
ABSTRACT
Porcine reproductive and respiratory syndrome virus (PRRSV) is a typical immunosuppressive virus devastating the global swine industry. DEAD-box helicases (DDXs) are a family of ATP-dependent RNA helicases that are predominantly implicated in modulating cellular RNA metabolism. Meanwhile, a growing number of studies have suggested that some DDXs are associated with innate immunity and virus infection, so they are considered potential antiviral targets. Herein, we screened 40 DDXs and found that ectopic expression of DDX10 exhibited a significant anti-PRRSV effect, while DDX10 knockdown promoted PRRSV proliferation. Further analysis revealed that DDX10 positively regulates type I interferon production, which may contribute to its anti-PRRSV effect. Interestingly, PRRSV infection promoted DDX10 translocation from the nucleus to the cytoplasm for macroautophagic/autophagic degradation to block the antiviral effect of DDX10. By screening PRRSV-encoded proteins, we found that the viral envelope (E) protein interacted with DDX10. In line with the autophagic degradation of DDX10 during PRRSV infection, E protein could induce autophagy and reduce DDX10 expression in wild-type cells, but not in ATG5 or ATG7 knockout (KO) cells. When further screening the cargo receptors for autophagic degradation, we found that SQSTM1/p62 (sequestosome 1) interacted with both DDX10 and E protein, and E protein-mediated DDX10 degradation was almost entirely blocked in SQSTM1 KO cells, demonstrating that E protein degrades DDX10 by promoting SQSTM1-mediated selective autophagy. Our study reveals a novel mechanism by which PRRSV escapes host antiviral innate immunity through selective autophagy, providing a new target for developing anti-PRRSV drugs.Abbreviations: ACTB: actin beta; ATG: autophagy related; co-IP: co-immunoprecipitation; CQ: chloroquine; DDX10: DEAD-box helicase 10; E: envelope; EGFP: enhanced green fluorescent protein; hpi: hours post infection; hpt: hours post transfection; IFA: indirect immunofluorescence assay; IFN-I: type I IFN; IFNB/IFN-β: interferon beta; IRF3: interferon regulatory factor 3; ISGs: interferon-stimulated genes; KO: knockout; MAP1LC3B/LC3: microtubule associated protein 1 light chain 3 beta; mAb: monoclonal antibody; MOI: multiplicity of infection; NBR1: NBR1 autophagy cargo receptor; NFKB/NF-κB: nuclear factor kappa B; OPTN: optineurin; ORF: open reading frame; PRRSV: porcine reproductive and respiratory syndrome virus; SeV: sendai virus; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; TCID50: 50% tissue culture infective dose; WT: wild type.
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV), characterized by causing reproductive disorders in sows and respiratory distress in piglets, was first identified in the United States in 1987. PRRSV is an enveloped, single-stranded, positive-sense RNA virus, classified in the genus Arterivirus of the family Arteriviridae, with a genome of approximately 15 kb in length [Citation1,Citation2]. At least 10 open reading frames (ORFs) are contained in the PRRSV genome [Citation3,Citation4]. ORF1a and 1b encode two polyproteins, pp1a and pp1ab, which are cleaved into at least 16 mature nonstructural proteins (nsps) by viral proteases [Citation5–7]. The remaining ORFs produce eight structural proteins, namely the glycoprotein (GP)2, GP3, GP4, GP5, GP5a, envelope (E), matrix (M), and nucleocapsid (N) proteins [Citation8,Citation9].
As a typical immunosuppressive virus, PRRSV has evolved elaborate immune evasion strategies to establish infection. At least six PRRSV-encoded proteins, including the nsp1α [Citation10,Citation11], nsp1β [Citation12], nsp2 [Citation13], nsp4 [Citation14], nsp11 [Citation15], and N protein [Citation16], have been demonstrated to antagonize interferon (IFN) production and signaling transduction. In addition to acting as IFN antagonists, some PRRSV-encoded proteins are also capable of facilitating virus infection by targeting host restriction factors. For example, nsp4 cleaves ZC3HAV1/ZAP (zinc finger CCCH-type containing, antiviral 1) and DCP1A (decapping mRNA 1A) to decrease their anti-PRRSV activities [Citation17,Citation18]; nsp3 degrades IFITM1 (interferon induced transmembrane protein 1) through the proteasome pathway to inhibit its anti-PRRSV effect; and E protein degrades host restriction factor porcine (Sus domesticus) CH25H (cholesterol 25-hydroxylase; SdCH25H) by inducing SdCH25H ubiquitination [Citation19]. Furthermore, apoptosis and autophagy are also exploited by PRRSV to enhance virus infection. PRRSV infection activates the unfolded protein response (UPR) pathway to induce apoptosis [Citation20,Citation21], which dysregulates cytokine production to benefit viral infection [Citation22,Citation23]. PRRSV infection also induces autophagy, and inhibiting autophagosome formation decreases the PRRSV yield [Citation24–28], suggesting that autophagy induction by PRRSV promotes virus replication.
The DEAD-box helicases (DDXs) are known for their extremely conserved amino acid motif D-E-A-D (Asp-Glu-Ala-Asp) and belong to helicase superfamily II, which is the largest subfamily of RNA helicases and is present in almost all living organisms [Citation29]. At least 40 DDXs have been identified in humans. They share 13 conserved motifs (Q, I, Ia, Ib, Ic, II, III, IV, IVa, V, Va, Vb, and VI) that are thought to be involved in RNA binding and decay and in ATP binding and hydrolysis [Citation30–32]. However, increasing evidence suggests that, in addition to their functions in modulating cellular RNA metabolism, DDXs are also involved in virus infection via regulating innate immunity or interacting with viral proteins. For example, DDX3X stimulates IFNB/IFN-β transcription via IRF3 (interferon regulatory factor 3) and NFKB/NF-κB (nuclear factor kappa B) signaling branches in dengue virus (DENV)-infected cells, leading to the inhibition of DENV replication [Citation32]; DDX60 enhances RIGI/DDX58 (RNA sensor RIG-I) signaling and significantly increases type I IFN (IFN-I) production in vivo to suppress the proliferation of hepatitis C virus (HCV) and vesicular stomatitis virus (VSV) [Citation33]; and DDX17 has been reported as a cofactor for ZC3HAV1 that is involved in the breakdown of human immunodeficiency virus (HIV-1) mRNA [Citation34,Citation35], and the knockdown of DDX17 results in ZC3HAV1 functioning being 50% less efficient [Citation34]. In terms of PRRSV, several DDXs have been reported to regulate PRRSV infection. For example, DDX3X has been demonstrated to enhance MAVS (mitochondrial antiviral signaling protein)-, TBK1 (TANK binding kinase 1)-, and IKBKE/IKKε (inhibitor of kappa B kinase subunit epsilon)-induced IFNB production to suppress PRRSV proliferation [Citation36]; and PRRSV infection induces DDX5 translocation from the nucleus to the cytoplasm where DDX5 interacts with viral nsp9 to act as a cofactor, thereby promoting PRRSV replication [Citation37]. Recently, our group demonstrated that DDX21 positively regulates PRRSV replication in association with its inhibitory effect on IFNB production [Citation38]. However, studies on the relationship between PRRSV and DDXs remain limited, and there is a particular dearth of systematical research on this subject.
In the present study, we used a high-throughput method to screen the roles of 40 DDXs in PRRSV infection and found that DDX10, DDX49, and RIGI inhibited PRRSV infection, while DDX4, DDX18, DDX21, and DDX27 promoted PRRSV proliferation. We further focused on DDX10 and revealed that DDX10 positively regulated IFN-I production and the expression of interferon-stimulated genes (ISGs), thereby inhibiting PRRSV proliferation. More importantly, we demonstrated that PRRSV facilitated DDX10 degradation via the autophagy pathway. Mechanistically, PRRSV E protein induced DDX10 autophagic degradation in an SQSTM1-dependent manner, which in turn antagonized the antiviral activity of DDX10.
Results
Screening for DDXs that regulate PRRSV proliferation
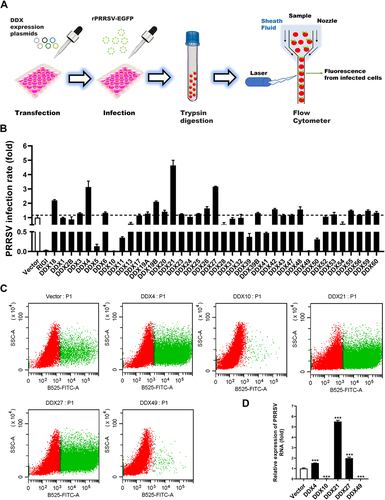
To comprehensively gauge the potential impacts of various DDXs on PRRSV infection, groups of MARC-145 cells were transfected with expression plasmids encoding one each of 40 different DDXs and subsequently infected with rPRRSV-EGFP [Citation39], a recombinant PRRSV stably expressing enhanced green fluorescent protein (EGFP). A flow cytometry assay was then conducted to measure the expression levels of EGFP as a readout for PRRSV multiplication (). RIGI, a well-known antiviral DDX [Citation40], and DDX18, a DDX that has been demonstrated to promote PRRSV infection [Citation41], were set as the positive control and negative control, respectively. Consistent with the results of previous studies [Citation39,Citation40], the levels of PRRSV replication were observed to be higher in DDX18-transfected cells and lower in RIGI-transfected cells as compared with those in empty vector-transfected cells. Significantly, we found that DDX10 and DDX49 remarkably inhibited the proliferation of PRRSV, while DDX4, DDX21, and DDX27 substantially promoted PRRSV proliferation (). Subsequently, qRT-PCR quantifying the levels of viral genome RNA was conducted to confirm the effects of these five DDXs on PRRSV infection, and the results were in good agreement with the flow cytometry data (). The pro-viral effects of DDX21 and DDX18 on PRRSV and the underlying mechanisms have been reported by our and other groups [Citation38]. Both DDX10 and DDX49 exhibit potential anti-PRRSV activity to a degree similar to that of the positive control RIGI, and the roles of DDX10 and DDX49 in virus infections have rarely been reported, however, we failed to clone the full-length cDNA of porcine DDX49 according to the predicted sequence. Thus, we focused on DDX10 in our subsequent experiments.
Figure 1. Screening for DDXs that regulate PRRSV infection. (A) Schematic diagram of the flow cytometry screening for DDXs that affect PRRSV proliferation. (B–D) MARC-145 cells were transfected with eukaryotic expression plasmids encoding various DDXs or empty vector using jetPRIME. At 30 hpt, the cells were infected with rPRRSV-EGFP (MOI = 0.5). At 24 hpi, the cells were collected for flow cytometry to detect the PRRSV infection rate (B and C) or for qRT-PCR to detect the copy number of PRRSV genome RNA (D). These experiments were repeated at least three times, and the data shown are the means ± SD (n = 3). (***P < 0.001).
DDX10 negatively regulates PRRSV infection
To further clarify the role of DDX10 in PRRSV proliferation, iPAMs, which are immortalized porcine alveolar macrophages (PAMs), the target cells of PRRSV infection in vivo, were transfected with the eukaryotic expression construct encoding Flag-tagged DDX10 (pCAGGS-Flag-DDX10) for 30 h and then infected with PRRSV. At 12, 24, and 36 h post infection (hpi), the cells were collected for qRT-PCR, western blotting, and TCID50 assays. The results showed that ectopic expression of DDX10 significantly downregulated the copy number of viral genome RNA (), expression level of PRRSV N protein (), and viral titer () at all tested time points. Furthermore, a dose-dependent inhibitory effect of DDX10 on PRRSV proliferation was also detected by indirect immunofluorescence assay (IFA) and western blotting (Fig. S1A and S1B), thus further confirming the anti-PRRSV effect of DDX10.
Figure 2. DDX10 negatively regulates the proliferation of PRRSV. (A–C) iPAMs were transfected with pCAGGS-Flag-DDX10 or empty vector for 30 h and then infected with PRRSV (MOI = 0.5). At 12, 24, and 36 hpi, the cells were collected for qRT-PCR (A), western blotting (B), and TCID50 assay (C). (D–F) iPAMs were transfected with siDDX10 or NC for 24 h and then infected with PRRSV (MOI = 0.5). At 12, 24, and 36 hpi, the cells were collected for qRT-PCR (D), western blotting (E), and TCID50 assay (F). ACTB (actin beta) served as a protein loading control. These experiments were repeated at least three times, and the data shown are the means ± SD (n = 3). (***P < 0.001; **P < 0.01; *P < 0.05).
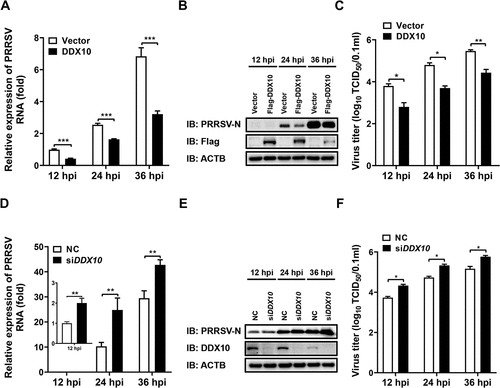
The effect of DDX10 knockdown on PRRSV infection was also investigated by using siRNA. Three DDX10-specific siRNAs were designed, and the siDDX10-2 (hereafter referred to as siDDX10) was chosen for use in subsequent experiments because of it having the highest knockdown efficiency as evidenced by qRT-PCR and western blotting results (Fig. S2A and S2B). iPAMs were transfected with siDDX10 or negative control siRNA (NC) for 24 h and then infected with PRRSV. At 12, 24, and 36 hpi, the cells were collected for qRT-PCR, western blotting, and TCID50 assays. The results showed that knockdown of DDX10 significantly upregulated the copy number of viral genome RNA (), expression level of PRRSV N protein (), and viral titer (), thus further demonstrating that DDX10 inhibits PRRSV replication.
DDX10 promotes IFNB production and ISG expression
Previous studies showed that some DDXs have antiviral activity that functions mainly through the regulation of IFN-I production or interaction with viral protein(s) [Citation42–45], and PRRSV is highly sensitive to IFN-I [Citation46]. Thus, the effect of DDX10 on IFNB production was assessed to preliminarily investigate the underlying anti-PRRSV mechanism of DDX10. To this end, iPAMs were transfected with pCAGGS-Flag-DDX10, followed by qRT-PCR and enzyme-linked immunosorbent assay (ELISA) to detect the mRNA and protein expression of porcine IFNB. The results showed that overexpression of DDX10 significantly upregulated the mRNA () and protein () levels of IFNB. We also detected the effect of DDX10 on the promoter activity of IFNB. iPAMs were co-transfected with pCAGGS-Flag-DDX10 and the firefly luciferase reporter plasmid for IFNB promoter (IFNB-Luc) together with the Renilla luciferase reporter plasmid pRL-TK (as an internal control) and then assessed in a dual-luciferase reporter assay. The results showed that overexpression of DDX10 strongly activated IFNB promoter activity (), further confirming that DDX10 induces IFNB production. The induction of IFNB requires the coordinated and cooperative actions of transcription factors IRF3 and NFKB [Citation46]. The results of a dual-luciferase reporter assay conducted with the luciferase reporter plasmids IRF3-Luc and NFKB-Luc revealed that ectopic expression of DDX10 activated IRF3 and NFKB (). Additionally, DDX10 significantly upregulated the phosphorylation levels of IRF3 (p-IRF3) and NFKB subunit RELA/p65 (p-RELA/p65) (), the hallmarks of IRF3 and NFKB activation [Citation47], thus further demonstrating that DDX10 positively regulates IFN-I production.
Figure 3. DDX10 positively regulates the production of IFNB and ISGs. (A and B) iPAMs cultured in 24-well plates were transfected with pCAGGS-Flag-DDX10 or empty vector for 30 h. The cells were harvested for qRT-PCR to detect the mRNA levels of IFNB (A), or for ELISA to detect the protein levels of IFNB (B). (C–E) iPAMs cultured in 24-well plates were transfected with pCAGGS-Flag-DDX10 or empty vector, together with reporter plasmids IFNB-Luc (C), IRF3-Luc (D), or NFKB-Luc (E) and internal reference plasmid pRL-TK for 24 h and then subjected to dual-luciferase reporter assays. (F) iPAMs were transfected with pCAGGS-Flag-DDX10 or empty vector for 30 h, followed by western blotting with primary antibodies against total IRF3, total RELA/p65, phosphorylated IRF3 (p-IRF3), phosphorylated RELA (p-RELA), Flag, and ACTB. (G–J) iPAMs were transfected with siDDX10 or NC for 24 h and then infected with PRRSV (MOI = 0.5). The cells were harvested at 12, 24, and 36 hpi for qRT-PCR to detect the mRNA expression levels of IFNB (G), MX1 (H), and IFIT2 (I) or for western blotting to detect the protein expression levels of ISG15, IFITM3, OAS1, PRRSV N, and DDX10 (J). ACTB served as a protein loading control. The experiments were repeated at least three times, and the data shown are the means ± SD (n = 3). (***P < 0.001; **P < 0.01; *P < 0.05).
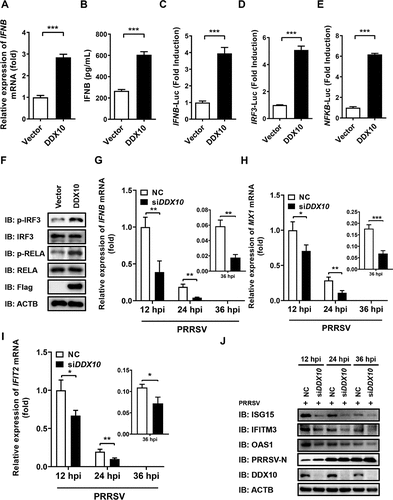
The antiviral effects of interferon alpha and IFNB are mainly due to the induction of a vast number of ISGs, so we further investigated the function of DDX10 in regulating IFNB and ISG induction upon PRRSV infection. First, iPAMs were transfected with siDDX10 or NC for 24 h and then infected with PRRSV for the indicated lengths of time. Consistently, the results of qRT-PCR and western blotting showed that DDX10 knockdown remarkably downregulated the mRNA expression levels of IFNB (), MX1 (), and IFIT2/ISG54 (), as well as the protein expression levels of ISG15, IFITM3, and OAS1 () at all tested time points after PRRSV infection. Taken together, these results suggested that the anti-PRRSV activity of DDX10 is at least partially associated with its ability to induce IFN-I production and ISG expression.
Because this is the first report regarding the IFN regulation function of DDX10, to strengthen this discovery, we examined the effects of human DDX10 on IFNB production and signaling transduction in HEK-293T cells. The results showed that overexpression of human DDX10 significantly increased the mRNA and protein levels of IFNB, as well as the promoter activities of IFNB, IRF3, and NFKB (Fig. S3A–S3E), while opposite effects occurred in cells transfected with human DDX10-specific siRNA (Fig. S3F–S3I). Furthermore, knockdown of human DDX10 with specific siRNA suppressed Sendai virus (SeV)-induced phosphorylation levels of IRF3 and RELA, as well as the expressions of ISG15, IFITM3, OAS1, MX1, and IFIT2 (Fig. S3J–S3L). On the basis of these data from different species, we concluded that DDX10 positively regulates IFN-I.
PRRSV infection downregulates DDX10 expression and promotes its cytoplasmic translocation
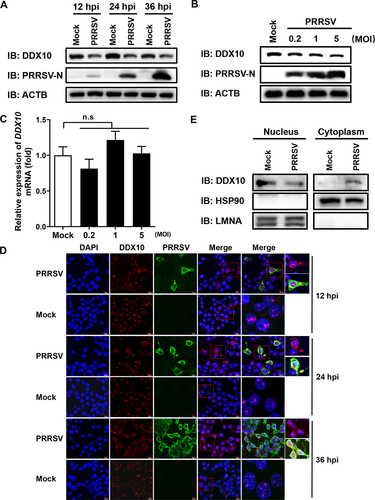
Because DDX10 exhibits strong anti-PRRSV activity, we wanted to know whether PRRSV infection also regulates DDX10. To this end, we began by analyzing the protein expression of DDX10 after PRRSV infection. iPAMs were infected with PRRSV, and the protein expression levels of endogenous DDX10 were detected by western blotting at 12, 24, and 36 hpi. The results showed that PRRSV infection downregulated DDX10 protein expression at all three time points (), and this downregulation occurred in a dose-dependent manner (). Similar results were observed in PRRSV-infected MARC-145 cells (Fig. S4A and S4B). Notably, we found that the mRNA expression levels of DDX10 were not reduced after PRRSV infection (), indicating that the decreased protein expression of DDX10 may be due to posttranslational modification or degradation. We then investigated the influence of PRRSV on the subcellular localization of DDX10. iPAMs were infected with PRRSV for 12, 24, or 36 h before being examined by IFA to detect the subcellular localization of endogenous DDX10. The results showed that PRRSV infection caused a partial translocation of DDX10 from the nucleus to the cytoplasm (). Similar results were also observed in PRRSV-infected MARC-145 cells (Fig. S4C). A nuclear and cytosolic fractionation assay was performed, and the results showed a higher cytoplasmic DDX10 expression level and a lower nuclear DDX10 expression level in PRRSV-infected cells than in mock-infected cells (), which was in line with the results of IFA (). Taken together, these results suggested that PRRSV infection decreases the protein expression of DDX10 and promotes its cytoplasmic translocation.
Figure 4. PRRSV infection facilities DDX10 translocation from the nucleus to the cytoplasm and decreases DDX10 protein expression. (A) iPAMs were infected with PRRSV (MOI = 0.5). At 12, 24, and 36 hpi, the cells were collected for western blotting. (B and C) iPAMs were infected with increasing doses of PRRSV (MOI = 0.2, 1, or 5). At 24 hpi, the cells were collected for western blotting (B) or qRT-PCR (C). The data shown are the means ± SD of three independent experiments. n.s., not significant. (D) iPAMs cultured on microscope coverslips were infected with PRRSV (MOI = 0.5). At 12, 24, and 36 hpi, the cells were fixed, and the subcellular localizations of DDX10 and the PRRSV N protein were analyzed by IFA. Nuclei were counterstained with DAPI. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoviewver.3.1; Olympus, Japan). (E) iPAMs grown in 100-mm plates were mock-infected or infected with PRRSV (MOI = 0.5). At 24 hpi, the cells were collected for a nuclear and cytosolic fractionation assay to detect DDX10 expression in the nucleus and cytoplasm.
PRRSV infection degrades DDX10 via the autophagy pathway
The apoptosis, autophagy, and ubiquitin-proteasome system are three major intracellular degradative pathways. To determine the pathway by which PRRSV degrades DDX10, iPAMs were infected with PRRSV and then treated with Z-VAD-FMK (inhibitor of the apoptosis pathway), chloroquine (CQ) and NH4Cl (inhibitors of the autophagy pathway), and MG132 (inhibitor of the ubiquitin-proteasome pathway), respectively. The western blotting results showed that treatment with CQ or NH4Cl almost completely restored DDX10 levels (abrogated the PRRSV-induced degradation), whereas no obvious differences in DDX10 levels were observed under the treatment with Z-VAD-FMK or MG132 as compared with the control dimethyl sulfoxide (DMSO) treatment (), suggesting that the autophagy pathway plays a major role in the degradation of DDX10 during PRRSV infection. To further solidify this conclusion, we examined the protein expression level of DDX10 in iPAMs exposed to rapamycin, an autophagy inducer. Consistent with the above conclusion, rapamycin treatment downregulated the DDX10 protein expression level in a dose-dependent manner (), and this level could be partially restored by NH4Cl (). Collectively, these results demonstrated that PRRSV infection degraded DDX10 via the autophagy pathway and DDX10 itself can be regulated by autophagy.
Figure 5. PRRSV degrades DDX10 via the autophagy pathway. (A–C) iPAMs were infected with PRRSV (MOI = 0.5) and then treated with Z-VAD-FMK (10 µM), MG132 (10 µM) (A), CQ (20 µM) (B), or NH4Cl (10 mM) (C). At 24 hpi, the cells were harvested for western blotting to detect DDX10 protein expression. (D) iPAMs were treated with various doses of rapamycin (2.5, 5, 10, or 20 µM) for 12 h and then harvested for western blotting. (E) iPAMs were mock-treated or treated with rapamycin (10 µM), together with H2O or NH4Cl (10 mM) for 12 h. Cell lysates were used for western blotting. (F) iPAMs transfected with pCAGGS-Flag-DDX10 were mock-treated or treated with rapamycin (10 µM) for 12 h, and then the cells were harvested and subjected to a nuclear and cytosolic fractionation assay. (G–H) iPAMs were transfected with GFP-LC3 expression plasmid (G) or LAMP1-DsRed expression plasmid (H) for 30 h and then infected with PRRSV (MOI = 0.5). At 24 hpi, cells were fixed for IFA to detect the endogenous DDX10 using anti-DDX10 antibody. Nuclei were counterstained with DAPI. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoviewver.3.1; Olympus, Japan).
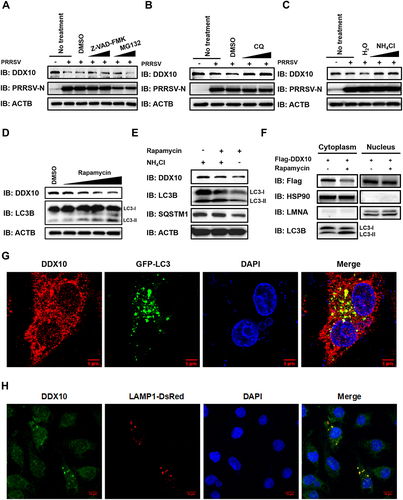
Because rapamycin-induced autophagy resulted in DDX10 degradation (), and PRRSV infection promoted DDX10 translocation from the nucleus into the cytoplasm for degradation, it seemed likely that the degradation of DDX10 occurs in the cytoplasm. To confirm this speculation, iPAMs were transfected with pCAGGS-Flag-DDX10 and then treated with rapamycin, followed by a nuclear and cytosolic fractionation assay. As shown in , the protein level of DDX10 in the cytoplasm was downregulated by rapamycin treatment, whereas there was no obvious change in the DDX10 level in the nucleus, demonstrating that the autophagic degradation of DDX10 occurred mainly in the cytoplasm.
The contents degraded by autophagy are first inside autophagosomes and then delivered to lysosomes for degradation. To further demonstrate that the degradation of DDX10 depends on autophagy, the potential colocalizations of DDX10 with the autophagosome marker (LC3), as well as the lysosome marker (LAMP1) were detected. As shown in , endogenous DDX10 partially colocalized with GFP-LC3 and LAMP1-DsRed in the cytoplasm, suggesting that DDX10 could colocalize with both autophagosomes and lysosomes for autophagic degradation.
PRRSV E protein interacts with DDX10 and induces DDX10 cytoplasmic translocation
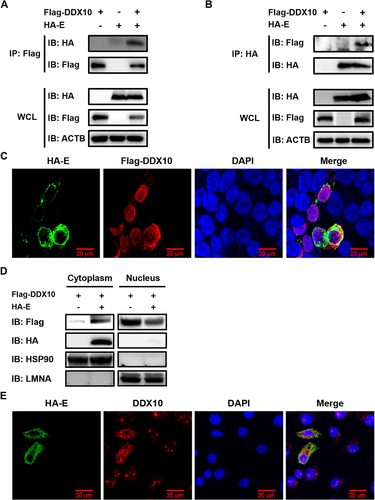
To further investigate the association between PRRSV and DDX10, we screened for the potential interaction of PRRSV-encoded proteins with DDX10. HEK-293T cells were co-transfected with pCAGGS-Flag-DDX10 and HA-tagged expression constructs encoding PRRSV proteins, followed by co-immunoprecipitation (co-IP) assays. The results showed that only E protein could be detected in the immunocomplexes isolated with anti-Flag antibody ( and Fig. S5). In a reverse co-IP experiment, DDX10 was also efficiently co-immunoprecipitated with E protein via an anti-HA antibody (). We further assessed whether DDX10 colocalizes with E protein, and the IFA results showed that some colocalization of the overexpressed DDX10 with E protein was visible in the cytoplasm. Notably, we also found that overexpression of E protein led to a partial translocation of DDX10 from the nucleus to the cytoplasm (), as observed in PRRSV-infected cells (). To further confirm this result, a nuclear and cytosolic fractionation assay was conducted. As shown in , co-expression of E protein and DDX10 resulted in an increase of cytoplasmic DDX10 and a decrease of nuclear DDX10, suggesting that E protein redistributes DDX10 from the nucleus to the cytoplasm. We further investigated the subcellular localization of endogenous DDX10 in cells expressing E protein. The results showed that E protein promoted the translocation of endogenous DDX10 from the nucleus to the cytoplasm where the translocated DDX10 colocalized with E protein ().
Figure 6. PRRSV E protein interacts with DDX10 and changes its localization from the nucleus to the cytoplasm. (A and B) HEK-293T cells were co-transfected with pCAGGS-Flag-DDX10 and pCAGGS-HA-E. The cells were lysed at 30 hpt and immunoprecipitated with an anti-Flag antibody (A) or anti-HA antibody (B). Whole-cell lysate (WCL) and immunoprecipitation (IP) complexes were analyzed by immunoblotting with anti-Flag, anti-HA, and anti-ACTB antibodies. (C) HEK-293T cells were co-transfected with pCAGGS-Flag-DDX10 and pCAGGS-HA-E. At 30 hpt, cells were fixed for IFA to detect the DDX10 and E proteins using anti-Flag and anti-HA antibodies, respectively. Nuclei were counterstained with DAPI. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoviewver.3.1; Olympus, Japan). (D) HEK-293T cells were co-transfected with pCAGGS-HA-E and pCAGGS-Flag-DDX10. Cells were then collected at 30 hpt for a nuclear and cytosolic fractionation assay. (E) iPAMs were transfected with pCAGGS-HA-E for 30 h. The cells were fixed for IFA to detect endogenous DDX10 and HA-tagged E protein using anti-DDX10 and anti-HA antibodies, respectively. Nuclei were counterstained with DAPI. Fluorescent images were acquired as described in (C).
PRRSV E protein facilitates the autophagic degradation of DDX10
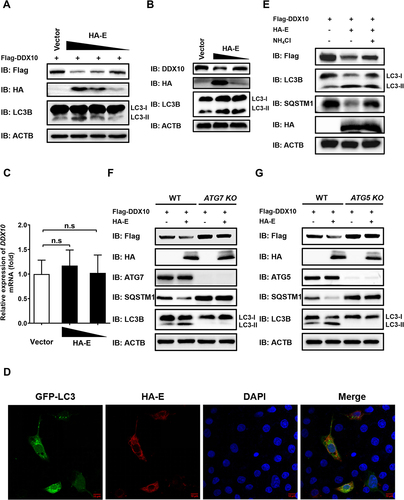
Because we found that PRRSV infection reduced DDX10 expression through the autophagy pathway (), we next examined the effect of PRRSV E protein on DDX10 expression and autophagy. The results showed that overexpression of E protein dose-dependently decreased the expressions of both exogenous and endogenous DDX10 at the protein level (), but not at the mRNA level (). Meanwhile, E protein was found to increase the levels of LC3-II (), and promote the formation of GFP-LC3 puncta (), indicating that E protein induces autophagy. Thus, we further explored whether autophagy is involved in E protein-mediated degradation of DDX10. First, we used NH4Cl to block the autophagy pathway, and the results revealed that E protein-induced reduction of DDX10 expression was largely rescued by NH4Cl (), suggesting the involvement of autophagy in E protein-induced degradation of DDX10. To further strengthen this conclusion, HEK-293T knockout cells for the essential autophagy genes ATG7 (autophagy related 7) and ATG5, hereafter referred to as ATG7 KO and ATG5 KO, respectively, were used, and we found that the DDX10 degradation induced by E protein was detectable in wild-type (WT) HEK-293T cells but not in either ATG7 KO or ATG5 KO cells (). Collectively, these data indicated that PRRSV E protein-mediated degradation of DDX10 was dependent on autophagy.
Figure 7. PRRSV E protein induces DDX10 degradation via the autophagy pathway. (A) iPAMs were transfected with pCAGGS-Flag-DDX10, together with empty vector or pCAGGS-HA-E (250, 500, or 1000 ng) for 30 h. The cells were harvested for western blotting. (B and C) iPAMs were transfected with emptor vector or pCAGGS-HA-E (500 or 1000 ng) for 30 h. The cells were then harvested for western blotting (B) or qRT-PCR (C). n.s., not significant. (D) iPAMs were co-transfected with pCAGGS-HA-E and GFP-LC3 expression plasmid for 30 h. The cells were fixed for IFA to detect the HA-tagged E protein using anti-HA antibody. Nuclei were counterstained with DAPI. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoviewver.3.1; Olympus, Japan). (E) iPAMs were transfected with pCAGGS-Flag-DDX10, together with empty vector or pCAGGS-HA-E for 12 h. Before harvesting, the cells were mock-treated or treated with NH4Cl (10 mM) for 12 h. The cell lysates were used for western blotting with the indicated antibodies. (F and G) WT, ATG7 knockout (KO) (F), or ATG5 KO HEK-293T (G) cells were co-transfected with pCAGGS-Flag-DDX10 and pCAGGS-HA-E for 30 h. The cell lysates were subjected to western blotting with the indicated antibodies.
PRRSV E protein induces the autophagic degradation of DDX10 in an SQSTM1-dependent manner
Accumulating evidence indicates that the autophagic clearance of protein aggregates is a highly selective process that depends on substrate recognition by cargo receptors [Citation48]. To determine which cargo receptor(s) is required for the autophagic degradation of DDX10, we performed co-IP assays to detect the interactions between DDX10 and cargo receptors SQSTM1, NBR1, OPTN, TOLLIP, and BNIP3L/NIX. As shown in , among these receptors, only SQSTM1 interacted with DDX10. In addition, the results of IFA showed that SQSTM1 colocalized with DDX10 in the cytoplasm (). We further analyzed the influence of these cargo receptors on DDX10 expression. Interestingly, overexpression of SQSTM1, NBR1, TOLLIP, and BNIP3L all promoted DDX10 degradation (Fig. S6A); however, overexpression of E protein potentiated the degradation of DDX10 mediated only by SQSTM1, but not other cargo receptors (Fig. S6B).
Figure 8. PRRSV E protein facilitates autophagic degradation of DDX10 depending on SQSTM1. (A) HEK-293T cells were transfected with pCAGGS-HA-DDX10 together with pCAGGS-Flag-SQSTM1, pCAGGS-Flag-NBR1, pCAGGS-Flag-OPTN, pCAGGS-Flag-TOLLIP, or pCAGGS-Flag-BNIP3L for 30 h. The cell lysates were immunoprecipitated using an anti-Flag antibody and then immunoblotted using the indicated antibodies. (B) iPAMs were infected with PRRSV (MOI = 0.5) for 24 h. The cells were fixed for IFA to detect the endogenous SQSTM1 and DDX10 using anti-SQSTM1 and anti-DDX10 antibodies, respectively. Nuclei were counterstained with DAPI. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoviewver.3.1; Olympus, Japan). (C) HEK-293T cells were co-transfected with pCAGGS-Flag-SQSTM1 and pCAGGS-HA-E for 30 h. The cell lysates were immunoprecipitated using an anti-Flag antibody and then immunoblotted using the indicated antibodies. (D) iPAMs were transfected with pCAGGS-HA-E for 30 h. The cells were fixed for IFA to detect the endogenous SQSTM1 and HA-tagged E protein using anti-SQSTM1 and anti-HA antibodies, respectively. Nuclei were counterstained with DAPI. Fluorescent images were acquired as described in (B). (E) HEK-293T cells were co-transfected with pCAGGS-HA-E and pCAGGS-Flag-DDX10. Cells were lysed at 30 hpt and immunoprecipitated with an anti-Flag antibody. The WCL and IP complexes were analyzed by immunoblotting with anti-HA, anti-Flag, anti-ubiquitin (Ub), and anti-ACTB antibodies. (F) WT or SQSTM1 KO HEK-293T cells were co-transfected with pCAGGS-Flag-DDX10 and pCAGGS-HA-E for 30 h. The cell lysates were subjected to western blotting with the indicated antibodies. (G and H) WT and SQSTM1 KO HEK-293T cells were separately transfected with pCAGGS-HA-E or empty vector for 30 h, and the cell lysates were subjected to western blotting with the indicated antibodies. (I) HEK-293T cells were co-transfected with pCAGGS-Flag-DDX10, pCAGGS-Flag-SQSTM1, and pCAGGS-HA-E, or co-transfected with pCAGGS-Flag-DDX10 and pCAGGS-Flag-SQSTM1 or pCAGGS-HA-E for 30 h. The cell lysates were subjected to western blotting using the indicated antibodies.
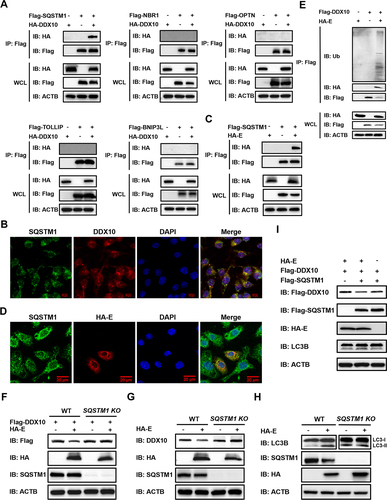
To further examine whether E protein-induced autophagic degradation of DDX10 is an SQSTM1-dependent event, we analyzed the relationship between E protein and SQSTM1 by performing co-IP assay and IFA. The results showed that E protein interacted and colocalized with SQSTM1 (), suggesting that SQSTM1 may be involved in the DDX10 degradation induced by E protein. Because SQSTM1 recognizes cargo primarily by its ubiquitin-associated (UBA) structural domain [Citation49,Citation50], we then investigated whether DDX10 undergoes ubiquitination and then interacts with SQSTM1. By detecting the amount of polyubiquitin linkages on DDX10 in the presence or absence of E protein, we found that the ubiquitination level of DDX10 was dramatically enhanced by E protein overexpression (), indicating that E protein promotes DDX10 ubiquitination for its recognition by SQSTM1. To further test this result, SQSTM1-deficient HEK-293T cells (SQSTM1 KO) were used, and western blotting results demonstrated that E protein-mediated degradation of DDX10 (both exogenous and endogenous) was almost entirely blocked in SQSTM1-deficient cells () but not in the WT cells, indicating that E protein-induced DDX10 degradation is indeed SQSTM1-dependent.
To further investigate whether SQSTM1-mediated selective autophagy is driven by E protein, we compared the autophagy levels induced by E protein in WT and SQSTM1 KO cells. As shown in protein induced detectable autophagy in WT cells but not in SQSTM1-deficient cells, suggesting that E protein promotes selective autophagy via SQSTM1. Subsequently, we investigated whether the SQSTM1-mediated degradation of DDX10 can be augmented by E protein. As shown in , co-expression of E protein and SQSTM1 further enhanced the degradation of DDX10 as compared with the overexpression of either E protein or SQSTM1 alone. Collectively, these results suggested that E protein degraded DDX10 by promoting SQSTM1-mediated selective autophagy.
Discussion
In recent years, the interactions between viruses and DDXs have received more attention because many DDXs have been identified as key regulators of antiviral innate immune responses, in addition to their roles in modulating host RNA metabolism. For example, RIGI [Citation51], DDX3X [Citation52], DDX41 [Citation53], and DDX60 [Citation54] each trigger IFN-I production to exert antiviral effects. In the present study, we screened 40 DDXs and found that DDX4, DDX18, DDX21, and DDX27 significantly promoted PRRSV proliferation, while DDX10, DDX49, and RIGI inhibited PRRSV proliferation. Among these anti-PRRSV or pro-PRRSV DDXs, RIGI is a well-known potent antiviral DDX, and the pro-PRRSV effects of DDX18 and DDX21 have been reported in previous studies [Citation38,Citation41], which indicates that the results of our high-throughput screening are reliable. It should be noted that DDX10 exhibited an anti-PRRSV effect as large as that displayed by RIGI, the positive control used in this study. Furthermore, there are very few reports of the relationship between virus and DDX10; thus, we chose DDX10 for further study. We then found that PRRSV infection resulted in the translocation of DDX10 from the nucleus to the cytoplasm and degraded DDX10 via SQSTM1-dependent selective autophagy to antagonize its antiviral role, which reveals a novel immune evasion strategy developed by PRRSV.
DDXs can inhibit virus infection via at least two basic mechanisms, regulating antiviral innate immunity or interacting with viral protein(s) [Citation32,Citation54,Citation55]. In this study, we have demonstrated that DDX10 positively regulates IFN-I production and signaling transduction, which may be one mechanism utilized by DDX10 to inhibit PRRSV infection. When we investigated how DDX10 promotes IFN-I production by examining individual knockdowns of RIGI, MAVS, MYD88, and TRIF, we found that none of these knockdowns attenuated DDX10-induced IFNB production (data not shown), suggesting that DDX10 induced IFN-I production independently of the classical RIGI-like receptor (RLR) and Toll-like receptor (TLR) pathways. The exact mechanism by which DDX10 activates IFN-I needs further study. In addition to demonstrating that DDX10 induces IFN-I, we also investigated whether DDX10 interacts with PRRSV-encoded proteins to inhibit viral proliferation. Although we found that DDX10 interacts with E protein, it appears not to utilize this strategy to inhibit PRRSV infection, given that DDX10 did not decrease the expression of E protein but rather E protein downregulated DDX10 expression. Whether or not other unidentified mechanisms are utilized by DDX10 to inhibit PRRSV infection remains a topic for further investigations.
Although DDX10 exhibits anti-PRRSV effects, PRRSV infection degrades DDX10 via SQSTM1-dependent selective autophagy to antagonize its antiviral activity. To date, numerous viruses have been reported to evade host antiviral immunity by degrading host proteins, particularly the activators of IFN-I signaling pathway, via the induction of selective autophagy through cargo receptors (e.g., SQSTM1, OPTN, and NBR1) [Citation56–62]. For example, severe acute respiratory syndrome coronavirus 2/SARS-CoV-2 nsp13 inhibits IFN-I production by recruiting TBK1 to SQSTM1 for autophagic degradation, enabling the virus to escape host innate immune responses [Citation63]. Infectious bursal disease virus (IBDV) VP3 protein induces TRAF6 autophagic degradation in a SQSTM1-dependent manner, thus inhibiting TRAF6-mediated NFKB activation and IFNB production, which facilitates viral replication [Citation64]. H7N9 influenza virus PB1 protein preferentially associates with NBR1, a receptor that recognizes ubiquitinated MAVS and delivers it to the autophagosome for degradation, thus facilitating H7N9 virus infection by blocking the RIGI–MAVS-mediated innate signaling pathway [Citation65]. In addition, SeV infection enhances the expression of CCDC50, a previously unknown autophagy receptor that was shown to specifically recognize and degrade K63-polyubiquitinated RIGI, thereby negatively regulating the IFN-I signaling pathway and inhibiting viral replication [Citation66]. As for PRRSV, it has been shown that PRRSV infection induces autophagy to promote PRRSV proliferation [Citation24–28]. However, the molecular mechanisms by which autophagy affects viral proliferation remain unclear. Herein, we demonstrated that PRRSV E protein predominantly induced SQSTM1-dependent selective autophagic degradation of the anti-PRRSV protein DDX10 to benefit viral proliferation. Notably, we also found that, in addition to SQSTM1, cargo receptors TOLLIP, NBR1, and BNIP3L downregulated DDX10 protein expression as well (Fig. S6A), even though there was no direct interaction between TOLLIP, NBR1, BNIP3L and DDX10 (). Interestingly, only SQSTM1, but not TOLLIP, NBR1, or BNIP3L, remained capable of reducing the protein expression levels of DDX10 in cells overexpressing E protein (Fig. S6B). Moreover, we found that E protein also interacted with NBR1, TOLLIP, and BNIP3L (Fig. S7). Similarly, IBDV VP3 promotes TRAF6 autophagic degradation in a mainly SQSTM1-dependent manner, but OPTN and NBR1 also partially contribute to the autophagic degradation of TRAF6. Furthermore, OPTN and NBR1 interact with VP3 but not with TRAF6 [Citation64]. Together, these findings reveal the complexity of the mechanisms employed by viruses for the autophagic degradation of host proteins.
Unrelated to the autophagy pathway, the apoptosis inhibitor Z-VAD-FMK was also found to weakly restore the PRRSV-induced reduction in DDX10 expression (). This result suggests that apoptosis may also partially be associated with DDX10 degradation to benefit PRRSV infection, which is consistent with the previous finding that PRRSV infection induces apoptosis to augment virus proliferation [Citation67]. Coincidently, our previous study showed that PRRSV E protein degrades pCH25H, a PRRSV antagonist, predominantly through the ubiquitin-proteasome pathway and, notably, that NH4Cl also partially restores the decrease of SdCH25H expression induced by E protein [Citation19], indicating that the autophagy pathway may be involved in E protein-mediated SdCH25H degradation as well.
Changes in the subcellular localization of DDXs are often associated with changes in their functions, half-life, and other properties. For example, DDX23 is translocated from the nucleus to the cytoplasm upon foot-and-mouth disease virus (FMDV) infection to inhibit the cap-dependent translation of FMDV. In response, FMDV 3C protease also promotes the translocation of DDX23 from the nucleus to the cytoplasm, where it degrades DDX23 to escape its antiviral effect [Citation68]. DDX10 is primarily localized in the nucleus and functions mainly in inducing transcription factor production and regulating ribosome biosynthesis and RNA splicing [Citation69]. Here, we observed the translocation of DDX10 from the nucleus to the cytoplasm in cells infected with PRRSV or overexpressing E protein, which subsequently caused the autophagic degradation of DDX10. Thus, the cytoplasmic translocation of DDX10 may be an immune evasion strategy utilized by PRRSV. However, the molecular mechanisms by which PRRSV or its E protein promotes the translocation of DDX10 from the nucleus to the cytoplasm require further investigation.
PRRSV E protein is a structural protein essential for the establishment of virus infection and has been demonstrated to function as an ion channel protein for viral shedding [Citation9,Citation70,Citation71]. Herein, we reported for the first time the role of E protein in modulating autophagy. Additionally, the involvement of PRRSV nsp2 in the autophagy process has also been identified previously [Citation72]. Notably, we observed that the protein expression levels of DDX10 appeared to be significantly attenuated by nsp2 (Fig. S5), suggesting that nsp2 may induce autophagic degradation of DDX10. To further confirm this speculation, HEK-293T cells were co-transfected with DDX10 expression construct and different doses of expression plasmids encoding HA-tagged nsp2. The results of western blotting revealed that nsp2 repressed DDX10 expression in a dose-dependent manner (Fig. S8A), which was significantly restored by NH4Cl treatment (Fig. S8B), demonstrating that nsp2 degrades DDX10 via the autophagy pathway. However, nsp2 still induced DDX10 degradation in SQSTM1 KO cells, suggesting that nsp2 degrades DDX10 independently of SQSTM1 (Fig. S8C). Considering that nsp2 induces autophagy in a manner dependent on the formation of aggresomes, the perinuclear inclusion bodies formed by active minus-end-directed transport of misfolded proteins on microtubules [Citation72], it is possible that nsp2 promotes the autophagic degradation of DDX10 through aggresome-related mechanisms.
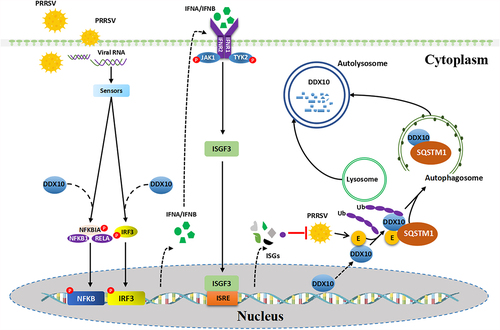
In summary, our present study reveals that DDX10 positively regulates the production of IFNB and induces the expression of ISGs, thereby inhibiting PRRSV proliferation. Significantly, to antagonize the antiviral activity of DDX10, PRRSV E protein interacts with DDX10 and facilitates its translocation from the nucleus to the cytoplasm where DDX10 is degraded via SQSTM1-dependent selective autophagy (). These findings fill the gap in our understanding of the mechanism by which PRRSV induces autophagy to facilitate its proliferation and provide new insights into the interaction between DDX10 and PRRSV for the development of anti-PRRSV drugs.
Figure 9. Proposed model for the autophagic degradation of DDX10 by PRRSV E protein to antagonize its antiviral activity. DDX10 activates the IFN-I signaling pathway and induces the expressions of ISGs to inhibit PRRSV proliferation. Conversely, PRRSV E protein facilitates the translocation of DDX10 from the nucleus to the cytoplasm and the SQSTM1-dependent selective autophagic degradation of DDX10, leading to immune escape.
Materials and methods
Cells and viruses
HEK-293T cells (American Type Culture Collection, CRL-11268) and MARC-145 cells (African green monkey kidney cells; American Type Culture Collection, CRL-12231) were cultured in Dulbecco’s Modified Eagle’s medium (DMEM; HyClone Laboratories Inc., SH30022.01) supplemented with 10% fetal bovine serum (FBS; PAN-Biotech, P30-1406) and 1% penicillin-streptomycin (Beyotime Biotechnology, C0222). iPAMs (an immortalized porcine alveolar macrophage cell line), PRRSV strain WUH3, and SeV have been described previously [Citation38,Citation73]. rPRRSV-EGFP, a recombinant PRRSV that stably expresses EGFP, was a kind gift from Dr. En-Min Zhou (Northwest A&F University, China) [Citation39].
Antibodies and reagents
Rabbit anti-ATG5 (ET1611-38) and anti-ATG7 (ET1610-53) monoclonal antibodies (mAbs) were obtained from Hangzhou HuaAn Biotechnology. Rabbit anti-SQSTM1 (16177S) and anti-LC3B (3868S) mAbs were obtained from Cell Signaling Technology. Mouse anti-SQSTM1 (MG432239) was purchased from Abmart. Rabbit anti-IRF3 (A2172), anti-p-IRF3 (AP0623), anti-RELA/p65 (A2547), anti-p-RELA (AP0123), anti-HSP90 (A0365), anti-LMNA (lamin A/C; A0249), and anti-ACTB (AC006) polyclonal antibodies were obtained from ABclonal Technology. Mouse anti-Flag (PM020) and anti-HA (M180-3) mAbs were purchased from MBL International Corporation. Mouse anti-ISG15 (sc-166,755), anti-IFITM3 (sc-100,768), and anti-OAS1 (sc-374,656) mAbs were purchased from Santa Cruz Biotechnology. Rabbit anti-DDX10 polyclonal antibody (A300-617A) was obtained from Bethyl Laboratories. Mouse anti-PRRSV N protein mAb (clone N4D2) was generated and stored in our laboratory.
Rapamycin (tlrl-rap) was obtained from InvivoGen. DMSO (276,855) and CQ (C6628) were purchased from Sigma-Aldrich. MG132 (S1748), Z-VAD-FMK (C1202), and NH4Cl (ST2030) were purchased from Beyotime Biotechnology.
Constructs
The Flag-tagged DDX10 eukaryotic expression plasmid containing the full-length cDNA of porcine DDX10 was constructed by PCR amplification of the cDNA from PK-15 cells, followed by its cloning into the pCAGGS vector (MiaoLingBio, P1267) with an N-terminal Flag tag (pCAGGS-Flag). The eukaryotic expression plasmids containing the genes encoding 40 different DDXs were constructed by cloning the individual DDX cDNA from Hela cells into the pCAGGS-Flag vector. The eukaryotic expression plasmids encoding PRRSV proteins were constructed by cloning each PRRSV gene into a pCAGGS-HA vector as described previously [Citation19]. Full-length cDNAs of autophagy receptors (SQSTM1, OPTN, NBR1, TOLLIP, and BNIP3L) were amplified from HEK-293T cells and separately inserted into pCAGGS-Flag vector to generate the eukaryotic expression plasmids pCAGGS-Flag-SQSTM1, pCAGGS-Flag-OPTN, pCAGGS-Flag-NBR1, pCAGGS-Flag-TOLLIP, and pCAGGS-Flag-BNIP3L, respectively. IFNB-Luc, NFKB-Luc, and IRF3-Luc have been described previously [Citation74]. LAMP1-DsRed was purchased from MiaoLingBio (P6764). GFP-LC3 was a gift from Dr. Hongbo Zhou at Huazhong Agricultural University, China. All constructed plasmids were verified by DNA sequencing.
Dual-luciferase reporter assay
Cells were cultured in 24-well plates. For transfection, 0.8 µg of the indicated expression plasmids or 50 pmol of siRNA, 0.1 µg of reporter plasmid (IFNB-Luc, IRF3-Luc, or NFKB-Luc), and 0.02 µg of pRL-TK reference plasmid (Promega Biotech Co., Ltd, E2241) were co-transfected with jetPRIME (Polyplus-transfection SA, 101000046) following the manufacturer’s instructions. At 24 h post transfection (hpt), the cells were stimulated with or without Sendai virus (SeV) (10 hemagglutinating activity units/well) for 12 h. Subsequently, the firefly luciferase and Renilla luciferase activities from lysed cells were evaluated through the Dual-Luciferase reporter assay system in accordance with the instructions of manufacturer (Promega Biotech Co., Ltd, E1980). Representative data from three independently conducted experiments are expressed as the relative firefly luciferase activities with normalization to the Renilla luciferase activities.
RNA interference
Small interfering (si) RNA targeting DDX10 (siDDX10), and negative control siRNA (NC) were designed by Suzhou GenePharma Co., Ltd, and the sequences used in our study are shown in Table S1. Cells that were 60% confluent were transfected with 50 pmol of siRNA by using jetPRIME (Polyplus-transfection SA, 101,000,046) in accordance with the manufacturer’s instructions.
RNA extraction and quantitative real-time (qRT)-PCR
Total cellular RNA was extracted using TRIzol reagent (Invitrogen, 15596018), and 1 µg of RNA from each sample was subsequently reverse transcribed to cDNA with the reverse transcription kit (Vazyme Biotech Co., Ltd, R223-01). The resulting cDNA was then used as the template in a SYBR green qPCR assay (Applied Biosystems, 4312704). Each sample’s abundance of individual mRNA transcripts was assayed three times, and the relative mRNA expression levels were normalized to the expression of GAPDH. All real-time PCR were performed in an ABI 7500 real-time PCR system (Applied Biosystems, USA). The qRT-PCR primers used in this study are shown in Table S2.
IFNB ELISA
Cells were collected and lysed, then the cell lysates were assayed for IFNB detection using IFNB ELISA kits (Solarbio Science & Technology Co., Ltd, SEKP-0046 for porcine IFNB; SEKH-0410 for human IFNB) according to the manufacturer’s instructions.
Indirect immunofluorescence assay (IFA)
The cells were seeded on microscope coverslips in 24-well plates. For examination by immunofluorescence microscopy, the cells were fixed with 4% paraformaldehyde for 10 minutes (min) at room temperature, permeabilized with methanol for 15 min, blocked in 10% bovine serum albumin for 30 min, and then incubated with primary antibodies. After being washed with PBS (HyClone Laboratories Inc., SH30256.01), the cells were incubated with the secondary antibodies Alexa Fluor 488-conjugated goat anti-mouse IgG (H + L) (Abbkine Scientific Co., Ltd, A23210) or Alexa Fluor 594-conjugated goat anti-rabbit IgG (H + L) (Abbkine Scientific Co., Ltd, A23420) for 1 h at 37°C. Finally, nuclei were stained with DAPI (Beyotime Biotechnology, C1002). Fluorescent images were acquired with a confocal laser scanning microscope (Fluoviewver.3.1; Olympus, Japan).
TCID50 assay
TCID50 assays to determine viral titers were performed as described previously [Citation75]. Briefly, MARC-145 cells were seeded in 96-well plates and then infected with serial 10-fold dilutions of supernatants in eight replicates. The plates were incubated for 72–96 h before virus titers were calculated. PRRSV titers were calculated using the Reed–Muench method and are shown as the TCID50 per milliliter [Citation76].
Western blotting assay
The cells were harvested with lysis buffer (Beyotime Biotechnology, P0013) supplemented with a protease inhibitor cocktail (Beyotime Biotechnology, P1010). Equal amounts of proteins were subjected to separation by 10% SDS-PAGE and then transferred to a polyvinylidene difluoride membrane, which was subsequently blocked with 10% nonfat milk in 1× Tris Buffered Saline with Tween 20 (TBST; 20 mM Tris, 150 mM NaCl, 0.1% Tween 20 [Shanghai Aladdin Biochemical Technology Co., Ltd, T104863]) and then treated with the indicated primary antibodies overnight at 4°C. After being washed three times with TBST, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Beyotime Biotechnology, A0208) or goat anti-mouse IgG (Beyotime Biotechnology, A0192) for 1 h at room temperature. After being washed, the membranes were visualized via treatment with enhanced chemiluminescence (ECL) reagents (Bio-Rad Laboratories Co., Ltd, 1705061).
Co-immunoprecipitation (co-IP) assay
At 30 hpt with expression plasmids, cells were collected and lysed using IP lysis buffer (50 mM Tris-HCl, pH 8.0, containing 150 mM NaCl and 1% Triton X-100 [Shanghai Aladdin Biochemical Technology Co., Ltd, T434386]). For immunoprecipitation, the resulting lysates were rapidly rotated on a rotary shaker for 30 min, and a portion of each supernatant from the lysed cells was used in the whole-cell extract assays. The remaining portions of the supernatants from the lysed cells were immunoprecipitated with the indicated antibodies overnight at 4°C and then treated with protein A + G agarose beads (Beyotime Biotechnology, P2055) for 4 h at 4°C. The beads containing the immunoprecipitation samples were washed three times with 1 ml of IP lysis buffer. Whole-cell extracts and immunoprecipitation samples were resuspended in SDS-PAGE loading buffer (Beyotime Biotechnology, P0286), boiled at 95°C for 10 min, and then subjected to western blot analysis.
Nuclear and cytosolic fractionation assay
Cells were harvested, lysed, and isolated using the Nuclear Cytosol Isolation Kit (Beyotime Biotechnology, P0028) in accordance with the manufacturer’s instructions and then subjected to western blot analysis.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8. Two-tailed unpaired t-tests were used to determine the significance of differences between groups and were considered statistically significant when the p-value was less than 0.05.
Supplemental Material
Download MS Word (13.1 MB)Acknowledgments
We would like to thank Dr. Jiyong Zhou (Zhejiang University) and Dr. Jun Cui (Sun Yat-sen University) for providing ATG7-KO-HEK-293T cells, ATG5-KO-HEK-293T cells, and SQSTM1-KO-HEK-293T cells. We also thank Dr. En-Min Zhou (Northwest A&F University) for providing rPRRSV-EGFP and Dr. Hongbo Zhou (Huazhong Agricultural University) for providing plasmid GFP-LC3.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data will be made available on request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2179844
Additional information
Funding
References
- Han J, Zhou L, Ge X, et al. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Microbiol. 2017Sep;209:30–47.
- Nelsen CJ, Murtaugh MP, Faaberg KS. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol. 1999 Jan;73(1):270–280.
- Han M, Yoo D. Engineering the PRRS virus genome: updates and perspectives. Vet Microbiol. 2014 Dec 5;174(3–4):279–295.
- Johnson CR, Griggs TF, Gnanandarajah J, et al. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J Gen Virol. 2011 May;92(Pt 5):1107–1116.
- Li Y, Tas A, Snijder EJ, et al. Identification of porcine reproductive and respiratory syndrome virus ORF1a-encoded non-structural proteins in virus-infected cells. J Gen Virol. 2012 Apr;93(Pt 4):829–839.
- Snijder EJ, Kikkert M, Fang Y. Arterivirus molecular biology and pathogenesis. J Gen Virol. 2013 Oct;94(Pt 10):2141–2163.
- Lalonde C, Provost C, Gagnon CA. Whole-genome sequencing of porcine reproductive and respiratory syndrome virus from field clinical samples improves the genomic surveillance of the virus. J Clin Microbiol. 2020 Oct 21;58(11):e00097–20.
- Wissink EHJ, Kroese MV, Maneschijn-Bonsing JG, et al. Significance of the oligosaccharides of the porcine reproductive and respiratory syndrome virus glycoproteins GP2a and GP5 for infectious virus production. J Gen Virol. 2004 Dec;85(Pt 12):3715–3723.
- Wissink EH, Kroese MV, van Wijk HA, et al. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. J Virol. 2005 Oct;79(19):12495–12506.
- Kim O, Sun Y, Lai FW, et al. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology. 2010 Jul 5;402(2):315–326.
- Han M, Du Y, Song C, et al. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1α subunit. Virus Res. 2013 Mar;172(1–2):54–65.
- Beura LK, Sarkar SN, Kwon B, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J Virol. 2010 Feb;84(3):1574–1584.
- Frias-Staheli N, Giannakopoulos NV, Kikkert M, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007 Dec 13;2(6):404–416.
- Huang C, Zhang Q, Guo XK, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator. J Virol. 2014 Sep;88(18):10934–10945.
- Yang L, He J, Wang R, et al. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus induces STAT2 degradation to inhibit interferon signaling. J Virol. 2019 Nov 15;93(22):e01352–19.
- Zhao K, Li LW, Jiang YF, et al. Nucleocapsid protein of porcine reproductive and respiratory syndrome virus antagonizes the antiviral activity of TRIM25 by interfering with TRIM25-mediated RIG-I ubiquitination. Vet Microbiol. 2019Jun;233:140–146.
- Zhao Y, Song Z, Bai J, et al. Porcine reproductive and respiratory syndrome virus Nsp4 cleaves ZAP to antagonize its antiviral activity. Vet Microbiol. 2020Nov;250:108863.
- Tao R, Fang L, Bai D, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 cleaves porcine DCP1A to attenuate its antiviral activity. J Immunol. 2018 Oct 15;201(8):2345–2353.
- Ke W, Fang L, Tao R, et al. Porcine reproductive and respiratory syndrome virus E protein degrades porcine cholesterol 25-hydroxylase via the ubiquitin-proteasome pathway. J Virol. 2019 Oct 15;93(20):e00767–19.
- Huo Y, Fan L, Yin S, et al. Involvement of unfolded protein response, p53 and Akt in modulation of porcine reproductive and respiratory syndrome virus-mediated JNK activation. Virology. 2013 Sep;444(1–2):233–240.
- Yuan S, Zhang N, Xu L, et al. Induction of apoptosis by the nonstructural protein 4 and 10 of porcine reproductive and respiratory syndrome virus. PLoS One. 2016 Jun 16;11(6):e0156518.
- Chen WY, Schniztlein WM, Calzada-Nova G, et al. Genotype 2 Strains of Porcine Reproductive and respiratory syndrome virus dysregulate alveolar macrophage cytokine production via the unfolded protein response. J Virol. 2018 Jan 15;92(2):e01251–17.
- Gao P, Chai Y, Song J, et al. Reprogramming the unfolded protein response for replication by porcine reproductive and respiratory syndrome virus. PLoS Pathog. 2019 Nov;15(11):e1008169.
- Chen Q, Fang L, Wang D, et al. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res. 2012 Feb;163(2):650–655.
- Liu Q, Qin Y, Zhou L, et al. Autophagy sustains the replication of porcine reproductive and respiratory virus in host cells. Virology. 2012 Aug 1;429(2):136–147.
- Li S, Zhou A, Wang J, et al. Interplay of autophagy and apoptosis during PRRSV infection of Marc145 cell. Infect Genet Evol. 2016Apr;39:51–54.
- Zhou A, Li S, Khan FA, et al. Autophagy postpones apoptotic cell death in PRRSV infection through Bad-Beclin1 interaction. Virulence. 2016 Dec 15;7(2):98–109.
- Wang G, Yu Y, Cai X, et al. Effects of PRRSV Infection on the Porcine Thymus. Trends Microbiol. 2020 Mar;28(3):212–223.
- Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010 Jun;20(3):313–324.
- De Bortoli F, Espinosa S, Zhao R. DEAH-Box RNA helicases in Pre-mRNA splicing. Trends Biochem Sci. 2021 Mar;46(3):225–238.
- Meier-Stephenson V, Mrozowich T, Pham M, et al. DEAD-box helicases: the Yin and Yang roles in viral infections. Biotechnol Genet Eng Rev. 2018 Apr;34(1):3–32.
- Li G, Feng T, Pan W, et al. DEAD-box RNA helicase DDX3X inhibits DENV replication via regulating type one interferon pathway. Biochem Biophys Res Commun. 2015 Jan 2;456(1):327–332.
- Oshiumi H, Miyashita M, Okamoto M, et al. DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep. 2015 May 26;11(8):1193–1207.
- Chen G, Guo X, Lv F, et al. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc Natl Acad Sci U S A. 2008 Mar 18;105(11):4352–4357.
- Zhu Y, Chen G, Lv F, et al. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc Natl Acad Sci U S A. 2011 Sep 20;108(38):15834–15839.
- Chen Q, Liu Q, Liu D, et al. Molecular cloning, functional characterization and antiviral activity of porcine DDX3X. Biochem Biophys Res Commun. 2014 Jan 24;443(4):1169–1175.
- Zhao S, Ge X, Wang X, et al. The DEAD-box RNA helicase 5 positively regulates the replication of porcine reproductive and respiratory syndrome virus by interacting with viral Nsp9 in vitro. Virus Res. 2015 Jan;2(195):217–224.
- Li J, Wang D, Fang P, et al. DEAD-box RNA helicase 21 (DDX21) positively regulates the replication of porcine reproductive and respiratory syndrome virus via multiple mechanisms. Viruses. 2022 Feb 24;14(3):467.
- Wang C, Huang B, Kong N, et al. A novel porcine reproductive and respiratory syndrome virus vector system that stably expresses enhanced green fluorescent protein as a separate transcription unit. Vet Res. 2013 Oct 31;44(1):104.
- Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011 May 27;34(5):680–692.
- Jin H, Zhou L, Ge X, et al. Cellular DEAD-box RNA helicase 18 (DDX18) Promotes the PRRSV Replication via Interaction with Virus nsp2 and nsp10. Virus Res. 2017 Jun;15(238):204–212.
- Zhang K, Zhang Y, Xue J, et al. DDX19 inhibits type I interferon production by disrupting TBK1-IKKε-IRF3 interactions and promoting TBK1 and IKKε degradation. Cell Rep. 2019 Jan 29;26(5):1258–1272.e4.
- Soulat D, Bürckstümmer T, Westermayer S, et al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. Embo J. 2008 Aug 6;27(15):2135–2146.
- Zan J, Xu R, Tang X, et al. RNA helicase DDX5 suppresses IFN-I antiviral innate immune response by interacting with PP2A-Cβ to deactivate IRF3. Exp Cell Res. 2020 Nov 15;396(2):112332.
- Zhang Z, Yuan B, Bao M, et al. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011 Sep 4;12(10):959–965.
- Overend C, Mitchell R, He D, et al. Recombinant swine beta interferon protects swine alveolar macrophages and MARC-145 cells from infection with Porcine reproductive and respiratory syndrome virus. J Gen Virol. 2007 Mar;88(Pt 3):925–931.
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006 Feb;7(2):131–137.
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014 Jun;16(6):495–501.
- Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005 Nov 21;171(4):603–614.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007 Aug 17;282(33):24131–24145.
- Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005 Oct 20;437(7062):1167–1172.
- Wang H, Kim S, Ryu WS. DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J Virol. 2009 Jun;83(11):5815–5824.
- Parvatiyar K, Zhang Z, Teles RM, et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol. 2012 Dec;13(12):1155–1161.
- Miyashita M, Oshiumi H, Matsumoto M, et al. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol. 2011 Sep;31(18):3802–3819.
- Su YS, Tsai AH, Ho YF, et al. Stimulation of the internal ribosome entry site (IRES)-dependent translation of enterovirus 71 by DDX3X RNA helicase and viral 2A and 3C proteases. Front Microbiol. 2018;9:1324.
- Wang R, Zhu Y, Zhao J, et al. Autophagy promotes replication of influenza A virus in vitro. J Virol. 2019 Feb 15;93(4):e01984–18.
- Chan ST, Ou JJ. Hepatitis C virus-induced autophagy and host innate immune response. Viruses. 2017 Aug 12;9(8):224.
- Hou P, Wang X, Wang H, et al. The ORF7a protein of SARS-CoV-2 initiates autophagy and limits autophagosome-lysosome fusion via degradation of SNAP29 to promote virus replication. Autophagy. 2022Jun;19:1–19.
- Du Y, Duan T, Feng Y, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. Embo J. 2018 Feb 1;37(3):351–366.
- Liu J, Wu X, Wang H, et al. HFE inhibits type I IFNs signaling by targeting the SQSTM1-mediated MAVS autophagic degradation. Autophagy. 2021 Aug;17(8):1962–1977.
- He X, Zhu Y, Zhang Y, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. Embo J. 2019 Jul 15;38(14):e100978.
- Wu Y, Jin S, Liu Q, et al. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy. 2021 Jun;17(6):1379–1392.
- Sui C, Xiao T, Zhang S, et al. SARS-CoV-2 NSP13 inhibits type I IFN production by degradation of TBK1 via p62-dependent selective autophagy. J Immunol. 2022 Feb 1;208(3):753–761.
- Deng T, Hu B, Wang X, et al. TRAF6 autophagic degradation by avibirnavirus VP3 inhibits antiviral innate immunity via blocking NFKB/NF-κB activation. Autophagy. 2022Mar;10:1–18.
- Zeng Y, Xu S, Wei Y, et al. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021 Feb;17(2):e1009300.
- Hou P, Yang K, Jia P, et al. A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. 2021 Jan;31(1):62–79.
- Wang G, Yu Y, Tu Y, et al. Highly pathogenic porcine reproductive and respiratory syndrome virus infection induced apoptosis and autophagy in thymi of infected piglets. PLoS One. 2015;10(6):e0128292.
- Abdullah SW, Han S, Wu J, et al. The DDX23 negatively regulates translation and replication of foot-and-mouth disease virus and is degraded by 3C proteinase. Viruses. 2020 Nov 25;12(12):1348.
- Savitsky K, Ziv Y, Bar-Shira A, et al. A human gene (DDX10) encoding a putative DEAD-box RNA helicase at 11q22-q23. Genomics. 1996 Apr 15;33(2):199–206.
- Snijder EJ, van Tol H, Pedersen KW, et al. Identification of a novel structural protein of arteriviruses. J Virol. 1999 Aug;73(8):6335–6345.
- Veit M, Matczuk AK, Sinhadri BC, et al. Membrane proteins of arterivirus particles: structure, topology, processing and function. Virus Res. 2014;194:16–36.
- Cao S, Liu J, Ding G, et al. The tail domain of PRRSV NSP2 plays a key role in aggrephagy by interacting with 14-3-3ε. Vet Res. 2020 Aug 18;51(1):104.
- Fang P, Fang L, Ren J, et al. Porcine Deltacoronavirus Accessory Protein NS6 Antagonizes Interferon Beta Production by Interfering with the Binding of RIG-I/MDA5 to Double-Stranded RNA. J Virol. 2018 Aug 1;92(15):e00712–18.
- Wang D, Fang L, Shi Y, et al. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J Virol. 2016 Feb 15;90(4):2090–2101.
- Ke W, Fang L, Jing H, et al. Cholesterol 25-hydroxylase inhibits porcine reproductive and respiratory syndrome virus replication through enzyme activity-dependent and -independent mechanisms. J Virol. 2017 Oct 1;91(19):e00827–17.
- Pizzi M. Sampling variation of the fifty percent end-point, determined by the reed-muench (Behrens) method. Hum Biol. 1950 Sep;22(3):151–190.