
ABSTRACT
Mcroautophagy/autophagy plays an important role in maintaining homeostasis during nutrient starvation. However, whether epitranscriptomic events are involved in this process remains unclear. Our recent findings suggest that m6A reader YTHDF3 has an essential role in autophagy induction. Elevated m6A modifications installed by METTL3 enable YTHDF3 to promote autophagosome formation and lysosomal function upon nutrient deficiency. This is due to YTHDF3 binding to the m6A modifications at the coding DNA sequence (CDS) and 3’ untranslated region (UTR) around the stop codon of Foxo3 mRNA, recruiting EIF3A and EIF4B to facilitate FOXO3 translation, thus boosting autophagy. In this punctum, we discuss our finding for how YTHDF3 responds to nutrient starvation to promote autophagy flux, providing insights into RNA post-transcriptional modifications linking nutrient cues to autophagic upcycling.
Autophagy is an important protective mechanism for cells to adapt to metabolic disorders such as nutrient deficiency. However, whether and how epitranscriptomic changes play a role in this process remains unclear. Given that the dynamic reversibility of RNA modification makes rapid gene regulation possible, we think that it has profound implications for autophagy regulation in response to nutrient cues; and some epitranscriptome participants may play a key role in this process.
In our recent study [Citation1], we profiled global proteome changes in nutrient-starved mouse embryonic fibroblasts (MEFs) and determined that YTHDF3, an m6A reader, serves as a nutrient responder to boost autophagy. Upon nutrient deficiency, YTHDF3 greatly increases, while levels of other YTH family proteins are not obviously affected. YTHDF3 loss dampens LC3-II elevation and SQSTM1/p62 degradation during nutrient starvation, whereas those can be restored by YTHDF3 re-expression and potentiated by YTHDF3 overexpression. These findings were confirmed by GFP-LC3 assays as well as electron microscopy of ythdf3−/− MEFs, suggesting YTHDF3 is necessary for, and a positive regulator of, autophagy induction. To probe in which steps YTHDF3 is involved, we used a tandem mCherry-GFP-LC3 reporter and found that both autophagosome and autolysosome formation are suppressed. Knocking down YTHDF3 impairs the puncta formation of ULK1, ATG13, ATG14, and ZFYVE1/DFCP1, suggesting that early steps including initiation and nucleation are compromised. Through different fluorescent reporters, we showed that silencing YTHDF3 can also increase pH and attenuate the hydrolytic activity of lysosomes. Furthermore, we examined the effect of YTHDF3 on MTORC1 and AMPK, two main upstream pathways for autophagy induction, and found they are not affected.
Does YTHDF3 boost autophagy by recognizing m6A? We found that during starvation, YTHDF3 and m6A signal accumulate in the cytoplasm. Meanwhile, the m6A level of mRNAs is significantly increased, as well as METTL3, a core catalytic subunit of the m6A methyltransferase, in protein level and catalyzing activity. Knocking down METTL3 abolishes cytoplasmic accumulation and diminishes levels of m6A, suggesting METTL3 is involved in these changes. In YTHDF3-overexpressing MEFs, we showed that METTL3 depletion dampens nutrient starvation-enhanced LC3-II accumulation and SQSTM1 degradation, which can then be restored by reintroducing wild-type rather than catalytic-mutant METTL3. In MEFs without ectopic YTHDF3 expression, METTL3 loss causes a similar autophagy defect, which can also be rescued by catalytically-active METTL3. These findings are also confirmed by mCherry-GFP-LC3 reporters, suggesting that starvation-induced autophagosome and autolysosome generation is METTL3-m6A-dependent. Interestingly, solely overexpressing METTL3 has barely any effect on autophagy, indicating METTL3 is generally redundant.
How is METTL3 increased upon starvation? Our experiments show the enhanced protein stability of METTL3 is the main reason for its upregulation. Impressively, nutrient starvation leads to METTL3 deubiquitination. We performed a proteome profiling of METTL3 interactors and identified three proteins, RPS27A, RPL23, and RPL11, that have regulatory roles in ubiquitination. Interestingly, their expressions do not change during starvation, but rather there is a change in their binding to METTL3. Upon starvation, the interaction of METTL3 with RPL11 and RPL23 increases, while the interaction with RPS27A decreases. siRNA knockdowns demonstrate that only siRps27a causes a METTL3 increment, reducing METTL3 ubiquitination. These results suggest that the impaired RPS27A-METTL3 interaction causes METTL3 deubiquitination, thus promoting METTL3 stabilization.
How does YTHDF3 promote autophagy? We noted after starvation, mRNA peaks with altered binding to YTHDF3 are enriched in the GGAC motif and distributed around the stop codon, coinciding with the general features of m6A. Comparing these peaks with the starvation-induced hyper-m6A peaks, we find 7 genes annotated to autophagy. Validation results indicate FOXO3 as the best candidate. The interaction of YTHDF3 and FOXO3 mRNA is prominent, especially after starvation. Their binding is dependent on METTL3-mediated m6A, which are distributed in the CDS and 3ʹUTR around the stop codon of Foxo3 mRNA. Silencing YTHDF3 reduces FOXO3 expression and causes downregulation of FOXO3 targets involved in autophagy, whereas overexpressing YTHDF3 leads to the opposite effects. By evaluating FOXO3 phosphorylation, we suggested YTHDF3 plays a major role in regulating FOXO3 expression rather than its posttranslational modification. Importantly, altering FOXO3 expression can reverse the effect of YTHDF3 on autophagy flux, suggesting an important role for FOXO3 in YTHDF3-regulated autophagy.
How does YTHDF3 promote FOXO3 expression? Our experiments showed that YTHDF3 enhanced FOXO3 expression by promoting its translation rather than altering its mRNA stability. The m6A sites near the stop codon in the FOXO3-CDS and 3′UTR play a critical role in this process. In addition, our co-IP assays show EIF3A and EIF4B are two prominent proteins that increase binding to YTHDF3 upon nutrient starvation, whereas their knockdown impairs FOXO3 expression, suggesting YTHDF3 may interact with EIF3A and EIF4B to initiate FOXO3 translation.
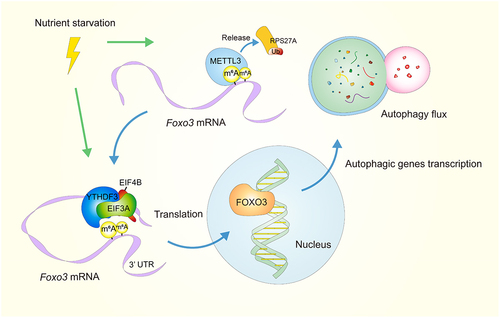
This study provides evidence for a link between m6A and autophagy. Upon nutrient deprivation, the increased METTL3 amount and activity ensure proper m6A in mRNAs. YTHDF3 increases and identifies the hyper-m6A near the stop codon in Foxo3 transcripts, recruiting EIF3A and EIF4B to promote FOXO3 translation, subsequently boosting autophagy (). However, why YTHDF3 increases during starvation remains a mystery. What signals or metabolites cause this change needs further study.
Figure 1. Upon nutrient starvation, the m6A reader YTHDF3 is upregulated, as well as the m6A writer METTL3, which is deubiquitinated and stabilized by releasing RPS27A. The m6A modifications at the CDS and 3’ UTR around the stop codon of Foxo3 are promoted by METTL3. Then YTHDF3 promotes FOXO3 translation by binding to these sites and recruiting EIF3A and EIF4B to the 5’ end of Foxo3 mRNA, forming mRNA loops. FOXO3 then transcriptionally activates core genes involved in autophagy, boosting this process.
One noteworthy question is how YTHDF3 modulates metabolism in response to nutrient scarcity under in vivo conditions. We observed that ythdf3−/− mice exhibit less body weight loss after fasting. Meanwhile, ythdf3−/− mice show more depositions of glycogen and lipid droplets within the liver upon fasting, which can be interpreted as impaired autophagy leading to blunt metabolic regulation. In light of this understanding, it will be interesting to expand insight into the role of epitranscriptomics in nutrient metabolism and homeostasis.
It will also be interesting to understand the role of other epitranscriptome participants in autophagy induction. We noted the effect of silencing YTHDF1 on autophagy flux was similar to but less significant for YTHDF3, whereas YTHDF2 loss had the opposite effect. Interestingly, YTHDF1 but not YTHDF2 promote FOXO3 translation, neither of which affect Foxo3 mRNA stability. Nutrient starvation increases YTHDF1 but decreases YTHDF2 binding to Foxo3 mRNA. These findings suggest that YTHDF1 may cooperate with YTHDF3, and compete with YTHDF2, binding Foxo3 mRNA, thereby promoting FOXO3 translation and affecting autophagy. In METTL14-deficient MEFs, decreased autophagy flux is also observed. However, we cannot exclude METTL3 effects, as METTL14 is required for METTL3 catalytic activity. Such effects and mechanisms for more epitranscriptome participants merit further exploration.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Hao W, Dian M, Zhou Y, et al. Autophagy induction promoted by m(6)A reader YTHDF3 through translation upregulation of FOXO3 mRNA. Nat Commun. 2022;13(1):5845.