cycle intermediates. In following with the lack of effect on tumor growth, tumor-specific Atg5 knockdown mice show no effect on glucose carbon flux to tumor TCA cycle metabolites. This finding emphasizes the importance of systemic autophagy, not tumor-specific autophagy on supporting KP lung tumor growth.
Our previous work demonstrated that glucose contributes carbon to the tumor TCA cycle via circulating lactate. We therefore performed in vivo [U13C3]-lactate tracing on mice bearing KP lung tumors followed by LC-MS and metabolic flux analyses on KP lung tumors. We found that acute systemic Atg5 knockdown significantly reduces uptake of lactate in tumors but does not affect lactate carbon flux to tumor TCA cycle metabolites. However, the lactate carbon flux to liver TCA cycle intermediates is significantly lower than in the WT control mice. The effects of tumor-specific autophagic loss do not result in a change of lactate carbon flux to the tumor or liver TCA cycle intermediates.
Cancer has been reported to hijack key gluconeogenic enzymes to engage in truncated gluconeogenesis for biosynthesis and redistributing glucose flux for antioxidant production. Lactate is one of the gluconeogenic precursors. Using an in vivo [U13C3]-lactate tracing study, we observed that the labeling fraction of glucose-6-phosphate, fructose-6-phosphate and serine from lactate in KP lung tumors is significantly reduced by systemic Atg5 knockdown, suggesting that systemic autophagy can support KP lung tumor growth by promoting gluconeogenesis for serine biosynthesis.
In a clinical setting, patients with NSCLC have better prognoses with higher tumor infiltrating lymphocytes. Compared to the tumor-specific Atg5 knockdown mice, systemic Atg5 knockdown mice tumors present with much higher lymphocyte infiltration following acute Atg5 knockdown (4 weeks), including CD4 and CD8 T cells and CD4 memory T cells. Of key interest, when we characterized circulating lymphocytes, nearly 40% of all PTPRC/CD45+ lymphocytes present with no GFP signal, suggesting that following systemic Atg5 knockdown, autophagy intact PTPRC/CD45+ cells are being regenerated. We further demonstrated that systemic autophagy supports KP lung tumor growth by preventing T-cell mediated tumor killing.
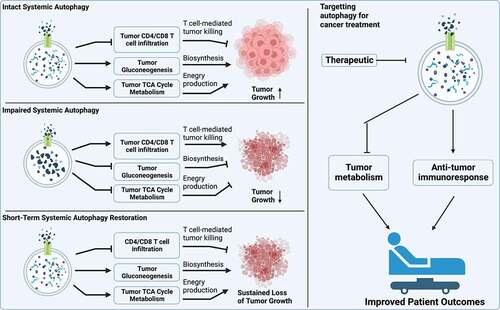
Taken together, our findings point to the inhibition of autophagy as a potential and valid target for KRAS-driven lung cancer therapy. We therefore mimicked clinical practice to toggle autophagy off-on-off in mice bearing established KP lung tumors. We found that intermittent systemic autophagy knockdown significantly extends the mouse lifespan. Most importantly, impaired tumor metabolism and increased T cell infiltration are sustained when autophagy is restored in a short term. These findings cement the possibility of short-term autophagy inhibition as a novel and exciting cancer therapy for patients with KP NSCLC ().
Figure 1. Systemic autophagy supports established KP lung tumor growth by maintaining tumor gluconeogenesis and TCA cycle metabolism for energy production and biosynthesis and promoting immune evasion. Targeting autophagy is a valid approach to cancer therapy.
Financial supports
K22CA190521, R01CA237347-01A1, ACS 134036-RSG-19-165-01-TBG, GO2 Foundation for Lung Cancer, Ludwig Princeton Branch of the Ludwig Institute for Cancer Research and New Jersey Health Foundation to J.Y.G.
Acknowledgments
The authors would like to thank to Vrushank Bhatt for his reading and suggestions of this manuscript. This work was supported by grants from NIH/NCI K22CA190521, NIH/NCI R01CA237347-01A1, ACS 134036-RSG-19-165-01-TBG as well as the GO2 Foundation for Lung Cancer, subaward from Ludwig Princeton Branch of the Ludwig Institute for Cancer Research and New Jersey Health Foundation to J.Y.G.
Disclosure statement
No potential conflicts of interest are reported by the authors(s).
Additional information
Funding
Reference
- Khayati K, Bhatt V, Lan T, et al. Transient systemic autophagy inhibition is selectively and irreversibly deleterious to lung cancer. Cancer Res. 2022. doi:10.1158/0008-5472.CAN-22-1039.