
ABSTRACT
Selenoprotein GPX4 (glutathione peroxidase 4), originally known as PHGPX (phospholipid hydroperoxide glutathione peroxidase), is the main oxidoreductase in the use of glutathione as a reducing agent in scavenging lipid peroxidation products. There are three GPX4 isoforms: cytosolic (cGPX4), mitochondrial (mGPX4), and nuclear (nGPX4), with distinct spatiotemporal expression patterns during embryonic development and adult life. In addition to inducing the main phenotype of ferroptosis, the loss of GPX4 can in some cells trigger apoptosis, necroptosis, pyroptosis, or parthanatos, which mediates or accelerates developmental defects, tissue damage, and sterile inflammation. The interaction of GPX4 with the autophagic degradation pathway further modulates cell fate in response to oxidative stress. Impaired GPX4 function is implicated in tumorigenesis, neurodegeneration, infertility, inflammation, immune disorders, and ischemia-reperfusion injury. Additionally, the R152H mutation in GPX4 can promote the development of Sedaghatian-type spinal metaphyseal dysplasia, a rare and fatal disease in newborns. Here, we discuss the roles of classical GPX4 functions as well as emerging GPX4-regulated processes in cell death, autophagy, and disease.
Abbreviations: AA: arachidonic acid; cGPX4: cytosolic GPX4; CMA: chaperone-mediated autophagy; DAMPs: danger/damage-associated molecular patterns; mGPX4: mitochondrial GPX4; nGPX4: nuclear GPX4; GSDMD-N: N-terminal fragment of GSDMD; I/R: ischemia-reperfusion; PLOOH: phospholipid hydroperoxide; PUFAs: polyunsaturated fatty acids; RCD: regulated cell death; ROS: reactive oxygen species; Se: selenium; SSMD: Sedaghatian-type spondylometaphyseal dysplasia; UPS: ubiquitin-proteasome system
Introduction
Cell death takes many forms and is not only involved in normal tissue development and homeostasis, but also triggers pathological inflammatory responses [Citation1]. Regulated cell death (RCD) refers to a large class of cell death forms that can be controlled by genetic and pharmacological approaches [Citation2]. Uncontrolled oxidative stress is a common signal involved in the initiation or execution phases of cell death [Citation3]. One of the common manifestations of oxidative stress is damage to the plasma membrane and membrane-bound organelles, which not only affects cell function, but even leads to cell lysis [Citation4]. This process is associated with lipid peroxidation, which is one of the major consequences of free radical-mediated damage to membrane structures [Citation5]. In addition to macroautophagy (hereafter referred to as autophagy), some enzymatic and non-enzymatic antioxidants defend against oxidative damage to maintain cellular homeostasis and tissue development. However, uncontrolled autophagy can also cause cell death, which is referred to as autophagy-dependent cell death [Citation6]. Understanding the context-dependent relationship between the autophagy pathway and cell death and the key regulator of this interplay may establish new approaches for the treatment of human diseases.
GPX (glutathione peroxidase) is a protein superfamily that carries cysteine as a redox-active residue in the catalytic site [Citation7]. The human GPX family contains eight isoforms with distinct tissue expression, structure, substrate specificity, and function [Citation8]. Mammalian GPX1, GPX2, GPX3, and GPX4 are selenoenzymes; in humans, GPX6 is a selenoprotein, whereas in rodents the protein is present as a cysteine-containing homolog [Citation9] (). GPX4, originally known as PHGPX (phospholipid hydroperoxide glutathione peroxidase), was initially purified in porcine liver or heart in 1982 as a peroxidation-inhibiting protein on liposomes and biological membranes in the presence of glutathione (GSH) [Citation10]. We now know that GPX4 reduces a variety of oxidative substrates (e.g., organic and hydroperoxides and hydrogen peroxide), thereby protecting cells from oxidative damage [Citation8]. gpx4 global knockout mice exhibit embryonic lethality, whereas gpx4-related conditional knockout mice also display selective developmental defects, increased cell death, or disease susceptibility ().
Figure 1. Differences among GPX family members. Briefly, human GPX members can be divided into groups of selenoproteins (GPX1, 2, 3, 4, and 6) and non-selenoproteins (GPX5, 7, and 8) with different subcellular locations, spatial structures, and molecular masses.
Table 1. Developmental and disease phenotypes associated with GPX4 deficiency in mice.
In this review, we first describe the structure and function of GPX4, then outline the novel roles of GPX4 in cell death and autophagy, and finally discuss the impact of GPX4 on embryonic development and disease.
GPX4 structure, isoform, and function
Structurally, GPX4 has a typical thioredoxin motif, comprised of four α-helices and 7 b-strands located near the protein surface, 5 of which cluster together to form a central b-sheet. GPX4 contains the conserved catalytically active tetrad observed in other GPX selenozymes this is comprised of selenocysteine (Sec46), glutamine (Gln81), tryptophan (Trp136), and asparagine (Asn137) [Citation42]. The stability of Sec46 is controlled by Gln81 and Trp136 via hydrogen-bonding and electrostatic interactions. Although Asn137 is responsible for catalytic activity, mutation of any of the residues in the tetrad inhibits GPX4 activity [Citation42]. A mutation of selenocysteine to cysteine reduces the activity of GPX4 by 90%. The structure of the GPX4 monomer lacks an internal extension of 20 amino acids that is conserved in the tetrameric GPX enzyme, which encodes a loop around the active site to prevent the entry of complex substrates into the active site [Citation8]. Therefore, the activity of GPX4 is not only affected by a single amino acid, but also by its spatial structure.
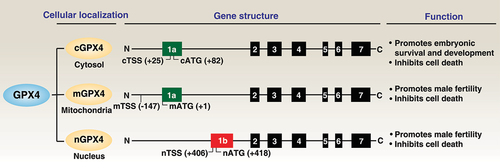
GPX4 can be expressed in a variety of tissues, with the highest content in the testis, affecting the development and function of sperm [Citation43]. GPX4 is present as three physiological isoforms: cytosolic (cGPX4), mitochondrial (mGPX4), and nuclear (nGPX4; ). The structural integrity of mammalian sperm chromatin depends on nGPX4, whereas cGPX4 is required for embryonic survival and development [Citation18,Citation22,Citation23,Citation26]. In addition to protecting from infertility, mGPX4 limits cell death induced by mitochondrial reactive oxygen species (ROS) or oxidized α-ketoisocaproic acid (a metabolic intermediate in the tricarboxylic acid/TCA cycle) [Citation44,Citation45]. cGPX4 can be translocated into the nucleus [Citation11]; mGPX4 contains an N-terminal 27 amino acid mitochondria-targeting sequence [Citation46]; and nGPX4 has a canonical nuclear localization signal, but more importantly lysine/arginine rich domains (similar to that in protamines) which facilitates nGPX4 binding to sperm DNA enabling oxidation of cysteines in protamines [Citation47]. In the liver tissue of humans and mice with metabolic-associated fatty liver disease, lipid stress induces the expression of a non-canonical transcript variant of GPX4, known as iGPX4 [Citation48]. This variant plays an opposing role by promoting oxidative stress and ferroptosis, due to its ability to maintain GPX4 in inactive oligomers [Citation48]. Further understanding of the localization sequences and regulatory signals of different GPX4 isoforms may lead to new therapeutic strategies to protect against oxidative damage.
Figure 2. GPX4 isoenzymes. The GPX4 protein has three isoforms (cGPX4, mGPX4, and nGPX4) encoded by distinct 1a and 1b exons. They exhibit overlapping and distinct functions in regulating development and cell death.
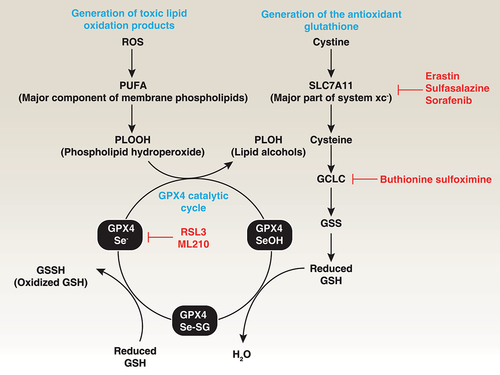
The main function of GPX4 is to use GSH as a co-factor to resist lipid peroxidation, thereby protecting the integrity of the membrane (). GPX4 serves as a critical antioxidant peroxidase, reducing phospholipid hydroperoxide (PLOOH), even when the latter is incorporated in membranes and lipoproteins. In addition to using GSH to eliminate toxic PLOOH, GPX4 can reduce thymine hydroperoxide, cholesterol hydroperoxide, and fatty acid hydroperoxide, protecting cells from oxidative damage [Citation49,Citation50]. Structural and biochemical studies have demonstrated that GPX4 interacts with the polar head of phospholipids, allowing it to bind to bilayer membranes [Citation51]. In addition to inhibition of hydroperoxide-dependent reactions, GPX4 has the additional function of inhibiting the activity of the ALOX (arachidonate lipoxygenase) family [Citation52]. However, whether ALOX is a key mediator of lipid peroxidation induced by GPX4 deficiency is debated [Citation20,Citation27]. Consequently, the dysfunction of GPX4 in preventing lipid peroxidation can trigger various types of RCD and tissue damage (discussed below).
Figure 3. Role of GPX4 in lipid peroxidation. Polyunsaturated fatty acids (PUFAs) are major components of cell membrane phospholipid bilayers and are susceptible to oxidative damage that produces the toxic lipid oxidation product PLOOH. In contrast, membrane system xc− is a heterodimeric complex composed of SLC7A11 and SLC3A2, which mediates cystine uptake into cells that is followed by GSH synthesis via the GCLC-GSS pathway. GSH is a co-factor in the catalytic cycle of GPX4 that detoxifies PLOOH to the lipid alcohol PLOH. Therefore, the pharmacological inhibition of GCLC, SLC7A11, and GPX4 using indicated small-molecular compounds can promote or enhance lipid peroxidation.
Another unique function of GPX4 compared to other GPX family members is the regulation of embryonic development in mammals. In mouse models, gpx4-/- embryos die in utero by mid-gestation (E7.5) [Citation11,Citation12]. In contrast, the global depletion of GPX1, GPX2, or GPX3 in mice produces no embryonic lethality. Because GPX4 ablation or inaction is embryonically lethal, only heterozygous or conditional knockout mice can be analyzed (). There are two reported Gpx4 genes, Gpx4a and Gpx4b. The dynamics of GPX4B expression suggest its proper role in fundamental developmental processes compared to GPX4A [Citation53]. However, GPX4B inhibits dorsal organizer formation via interacting with LEF1 (lymphoid enhancer binding factor 1) and decreasing conserved Wnt-Ctnnb1/β-catenin signaling in zebrafish embryos, which is independent of its selenoprotein activation [Citation54]. In addition, mGPX4 deficiency impairs hindbrain development and induces cerebral apoptosis [Citation55]. Silencing the expression of nGPX4 leads to retardation in atrium formation [Citation55]. Thus, GPX4 has unique protective functions in development and life that other GPX members may not be able to replace.
GPX4 expression and modification
GPX4 contains eight exons, including alternative 1a and 1b (). The first 5“translational start site is present in exon 1a producing the long form mGPX4; a second translation start site exists closer to the 3” end and is used to generate the short form cGPX4. The translational start site for nGPX4 is present within the alternative exon 1b. All GPX4 isoforms share exon 2, 3, 4, 5, 6, and 7. Of note, the in-frame TGA opal stop codon that codes for Sec insertion is contained within exon 3. GPX4 expression is dependent on the availability of Sec, which is encoded by the stop codon UGA. The 3’ UTR of selenoprotein mRNA contains a conserved stem loop structure called the Sec insertion sequence/SECIS element; the Sec insertion sequence is required for UGA to be recognized as a Sec codon instead of a stop [Citation56]. The re-coding of highly specific UGA codons from stop signals to Sec is driven by selenocysteinyl-tRNASec, which is synthesized from selenophosphate [Citation57].
Statins are inhibitors of isopentenyl pyrophosphate biosynthesis and are used as anticholesterol drugs that preferentially affect selenocysteinyl-tRNASec-mediated GPX4 biosynthesis [Citation58]. Selenium released as a product from the degradation of other selenoproteins may increase Gpx4 mRNA stability and protein synthesis under conditions of selenium deficiency [Citation59]. Furthermore, GPX4 expression is regulated by multiple transcription factors, such as SP1 (Sp1 transcription factor) and TFAP2A/AP2 (transcription factor AP-2 alpha), in specific cells or conditions [Citation9] ().
Figure 4. Expression and modification of GPX4. (A) Multiple transcription factors are required for GPX4 expression. As a selenoprotein, Sec-tRNA[Ser]Sec requires a Sec insertion sequence element in Gpx4 mRNA. (B) GPX4 can undergo ubiquitination (Ub), phosphorylation (P), sumoylation (Su) and alkylation (Alk) at specific sites. (C) Both the UPS and autophagy are involved in GPX4 protein degradation.
![Figure 4. Expression and modification of GPX4. (A) Multiple transcription factors are required for GPX4 expression. As a selenoprotein, Sec-tRNA[Ser]Sec requires a Sec insertion sequence element in Gpx4 mRNA. (B) GPX4 can undergo ubiquitination (Ub), phosphorylation (P), sumoylation (Su) and alkylation (Alk) at specific sites. (C) Both the UPS and autophagy are involved in GPX4 protein degradation.](/cms/asset/20575311-76f4-45d6-8536-9688850c856a/kaup_a_2218764_f0004_oc.jpg)
Posttranslational modifications play complex roles in regulating protein expression and activity. GPX4 can be modified by ubiquitination on Lys47, Lys80, Lys107, Lys135, Lys162, and Lys167 or by phosphorylation on Ser40, Ser13, and Tyr 96 or by succination at Cys93 [Citation60] or by alkylation at Cys107 [Citation61] (). However, whether these posttranslational modifications are critical for GPX4 function remains uncertain. Regardless, a variety of small-molecule compounds or drugs can affect the expression of GPX4, most of which cause GPX4 degradation [Citation62]. Further understanding the signals and mechanisms of GPX4 degradation may help in developing specific GPX4 inhibitors or activators.
The ubiquitin-proteasome system (UPS) is the primary mechanism for the degradation of short-lived proteins and acts through a multilevel cascade consisting of E1, E2, and E3 enzymes. Conversely, deubiquitinases/DUBs remove ubiquitin chains from substrate proteins, counteracting the activity of E3 ligases, thereby stabilizing the targeted proteins. The alladium pyrithione complex is a broad-spectrum inhibitor of deubiquitinases that induces apoptosis and ferroptosis in human non-small cell lung cancer cells in part through GPX4 degradation [Citation63]. Similarly, increased GPX4 ubiquitination by DMOCPTL, a natural product parthenolide derivative, induces ferroptosis and apoptosis in human triple-negative breast cancer cells [Citation64]. DMOCPTL can bind to the GPX4 active site (Sec73Cys) [Citation64,Citation65], highlighting that DMOCPTL results in the direct degradation of GPX4. Another natural small molecule, bufatalin, also promotes ubiquitination and degradation of GPX4 and subsequent ferroptosis in human non-small cell lung cancer cells [Citation66]. Environmental carcinogens (e.g., benzo[a]pyrenediol epoxide/BPDE [Citation67]) and electrophilic dopamine quinone [Citation68] are potential factors to induce UPS degradation of GPX4. Another nonclassical degradation inducer of the GPX4 protein is a missense mutation in the gene encoding its synthase and its pathogenic variant (the R152H variant), suggesting that Lys125/Lys127 are sites for ubiquitin ligation and subsequent degradation [Citation69,Citation70]. Besides the UPS, the activation of autophagy, including chaperone-mediated autophagy [Citation71], mediates GPX4 degradation to suppress tumor growth or cause tissue damage by activating ferroptosis [Citation71–76] (discussed below, ).
Some binding proteins affect GPX4 protein degradation and activity. For example, unlike the autophagic GPX4 degradation mediated in breast cancer cells by the binding of HSP90 (heat shock protein 90) to GPX4 [Citation71], the upregulation of HSPA5/BIP (heat shock protein family A (Hsp70) member 5) prevents GPX4 degradation through protein-protein interactions in pancreatic cancer cells [Citation77]. A small-scale RNAi screen identified SMG9 (SMG9 nonsense mediated MRNA decay factor), a component of the nonsense-mediated mRNA decay/NMD machinery, that promotes ferroptosis by binding and promoting GPX4 degradation in cancer cells through a nonsense-mediated mRNA decay-independent manner [Citation78]. The linear ubiquitination assembly complex, consisting of RNF31/HOIP (ring finger protein 31) and SHARPIN (SHANK associated RH domain interactor), binds and stabilizes GPX4 protein in mouse fibroblasts under normal or oxidative stress conditions [Citation79]. Furthermore, mass spectrometry identified the redox-regulated adaptor protein YWHAE/14-3-3ε as a protein that competitively inhibits the interaction of GPX4 with its small-molecule inhibitor RSL3 through a conformational transition of GPX4 [Citation80]. These findings also raise the hypothesis of whether changes in the spatial conformation of GPX4 affect its protein degradation [Citation81]. Although the mechanisms that enable GPX4 expression or degradation in response to drug treatment are well understood, much more is to be learned of the complex regulatory processes that control GPX4 levels during pathological conditions.
GPX4 metabolic mechanism
As first reported in 1963 [Citation82], lipid peroxidation generally refers to the process by which oxidants (such as free radicals) attack the unsaturated carbon bonds of lipids, especially those found in polyunsaturated fatty acids (PUFAs) [Citation83]. GPX4 has broader substrate recognition specificity than other GPX enzymes in response to lipid peroxidation on PUFAs. Below, we discuss the three main types of metabolic pathways that GPX4 is involved in.
GSH metabolism
GSH is the most abundant low-molecular weight thiol in mammalian cells and plays a well-known antioxidant function [Citation84]. Two cytosolic enzymes, GCLC/γ-GCS (glutamate-cysteine ligase catalytic subunit) and GSS (glutathione synthetase), sequentially catalyze the synthesis of GSH from glycine, cysteine and glutamate in the presence of adenosine 5’-triphosphate/ATP () [Citation84]. Buthionine sulphoximine/BSO is an inhibitor of GCLC and therefore lowers GSH concentrations. In addition, enteral or parenteral cystine, methionine, N-acetyl-cysteine, and L-2-oxothiazolidine-4-carboxylate are effective precursors of cysteine for tissue GSH synthesis [Citation84]. With regard to degradation, GSH is degraded in an extracellular manner followed by GSH export and is hydrolyzed by GGT/gamma-glutamyltranspetidase. GPX4 exhibits catalytic oxidation-reduction steps involving redox shuttling of the Sec active site between oxidized and reduced states. This redox state change of GPX4 is associated with a change between the reduced form GSH or the oxidized GSH disulfide form/GSSH, which ultimately converts toxic PLOOH to chemically inert lipid alcohols/PLOH [Citation85]. Therefore, the depletion of GSH typically results in inactivity of GPX4 [Citation86]. This dimerization of GPX4 and GSH is reversible by reduction. The amino acid antiporter system xc− is a heterodimeric complex composed of SLC7A11 (solute carrier family 7 member 11) and SLC3A2 (solute carrier family 3 member 2) that typically mediates the exchange of extracellular cystine and intracellular glutamate across cell membranes. System xc−-mediated cystine uptake promotes not only GSH synthesis, but also GPX4 protein synthesis through activating MTOR (mechanistic target of rapamycin kinase), a regulator of lipid metabolism and autophagy, indicating a crosstalk between autophagy and ferroptosis [Citation87,Citation88].
Arachidonic acid metabolism
The lipid arachidonic acid (AA) is a major component of cell membranes, is a PUFA and is primarily metabolized by three families of enzymes: by PTGS1/COX (prostaglandin-endoperoxide synthase 1) to form prostaglandins and thromboxanes, by ALOX to form leukotrienes, and by CYP (cytochrome P450 family) enzymes to form epoxides [Citation89]. These three pathways allow AA to be converted into a range of metabolites that can trigger different inflammatory responses [Citation90]. GPX4 promotes inflammation resolution by eliminating oxidative species produced via an AA metabolic network [Citation91]. An ω-6 PUFA AA-enriched Western diet was recently found to be a risk for inflammatory bowel disease [Citation92]. Cytokines triggered by PUFAs and AAs are restricted to GPX4, which suppresses ferroptosis (discussed below) rather than controls AA metabolism [Citation93]. Collectively these findings suggest that the activation of GPX4 can be used as an anti-inflammatory strategy, although there is little evidence that GPX4 can be released into the extracellular space as a danger signal.
Selenium metabolism
There exist at least 25 selenoproteins containing selenium (Se) as the amino acid Sec [Citation94]. The synthesis of Sec requires serine and inorganic Se (Se2-); it is encoded by the UGA stop codon and its incorporation involves a unique tRNA. The main form of Se found in food is selenomethionine in which Se substitutes for sulfur in methionine [Citation95]. Selenomethionine is not made in animals, which cannot distinguish it from methionine, resulting in its nonspecific incorporation into a wide range of Se-containing proteins [Citation95]. GPX4 is a selenoprotein that links selenium to cellular homeostasis by inhibiting ferroptosis [Citation34]. Decreased selenium levels increase cellular ferroptosis vulnerability and cause ribosome stalling due to inefficient translation at the GPX4 Sec UGA codon; this leads to an increase in ribosomal collisions, and premature termination of translation as well as UPS-dependent clearance of the N-terminal GPX4 fragment [Citation96].
GPX4 and cell death
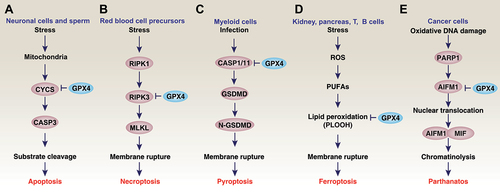
As one of the major antioxidant enzymes, GPX4 is a component in protecting cells and tissues from free radical damage and is therefore essential for cell survival. In contrast, GPX4 deficiency is associated with the course of several RCDs, especially ferroptosis, which will be covered in the following sections ().
Figure 5. GPX4 and cell death. Regulated cell death mechanisms have been studied in a wide range of oxidative stress models. The overexpression of GPX4 blocks cell death, whereas GPX4 depletion induces cell death in a cell-type and stimulus-dependent manner. For example, GPX4 has been demonstrated to be a negative regulator of apoptosis (A), necroptosis (B), pyroptosis (C), ferroptosis (D), or parthanatos (E).
Apoptosis
Apoptosis was originally described as a programmed cell death that occurs in multicellular organisms [Citation97]. CASPs (caspases) comprise a family of proteases that play a central role in apoptosis because of their activity to cleave specific target proteins, although caspase-independent apoptosis also occurs in some cases [Citation97]. In general, there are two main ways to initiate caspase-dependent apoptosis: through intrinsic and extrinsic pathways [Citation98]. The intrinsic pathway is controlled by the BCL2 protein family, which regulates apoptosis sensitivity through the control of mitochondrial membrane permeability and subsequent release of mitochondrial proteins, such as CYCS (cytochrome C, somatic) and DIABLO/SMAC (diablo IAP-binding mitochondrial protein) [Citation99]. The extrinsic pathway is activated by the binding of a cell surface death receptor (such as FAS [Fas cell surface death receptor], TNFRSF1A/TNFR1 [TNF receptor superfamily member 1A], or TNFSF10/TRAIL [TNF superfamily member 10]) to its corresponding ligand, leading to CASP8 activation [Citation97]. Finally, intrinsic and extrinsic pathways can activate a common effector CASP (e.g., CASP3 or CASP7) to trigger apoptosis.
Numerous early studies have shown that GPX4 acts as an anti-apoptotic factor in vitro and in vivo [Citation100–102]. Mechanistically, the overexpression of GPX4 protects cells from mitochondrial pathway-mediated apoptosis by blocking mitochondrial release of CYCS, inactivating CASP3, and inhibiting hydroperoxide production [Citation15,Citation100,Citation102–105] (). In vivo, mGPX4 knockout mice exhibit a cone-rod dystrophy-like phenotype, where the loss of cone photoreceptors precedes the loss of rod photoreceptors due to apoptosis [Citation19]. Among the hydroperoxides, cardiolipin hydroperoxide is the most highly inhibited by GPX4 [Citation86,Citation106]. GPX4 depletion in mouse embryonic fibroblasts and in neurons causes lipid peroxidation and AIFM1/AIF (apoptosis-inducing factor, mitochondrion-associated 1)-mediated and caspase-independent apoptosis [Citation20]. However, CRISPR-mediated knockout of AIFM1 fails to protect cells from GPX4 depletion-induced ferroptosis [Citation107]. Functionally, GPX4 disruption results in short life span, the failure of embryonic brain development, and male infertility, in part due to increased apoptosis [Citation15,Citation23,Citation108,Citation109]. These studies establish a role for GPX4 in blocking CASP-dependent and -independent apoptosis, which is associated with tissue development. It is still uncertain how to distinguish CASP-independent apoptosis from non-apoptotic cell death when they share some common regulators, such as AIFM.
Necroptosis
Necroptosis is another mode of RCD that mimics the features of apoptosis and necrosis, as discovered in studies of TNF (tumor necrosis factor) signaling under CASP inhibition [Citation110]. Briefly, TNF can induce CASP-independent necrosis through a mechanism involving RIPK1 (receptor interacting serine/threonine kinase 1). Necrostatins are necrosis inhibitors that target RIPK1, providing evidence that TNF-induced necrosis is regulated by a kinase, and this process is termed necroptosis [Citation111]. Subsequent studies reveal that RIPK1 and RIPK3 (receptor interacting serine/threonine kinase 3) are key upstream kinases that initiate necroptosis through the phosphorylation of executor protein MLKL (mixed lineage kinase domain like pseudokinase) by multiple regulators [Citation112]. Finally, phosphorylated MLKL oligomerizes and translocates to membranes, leading to the disruption of cell membranes and organelle membranes, which results in cell death and leakage of intracellular contents [Citation110,Citation112]. Although many pathological stimuli can trigger necroptosis, the most typical pathway for inducing necroptosis is TNF and FAS signaling, in which CASP8 inhibition activates necrosomes (a multi-protein complex containing RIPK1, RIPK3, and MLKL) to trigger necroptosis [Citation113]. This process is regulated by oxidative stress signals, especially mitochondrial ROS [Citation114].
A transgenic animal study of the development of anemia using GPX4 depletion in erythrocyte precursors provides the first evidence of GPX4 in inhibiting necroptosis, but not ferroptosis [Citation30] (). Rescue experiments reveal that the administration of vitamin E, but not a ferroptosis inhibitor and an iron chelator, suppresses anemia in mice caused by the conditional depletion of GPX4 in red blood cell precursors [Citation30]. Furthermore, the knockout of Ripk3 prevents the development of anemia with decreased lipid peroxidation caused by the conditional depletion of GPX4 in erythrocyte precursors [Citation30]. However, the depletion of ALOX12 and ALOX15 does not affect GPX4 depletion-induced erythropoiesis. In addition to necroptosis, the role of GPX4 in erythropoiesis may also be related to mitophagy to clear damaged mitochondria [Citation30]. In other cases, the overactivation of mitophagy by the mitochondrial complex I inhibitor BAY 87–2243 triggers necroptosis and ferroptosis in melanoma cells [Citation115]. These findings raise additional questions about how dysfunctional mitochondria trigger both CASP-independent necroptosis and ferroptosis. Additional work may be required to elucidate whether lipid peroxidation products affect the posttranslational modification of necroptotic regulatory proteins.
Pyroptosis
Pyroptosis is a regulated form of necrosis that occurs primarily due to the activation of CASP1- and CASP11-dependent inflammasomes in macrophages and monocytes [Citation116]. Furthermore, the cleavage of GSDMD (gasdermin D) by CASP1 and CASP11 to produce an N-terminal fragment of GSDMD (GSDMD-N) is a key event driving infection-associated pyroptotic cell death [Citation117]. Increased GSDMD-N binds to lipids on the inner leaflet of the plasma membrane; the resulting pores cause cellular lysis. Other GSDM family members play cell-type-dependent roles in mediating pyroptosis after cleavage by different enzymes, not only CASP1 or CASP11 [Citation118]. Targeting pyroptosis is an attractive strategy to limit lethal infection or enhance antitumor immunity [Citation119].
gpx4Mye-/- mice (myeloid cell-specific gpx4-knockout mice) are susceptible to lethal infection [Citation33]. Although the antioxidant vitamin E protects against septic death, only the administration of caspase inhibitors (Z-VAD-FMK and wedelolactone), but not other cell death inhibitors (ferrostatin-1 or necrostatin-1) prevents septic death in gpx4Mye-/- mice [Citation33]. The knockout of Casp11 in myeloid cells also confers similar protection in septic gpx4Mye-/- mice [Citation33] (). CASP11-mediated noncanonical inflammasome activation in pyroptosis requires two signals. Signal 1 (also known as the priming signal) is mediated by the transcriptional expression of pro-IL1B (interleukin 1 beta) and pro-CASP11 through the activation of the NFKB/NF-κB (nuclear factor kappa B) pathway [Citation118]. Signal 2 relies on CASP11-dependent cleavage of GSDMD to generate activated GSDMD-N [Citation118]. The deletion of Gpx4 in myeloid cells produces lipid peroxidation that affects only signal 2, but not signal 1 [Citation33]. CASP1-dependent NLRP3 (NLR family pyrin domain containing 3) inflammasome is also inhibited by GPX4, indicating that GPX4 has a broad role in inhibiting pyroptosis [Citation33]. Recently, GPX4 was identified as one of the target genes of MIR1656 in the regulation of pyroptosis in kidney tissue of broiler chicks [Citation120]. Combined with other pyroptotic genes, GPX4 can be used to build a prognostic model for diseases including malignancies, requiring further clinical validation [Citation121,Citation122]. A major future challenge will be to determine exactly how GPX4 regulates membrane oxidative damage under different inflammatory conditions.
Ferroptosis
Currently, the most comprehensive understanding of GPX4’s role in cell death is through ferroptosis, which shares similar mechanisms with the initial concept of oxytosis [Citation123]. Ferroptosis was originally described as iron-dependent non-apoptotic cell death induced by compounds that selectively kill RAS-mutant cancer cells [Citation124]. This oxidative death is characterized by an increase in lipid hydroperoxide and occurs in both cancer cells and normal tissues [Citation125,Citation126]. The process requires ACSL4 (acyl-CoA synthetase long chain family member 4)-mediated incorporation of PUFAs into phospholipids [Citation127–129], which is further controlled by various antioxidant and oxidative pathways [Citation130]. The pharmacological induction of ferroptosis mainly involves inhibition of the SLC7A11-GPX4 pathway, whereas the compounds ferrostatin-1 or liproxstatin-1 exhibit strong antioxidant activities in blocking ferroptosis [Citation85,Citation131,Citation132]. The extrinsic ferroptotic pathway involves the use of erastin, sulfasalazine, or sorafenib to inhibit SLC7A11-mediated cysteine uptake in the cell membrane, and subsequent GSH synthesis in the cytoplasm. The intrinsic ferroptosis pathway is completed by directly inhibiting GPX4 activity using compound RSL3, ML210, or FIN56. Other ferroptosis inducers, such as FINO2, are involved in the activation of iron accumulation [Citation133]. Most ferroptosis inducers lead to GPX4 degradation, suggesting that the protein degradation pathway favors ferroptosis [Citation62].
Although both cGPX4 and mGPX4 can inhibit ferroptosis under different conditions [Citation45,Citation85], the first evidence that gpx4 gene deletion induces ferroptosis was reported after an induced disruption of GPX4 in mouse embryonic fibroblast cells (also known as Pfa1 cells) [Citation21]. Subsequent conditional knockout mouse models showed that GPX4 expression in different organs or cells, such as kidney [Citation21], pancreas [Citation36], T cells [Citation28], or B cells [Citation35], prevents ferroptosis-related tissue damage, sterile inflammation, or immune responses (). Our understanding of GPX4-related ferroptosis modulation in different cell types also involves hepatocyte degeneration, neuroinflammation, myocardial infarction, tumorigenesis, and tumor therapy [Citation134–136]. The importance of GPX4’s utilization of cysteine to suppress ferroptosis is further confirmed by embryonic and tissue development studies using GPX4cys/cys mice, a tool to study the difference between thiolate- and selenolate-based GPX4 catalysis [Citation34]. Given that many GPX4-independent antioxidant systems have been identified in ferroptosis [Citation132], synergistic or complementary functions involving these different antioxidant pathways need to be further assessed.
Parthanatos
Parthanatos is triggered by excessive oxidative damage caused by genotoxic stress or excitotoxicity, which leads to hyperactivation of poly (ADP-ribose) polymerase (PARP). The first identification of parthanatos was in 2006 when hyperglycemia was found to increase mitochondrial ROS production and DNA damage in diabetes models [Citation137]. The PARP-dependent nuclear translocation of AIFM1 from mitochondria induces DNA breakage, ultimately leading to cell death through the formation of an AIFM1-MIF complex. This process is now associated with various diseases, such as Parkinson’s disease, stroke, heart attack, and diabetes [Citation138]. Cyclophosphamide, an alkylating agent, induces degradation of GPX4, which results in AIFM1-dependent parthanatos that suppresses tumor growth [Citation139]. However, it does not trigger lipid peroxidation-mediated ferroptosis [Citation139] (). These findings offer further insights into the relationship between oxidative damage, GPX4 degradation, and cell death outcomes, which can be context-dependent. Further studies are needed to investigate whether the GPX4-mediated regulation of parthanatos is a widespread mechanism that also occurs in other disease models.
GPX4 and autophagy
Autophagy is an evolutionarily conserved degradation mechanism that includes three subtypes: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [Citation140]. Mammalian macroautophagy degrades various substrates, including damaged organelles, dysfunctional proteins, and invading pathogens [Citation141]. The autophagy machinery relies on the ATG (autophagy related) protein family, which forms distinct protein complexes to control the generation of the membrane compartments that are the morphological and functional hallmark of this process [Citation142]. These include phagophores that engulf various cargos and mature into double-membrane autophagosomes, which fuse with lysosomes to become autolysosomes; the latter degrade the cargos and release the breakdown products into the cytosol for reuse [Citation143]. In many cases, elevated autophagy is a survival-promoting defense mechanism, but unlimited autophagy causes oxidative stress-related cell death [Citation144]. Recently, ferroptosis has been considered a type of autophagy-dependent cell death due to the selective autophagic degradation of anti-ferroptosis proteins (e.g., ferritin [Citation145,Citation146], BMAL1/ARNTL [basic helix-loop-helix ARNT like 1] [Citation147], SLC40A1/ferroportin-1 [Citation39], or GPX4 [Citation71] as well as organelles [e.g., lipid droplets] [Citation148]). In addition to involving classic autophagy upstream kinases, such as the inhibition of MTOR [Citation88] and the activation of AMP-activated protein kinase/AMPK [Citation149,Citation150], the process of autophagy-dependent ferroptosis requires certain specialized proteins, such as TMEM164 (transmembrane protein 164) [Citation76] and HPCAL1 (hippocalcin like 1) [Citation151]. Below, we summarize the interplay between GPX4 and autophagy, which determines cell death sensitivity.
Regulation of GPX4 by autophagy
The transmembrane protein TMEM164 is located in the plasma membrane and that of various organelles. In certain cancer cells, TMEM164 is needed for the formation of the ATG12–ATG5-ATG16L1 complex and subsequent degradation of GPX4 induced by ferroptosis activators (such as erastin or RSL3) () [Citation76]. However, ATG9A, not TMEM164, mediates the ATG12–ATG5-ATG16L1 complex assembly induced by starvation [Citation76]. HPCAL1, rather than SQSTM1/p62 (sequestosome 1), is a specific autophagy receptor involved in CDH2 (cadherin 2) degradation for ferroptosis in cancer cells [Citation151]. Conversely, TAX1BP1 (Tax1 binding protein 1) specifically mediates autophagic degradation of GPX4 in copper-induced ferroptosis, unlike SQSTM1-mediated GPX4 degradation in response to erastin [Citation151]. Mechanistically, exogenous copper increases the ubiquitination and aggregate formation of GPX4 by directly binding to cysteines in GPX4, Cys107 and Cys148, leading to subsequent recognition by TAX1BP1 [Citation151]. While the precise mechanisms linking copper and ferroptosis are still being studied, autophagy plays a multifaceted role in regulating metal-dependent cell death [Citation152].
Figure 6. GPX4 and autophagy. Autophagy is a dynamic degradation process that involves the formation of multiple membrane structures. In addition to the classical autophagic machinery, the induction of autophagy-dependent ferroptosis requires specific proteins, such as TMEM164, which is responsible for the formation of phagophores for autophagosome generation. The selective degradation of pro-survival proteins or organelles by autophagy promotes ferroptosis, including the passive release of DAMPs. On the other hand, the early secretion of inflammatory mediators such as DCN during ferroptosis is involved in secretory autophagy mediated by the fusion of the plasma membrane. The autophagic degradation of GPX4 is mediated by different mechanisms. For example, copper and erastin can promote GPX4 degradation by using autophagic receptors TAXIBP1 and SQSTM1, respectively. In addition to macroautophagy, CMA also mediates GPX4 degradation in response to erastin. This process can be inhibited by phosphorylated CKB at T133, which leads to GPX4 phosphorylation at S104.
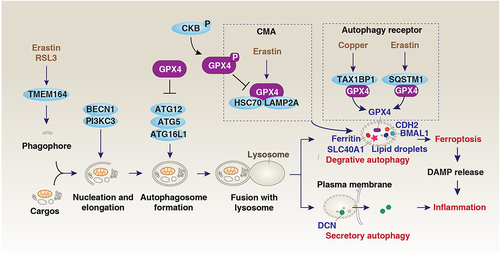
In addition to macroautophagy, interactions between GPX4 and HSPA8/HSC70 (heat shock protein family A (Hsp70) member 8) or LAMP2A (lysosomal associated membrane protein 2A) can promote CMA-mediated GPX4 degradation in HT1080 cancer cells [Citation71]. The inhibition of CMA-mediated GPX4 degradation can occur through CKB (creatine kinase B)-mediated phosphorylation of GPX4 at serine 104 residue [Citation153]. Because GPX4 is overexpressed in multiple cancers, inducing autophagy-dependent GPX4 degradation could be a different approach to anticancer therapy compared to inhibiting autophagy to trigger apoptosis.
Regulation of autophagy by GPX4
Because ROS can trigger different autophagy pathways, the antioxidant GPX4 is also a potential regulator of autophagy. The overexpression of GPX4 reduces cardiolipin peroxidation and prevents the formation of PLOOHs in the cytosol and mitochondria in response to photodynamic stress, thereby inhibiting autophagosome formation [Citation154]. L-carnitine, a compound produced in the body from lysine and methionine, reduces intracellular ROS and GSH levels and increases GPX4 mRNA levels, thereby enhancing autophagy and developmental capacity in aged oocyte embryos [Citation155]. In yeast, 12-hydroxyeicosatetraenoic acid (which is probably a product of GPX4 depletion in mammals) is a substrate for Atg8 (MAP1LC3 [microtubule-associated protein 1 light chain 3] in mammalian cells) lipidation required to activate autophagy [Citation156]. Further elucidation of the dynamic relationship between autophagy, lipid metabolism, and GPX4 is important for understanding cell fate [Citation140,Citation157].
GPX4 in disease
Impaired GPX4 activity is often positively associated with increased oxidative stress and RCD in disease. Therefore, targeting GPX4 for lipid homeostasis is a valuable approach to maintaining human health [Citation158]. In this section, we discuss the pathological functions and potential applications of GPX4 inhibition, most of which are related to studies on ferroptotic damage.
Cancer
On the one hand, tissue damage associated with cell death leads to chronic inflammation, which is a high risk for tumorigenesis. On the other hand, the induction of cell death is the main principle of anticancer therapy. Consistent with this concept, GPX4 plays a dual role in tumor initiation and progression (). Numerous studies have shown that GPX4 inhibitors enhance sensitivity to chemotherapy, radiotherapy, and immunotherapy by inducing ferroptosis [Citation159]. For example, enhanced ferroptosis by GPX4 inhibition increases the sensitivity of colorectal cancer to oxaliplatin, non-small-cell lung cancer to lapatinib, hepatocellular carcinoma to sorafenib, and Epstein-Barr virus-infected nasopharyngeal carcinoma to platinum [Citation160–163]. Of note, recent studies suggest that sorafenib may have no effect on triggering ferroptosis in cancer cells [Citation164] and ferroptosis is a non-immunogenic form of cell death [Citation165]. Inhibiting GPX4 is also a new way to kill cancer persister cells, the small subset of cancer cells that survive treatment and resume growth after the drug is removed [Citation166]. Recently, researchers developed GPX4 protein degradation strategies to promote ferroptotic cancer cell death. One is the use of a photodegradation targeting chimera/PDTAC, which selectively and efficiently degrades GPX4, resulting in ferroptosis and potent antitumor immunity [Citation167]. Another designed GPX4 degrader, namely dGPX4, which depletes GPX4 via proteasomal protein degradation, shows a 5-fold enhancement of efficiency in triggering ferroptosis compared to the GPX4 inhibitor ML162 [Citation168]. Because GPX4 can be expressed in antitumor immune cells (such as T cells [Citation28], dendritic cells [Citation169], and neutrophils [Citation170]) and immunosuppressive cells (e.g., Treg [Citation171]), nonselective targeting of GPX4 may cause immune side effects during preclinical cancer therapy. However, the completion of studies with human participants is needed to draw conclusions about the side effects.
Figure 7. Dual roles of GPX4 in cancer. The loss of GPX4 results in cell death and tissue damage. On the one hand, chronic inflammation may occur, leading to DAMP-mediated macrophage polarization in the tumor microenvironment, which can promote tumor growth. On the other hand, acute cell death caused by the loss of GPX4 can clear tumor cells and release DAMPs, activating anti-tumor immunity. GPX4 is widely expressed in cancer cells and immune cells, including DCs, CD8 T cells, and neutrophils. However, current GPX4 inhibitors are nonselective and can also cause immune cell death, leading to inhibition of anti-tumor immunity.
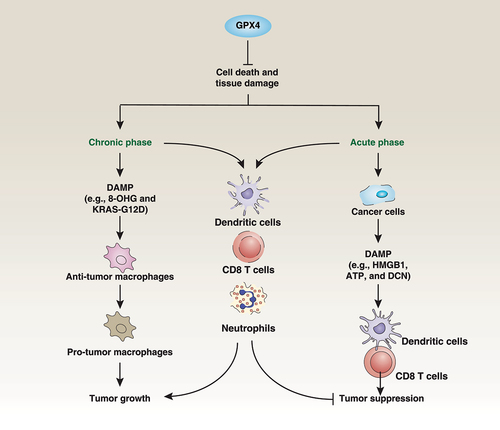
GPX4 depletion-induced ferroptosis accelerates KRAS-driven pancreatic tumorigenesis by activating certain inflammatory pathways, resulting in the polarization of macrophages for tumor growth [Citation37,Citation172]. This process is mediated by danger/damage-associated molecular patterns (DAMPs), which are endogenous danger-signaling molecules released from dead or dying cells. It has been reported that at least two DAMPs, namely mutant KRAS protein [Citation172] and the DNA oxidation product 8-hydroxyguanosine/8-OHG [Citation37], are released by ferroptotic cells to mediate macrophage polarization through the activation of AGER/RAGE (advanced glycosylation end-product specific receptor) and STING1/STING/TMEM173 (stimulator of interferon response cGAMP interactor 1) pathways, respectively. In early stages of erasin or RSL3 treatment of pancreatic cancer cells, the release of the proteoglycan DCN (decorin) is associated with secretory autophagy () [Citation173]. However, in later stages, DCN is released passively due to plasma membrane rupture. The release of DCN contributes to inflammation as well as antitumor immunity [Citation173]. While these studies are promising, researchers need to collect more data before they can recommend different strategies to target DAMPs for enhancing or inhibiting antitumor immunity.
Neurodegenerative disease
Neurodegeneration is an age-related pathological condition that results in the loss of neural structure and function, often accompanied by abnormal cell death, including apoptosis, necrosis, necroptosis, oxytosis, and ferroptosis. GPX4 facilitates neuronal differentiation in mice after birth [Citation20,Citation174,Citation175]. The ablation of GPX4 in forebrain neurons [Citation32] and spinal motor neurons [Citation29] promotes a loss of spatial learning and memory function, and accelerates the progression of motor neuron disease in mouse models. More specifically, a ferroptosis inhibitor and vitamin E diet ameliorate hippocampal neurodegeneration in forebrain neurons in mice that have undergone a conditional knockout of Gpx4 [Citation32]. In addition, conditional ablation of neuronal GPX4 via tamoxifen treatment in adult mice (3–4 months of age) results in a striking paralysis phenotype associated with motor neuron degeneration with ferroptosis-like features, whereas neurons in the cerebral cortex are unaffected [Citation29]. After brain injury, the induction of GPX4 can inhibit further cellular damage caused by ferroptosis [Citation29]. Thus, in this context, the induction of GPX4 expression and activity may be a strategy to protect against neurodegenerative diseases, such as Alzheimer and Parkinson diseases [Citation176,Citation177].
Inflammatory and autoimmune disorders
Sepsis is a potentially life-threatening disease caused by microbial infection and subsequent multiple organ dysfunction [Citation118]. As previously discussed, GPX4 expression in myeloid cells (especially macrophages) limits CASP1-CASP11-mediated inflammasome activation and GSDMD-dependent pyroptosis, thereby preventing a cytokine storm and septic death during bacterial infection [Citation33]. GPX4 also exhibits activity to inhibit inflammatory NFKB pathway activation and proinflammatory mediator production (e.g., production of IL1B and PTGS2/COX2 [prostaglandin-endoperoxide synthase 2]) in various experimental models, such as angiogenesis and hair follicle development [Citation25,Citation178]. A mouse model with conditional knockout of Gpx4 in T cells displays a phenotype of accumulated membrane lipid peroxides, leading to an intrinsic defect in protecting against acute lymphocytic choriomeningitis virus and Leishmania major parasite infections [Citation28]. Interestingly, peripheral T-cell survival, but not thymic T-cell survival and T-cell memory function, are affected by GPX4 depletion, supporting the idea that GPX4 is context-dependent for T-cell differentiation and function [Citation28]. B1 and marginal zone B cells play a key role in preventing autoimmunity by producing regulatory cytokines and natural antibodies, and die from ferroptosis in the absence of GPX4 [Citation35]. GPX4 depletion-mediated ferroptosis may be associated with COVID-19 [Citation179], an infectious disease caused by a recently identified coronavirus [Citation180]. A deficiency in GPX4 in the pancreas or small intestine can lead to pancreatitis [Citation36] and inflammatory bowel disease [Citation93]. Furthermore, the ablation of GPX4 in neutrophils exacerbates the development of autoimmune diseases, such as systemic lupus erythematosus, by inducing ferroptosis [Citation39]. Mechanistically, autoantibodies and type 1 interferon IFNA production during the latter disease reduce GPX4 expression through the transcriptional repressor CREM (cAMP response element modulator) [Citation39]. Overall, these conditional GPX4-depletion models suggest that GPX4 has a broad regulatory role in preventing inflammatory and autoimmune diseases.
Dietary selenium plays a functional role in inflammation and immunity, primarily through the incorporation of selenium into selenoproteins. Selenium deficiency can lead to GPX4 downregulation and oxidative immune stress in chicken thymus [Citation181]. Human follicular helper T cells are prone to ferroptosis due to GPX4 deficiency, and selenium supplementation reverses this process [Citation40]. The genetic depletion of the ferroptosis effector ACSL4 restores the viability of immune cells, such as CD8+ T cells [Citation182]. Therefore, the inhibition of ferroptosis by GPX4 may be a strategy for maintaining immune cell numbers during innate or adaptive immune responses. The involvement of GPX4 in immune cell differentiation requires further characterization.
The STING1 sensing pathway has emerged as a next-generation immunotherapy target in the fields of infection and immunity [Citation183]. Hyperactivation of STING1 is also a bridge between the autophagy response and cell death [Citation184]. A complex relationship exists between GPX4 and STING1, and key checkpoints of it remain unknown. On the one hand, STING1 mediates inflammation induced by GPX4 depletion-triggered ferroptosis injury [Citation37]. As a negative feedback mechanism, excessive lipid peroxidation can inhibit STING1 activation through lipid-aldehyde 4-hydroxynonenal/4-HNE binding to STING1, resulting in STING1 carbonylation at Cys88 [Citation185]. Cys88 is a key residue for final palmitoylation required for STING1 trafficking to the Golgi for STING1 activation [Citation185]. On the other hand, STING1-mediated autophagy and MFN1 (mitofusin1)-MFN2-dependent mitochondrial fusion can increase ferroptosis sensitivity to various stimuli, including GPX4 inhibitors [Citation186]. In the future, a comprehensive evaluation of the combination of STING1- and GPX4-targeted drugs for immune-related diseases is required.
Infertility
The process in which germ cells in the seminiferous tubules of the testis develop into haploid spermatozoa is referred to as spermatogenesis, and is physiologically finely regulated by oxidative stress. GPX4 (especially nGPX4 and mGPX4) is strongly expressed in testes and sperm, thereby ensuring male fertility. The nGPX4 is required for the structural integrity of mammalian sperm chromatin and is responsible for sperm maturation [Citation18,Citation47]. In addition, the nulling of mGPX4 also leads to male infertility in mice due to abnormal sperm maturation, but mGPX4 depletion fails to cause animal death [Citation23]. mGPX4 does not rescue the lethal phenotype in gpx4-null mice, but protects sterile mice expressing cGPX4 in the gpx4-null background [Citation22]. Furthermore, GPX4, especially mGPX4, is the major selenoenzyme for spermatogenesis in the testis [Citation23,Citation187]. Selenium deficiency increases GPX4-mediated lipid peroxidation and subsequent apoptotic cell death in germ cells [Citation109]. An inactive mutant form of GPX4 (the heterozygote of Gpx4_U46S) produces reduced litter size resulting from heterozygous reproduction and impaired sperm fertilization of oocytes in vitro [Citation26]. Overall, decreased GPX4 expression or impaired GPX4 activity cause sperm apoptosis, leading to male fertility. Accordingly, selenium is an essential nutrient for maintaining spermatogenesis.
Ischemia-reperfusion injury
Ischemia-reperfusion (I/R) injury is a pathological condition requiring physicians to manage local tissue damage and prevent systemic inflammation. DAMPs are immune mediators of cell death-induced I/R damage. Ferroptosis is also associated with I/R injury in multiple organs [Citation188]. Although it remains controversial whether ferroptosis is the ultimate causative factor for the condition, the overexpression of GPX4 prevents I/R damage in certain tissues by limiting lipid peroxidation [Citation189]. The depletion or inhibition of GPX4 expression can accelerate I/R damage. For example, the downregulation of GPX4 expression by Mir135b-3p promotes cardiomyocyte I/R injury [Citation190]. Statin-induced selective downregulation of selenoprotein (including GPX4) expression leads to increased sensitivity of hepatocytes to peroxides by preventing mevalonate-dependent maturation of a single human Sec-tRNA [Citation191]. As a gut microbiota metabolite, capsiate induces GPX4 expression and inhibits ferroptosis-related intestinal I/R injury [Citation40]. Reducing a high-fat diet prevents I/R damage mediated by GPX4 downregulation in the liver of obese mice [Citation192,Citation193]. Furthermore, bicyclol and glycyrrhizin prevent ferroptosis-induced liver and brain injury, respectively, by activating the GPX4 pathway in vivo [Citation194,Citation195]. The duration of ischemia determines the level of production of toxic oxidized lipids during liver I/R injury, such as phosphatidylcholine hydroperoxide/PC-OOH and phosphatidylethanolamine hydroperoxide/PE-OOH [Citation196]. Investigators must further evaluate the production of different lipid peroxidative metabolites and their impact on systemic inflammation during I/R injury.
Sedaghatian-type spondylometaphyseal dysplasia
Sedaghatian-type spondylometaphyseal dysplasia (SSMD) is a rare neonatal lethal disease characterized by severe metaphyseal chondrodysplasia with shortened limbs, cardiorespiratory deficits, and central nervous system abnormalities. Most of the affected infants die of respiratory failure within a few days of birth. Although more mutations in GPX4 (especially truncating ones) lead to SSMD, the development of SSMD is strongly associated with R152H mutations of GPX4, resulting in loss of enzymatic activity [Citation70]. In contrast, selenium supplementation, selective antioxidants, and deuterated PUFAs can alleviate the pathological changes of SSMD in mice [Citation69]. These genetic studies provide the only available evidence linking GPX4 mutations to human disease, making GPX4 a rare disease target. Additional studies are needed to determine how pediatricians can appropriately recommend these selenium-rich food strategies to patients with SSMD.
The implication of GPX4 inhibitors
Inhibitors of GPX4 have been developed and studied as potential cancer therapeutics, as well as for other ferroptosis-related diseases. GPX4 inhibitors are classified into two main types: GPX4 activity inhibitors and GPX4 protein degraders. RSL3, ML210, ML162, and FIN56 are among the most used GPX4 inhibitors to induce ferroptosis. For instance, RSL3 triggers ferroptotic cell death (with an IC50 of 0.01 µM in HRASG12V-expressing BJeHLT cells) by inducing a lethal accumulation of lipid hydroperoxides [Citation85]. RSL3 reduces tumor volume in various mouse xenograft models, with diffuse large B cell lymphomas and renal cell carcinomas being the most sensitive [Citation85]. ML210, a selective covalent inhibitor of cellular GPX4, is a potential anticancer agent in cancers that are resistant to conventional chemotherapy [Citation58,Citation197]. JKE-1716, a derivative of ML210, is a GPX4 inhibitor containing nitrolic acid, which effectively inhibits the growth of multiple cancer cell lines [Citation198]. ML162, which is selectively lethal to mutant RAS oncogene-expressing cells (with IC50s of 25 and 578 nM for HRASG12V-expressing and wild-type RAS-expressing BJeHLT cells, respectively), is another GPX4 inhibitor to suppress tumor cell proliferation [Citation197]. Recently, a study has suggested that RSL3 and ML162 may not directly inhibit GPX4, but instead target TXNRD1 (thioredoxin reductase 1) [Citation199]. The study used pure enzyme system and thermal stabilization assays to arrive at this conclusion, which challenges the previously reported mechanism of RSL3 and ML162 inducing ferroptosis [Citation199]. FIN56 induces ferroptosis in cancer cells by promoting the degradation of GPX4 protein and reducing the abundance of coenzyme Q10 [Citation200]. Moreover, some chemicals, nanomedicines, natural products, and clinical agents have been found to induce GPX4 degradation via either autophagy-dependent or UPS-dependent pathways, although these inhibitors may also impact other molecular targets [Citation62]. The use of GPX4 inhibitors in vivo (in living organisms) presents several challenges, particularly due to the risk of excessive systemic toxicity caused by nonselective inhibition of GPX4.
Conclusion and perspective
The production of oxidizing species, including ROS, can damage DNA, proteins, lipids, and carbohydrates, and even lead to cell death. Multiple protective mechanisms, including integrated antioxidant systems, are activated to remove free radicals and their toxic products. In this review, we summarized the function and modification of GPX4, a second selenoprotein found in animals, in preventing oxidative damage, particularly in various types of RCD. Selenium-dependent GPX4 plays a unique role in detoxifying lipid peroxides compared to other antioxidant enzymes in the body. Although lipid peroxidation plays a major role in mediating ferroptosis, this biochemical response to oxidative damage also occurs in other types of RCD [Citation201,Citation202]. While degradation of GPX4 by autophagy can enhance ferroptosis sensitivity, the drug and cellular selectivity of this pathway has largely remained unclear.
We also highlighted that GPX4 dysfunction is associated with a variety of developmental abnormalities and pathological conditions. In addition to common human diseases, such as cancer and neurogenerative diseases, GPX4 has been implicated in spermatogenesis and the pathogenesis of the rare genetic disease SSMD, as its mutation or downregulation reduces enzymatic activity. This knowledge may provide new strategies for targeting the GPX4 pathway to prevent developmental, inflammatory, or aging-related diseases.
Despite these advances in understanding GPX4 biology, several important questions remain to be answered. How does GPX4 have tissue or cell selectivity to prevent a different RCD? The process of cell death is a complex interconnected process involving multiple molecules. The absence of one molecule may briefly result in a phenotype, but from an evolutionary perspective, other molecules would have evolved to take over its function. Not all conditional GPX4 mouse models produce spontaneous disease phenotypes on a normal diet, although a vitamin E-depleted diet accelerates the pathogenic phenotype of cell death [Citation31]. While vitamin E is thought to act primarily as a chain-breaking antioxidant to prevent the spread of lipid peroxidation, other nutrient regulatory functions certainly occur [Citation203]. Indeed, in some cases, vitamin E supplementation does not completely prevent iron-dependent lipid peroxidation [Citation204]. Are GPX4-mediated antioxidant functions required for the physiological functions of all tissues? Is there a Se-independent function of GPX4 in oxidative damage? How can we selectively target GPX4 only in cancer cells, but not normal cells? How can we overcome the side effects of GPX4 inhibitors, including covalent inhibitors? No specific GPX4 activator is currently available. There is also a need to further understand the pathological role of autophagy-dependent GPX4 degradation in oxidative or reductive cell death [Citation205].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126.
- Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364.
- Ryter SW, Kim HP, Hoetzel A, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89.
- Kayagaki N, Kornfeld OS, Lee BL, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591:131–136.
- Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–425.
- Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–616.
- Flohe L, Toppo S, Orian L. The glutathione peroxidase family: discoveries and mechanism. Free Radic Biol Med. 2022;187:113–122.
- Weaver K, Skouta R. The selenoprotein glutathione peroxidase 4: from molecular mechanisms to novel therapeutic opportunities. Biomedicines. 2022;10:891.
- Brigelius-Flohe R, Flohe L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid Redox Signal. 2020;33:498–516.
- Ursini F, Maiorino M, Valente M, et al. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211.
- Yant LJ, Ran Q, Rao L, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
- Imai H, Hirao F, Sakamoto T, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–286.
- Ran Q, Van Remmen H, Gu M, et al. Embryonic fibroblasts from Gpx4± mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radic Biol Med. 2003;35:1101–1109.
- Scimeca MS, Lisk DJ, Prolla T, et al. Effects of gpx4 haploid insufficiency on GPx4 activity, selenium concentration, and paraquat-induced protein oxidation in murine tissues. Exp Biol Med (Maywood). 2005;230:709–714.
- Ran Q, Liang H, Ikeno Y, et al. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62:932–942.
- Garry MR, Kavanagh TJ, Faustman EM, et al. Sensitivity of mouse lung fibroblasts heterozygous for GPx4 to oxidative stress. Free Radic Biol Med. 2008;44:1075–1087.
- Conrad M, Moreno SG, Sinowatz F, et al. The nuclear form of phospholipid hydroperoxide glutathione peroxidase is a protein thiol peroxidase contributing to sperm chromatin stability. Mol Cell Biol. 2005;25:7637–7644.
- Puglisi R, Maccari I, Pipolo S, et al. The nuclear form of glutathione peroxidase 4 is associated with sperm nuclear matrix and is required for proper paternal chromatin decondensation at fertilization. J Cell Physiol. 2012;227:1420–1427.
- Azuma K, Koumura T, Iwamoto R, et al. Mitochondrial glutathione peroxidase 4 is indispensable for photoreceptor development and survival in mice. J Biol Chem. 2022;298:101824.
- Seiler A, Schneider M, Forster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248.
- Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191.
- Liang H, Yoo SE, Na R, et al. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J Biol Chem. 2009;284:30836–30844.
- Schneider M, Forster H, Boersma A, et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. Faseb J. 2009;23:3233–3242.
- Imai H, Hakkaku N, Iwamoto R, et al. Depletion of selenoprotein GPx4 in spermatocytes causes male infertility in mice. J Biol Chem. 2009;284:32522–32532.
- Sengupta A, Lichti UF, Carlson BA, et al. Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of COX-2. J Invest Dermatol. 2013;133:1731–1741.
- Ingold I, Aichler M, Yefremova E, et al. Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. J Biol Chem. 2015;290:14668–14678.
- Brutsch SH, Wang CC, Li L, et al. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid Redox Signal. 2015;22:281–293.
- Matsushita M, Freigang S, Schneider C, et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–568.
- Chen L, Hambright WS, Na R, et al. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–28106.
- Canli O, Alankus YB, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–148.
- Carlson BA, Tobe R, Yefremova E, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31.
- Hambright WS, Fonseca RS, Chen L, et al. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17.
- Kang R, Zeng L, Zhu S, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97–108 e4.
- Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409–22 e21.
- Muri J, Thut H, Bornkamm GW, et al. B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 2019;29:2731–44 e4.
- Liu K, Liu J, Zou B, et al. Trypsin-mediated sensitization to ferroptosis increases the severity of pancreatitis in mice. Cell Mol Gastroenterol Hepatol. 2022;13:483–500.
- Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11:6339.
- Piattini F, Matsushita M, Muri J, et al. Differential sensitivity of inflammatory macrophages and alternatively activated macrophages to ferroptosis. Eur J Immunol. 2021;51:2417–2429.
- Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;17:3361–3374.
- Yao Y, Chen Z, Zhang H, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol. 2021;22:1127–1139.
- Wang S, Li W, Zhang P, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75.
- Tosatto SC, Bosello V, Fogolari F, et al. The catalytic site of glutathione peroxidases. Antioxid Redox Signal. 2008;10:1515–1526.
- Roveri A, Flohe L, Maiorino M, et al. Phospholipid-hydroperoxide glutathione peroxidase in sperm. Methods Enzymol. 2002;347:208–212.
- Vuckovic AM, Venerando R, Tibaldi E, et al. Aerobic pyruvate metabolism sensitizes cells to ferroptosis primed by GSH depletion. Free Radic Biol Med. 2021;167:45–53.
- Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–590.
- Arai M, Imai H, Sumi D, et al. Import into mitochondria of phospholipid hydroperoxide glutathione peroxidase requires a leader sequence. Biochem Biophys Res Commun. 1996;227:433–439.
- Pfeifer H, Conrad M, Roethlein D, et al. Identification of a specific sperm nuclei selenoenzyme necessary for protamine thiol cross-linking during sperm maturation. Faseb J. 2001;15:1236–1238.
- Tong J, Li D, Meng H, et al. Targeting a novel inducible GPX4 alternative isoform to alleviate ferroptosis and treat metabolic-associated fatty liver disease. Acta Pharm Sin B. 2022;12:3650–3666.
- Maiorino M, Thomas JP, Girotti AW, et al. Reactivity of phospholipid hydroperoxide glutathione peroxidase with membrane and lipoprotein lipid hydroperoxides. Free Radic Res Commun. 1991;12-13(Pt 1):131–135.
- Thomas JP, Maiorino M, Ursini F, et al. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem. 1990;265:454–461.
- Cozza G, Rossetto M, Bosello-Travain V, et al. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: the polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic Biol Med. 2017;112:1–11.
- Schnurr K, Belkner J, Ursini F, et al. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J Biol Chem. 1996;271:4653–4658.
- Mendieta-Serrano MA, Schnabel D, Lomeli H, et al. Spatial and temporal expression of zebrafish glutathione peroxidase 4 a and b genes during early embryo development. Gene Expr Patterns. 2015;19:98–107.
- Rong X, Zhou Y, Liu Y, et al. Glutathione peroxidase 4 inhibits Wnt/beta-catenin signaling and regulates dorsal organizer formation in zebrafish embryos. Development. 2017;144:1687–1697.
- Borchert A, Wang CC, Ufer C, et al. The role of phospholipid hydroperoxide glutathione peroxidase isoforms in murine embryogenesis. J Biol Chem. 2006;281:19655–19664.
- Peng JJ, Yue SY, Fang YH, et al. Mechanisms affecting the biosynthesis and incorporation rate of selenocysteine. Molecules. 2021;26:7120.
- Yuan J, Palioura S, Salazar JC, et al. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc Natl Acad Sci U S A. 2006;103:18923–18927.
- Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457.
- Weitzel F, Ursini F, Wendel A. Phospholipid hydroperoxide glutathione peroxidase in various mouse organs during selenium deficiency and repletion. Biochim Biophys Acta. 1990;1036:88–94.
- Kerins MJ, Milligan J, Wohlschlegel JA, et al. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018;109:2757–2766.
- Grossman EA, Ward CC, Spradlin JN, et al. Covalent ligand discovery against druggable hotspots targeted by anti-cancer natural products. Cell Chem Biol. 2017;24:1368–76 e4.
- Chen X, Yu C, Kang R, et al. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021;28:1135–1148.
- Yang L, Chen X, Yang Q, et al. Broad spectrum deubiquitinase inhibition induces both apoptosis and ferroptosis in cancer cells. Front Oncol. 2020;10:949.
- Ding Y, Chen X, Liu C, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14:19.
- Sakamoto K, Sogabe S, Kamada Y, et al. Discovery of GPX4 inhibitory peptides from random peptide T7 phage display and subsequent structural analysis. Biochem Biophys Res Commun. 2017;482:195–201.
- Zhang W, Jiang B, Liu Y, et al. Bufotalin induces ferroptosis in non-small cell lung cancer cells by facilitating the ubiquitination and degradation of GPX4. Free Radic Biol Med. 2022;180:75–84.
- Tian P, Xu ZY, Guo JR, et al. BPDE induces human trophoblast cell ferroptosis by up-regulating iron metabolism and promoting GPX4 proteasomal degradatio. Ecotoxicol Environ Saf. 2021;228:113028.
- Hauser DN, Dukes AA, Mortimer AD, et al. Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic Biol Med. 2013;65:419–427.
- Fradejas-Villar N, Zhao W, Reuter U, et al. Missense mutation in selenocysteine synthase causes cardio-respiratory failure and perinatal death in mice which can be compensated by selenium-independent GPX4. Redox Biol. 2021;48:102188.
- Liu H, Forouhar F, Seibt T, et al. Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol. 2022;18:91–100.
- Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116:2996–3005.
- Thayyullathil F, Cheratta AR, Alakkal A, et al. Acid sphingomyelinase-dependent autophagic degradation of GPX4 is critical for the execution of ferroptosis. Cell Death Dis. 2021;12:26.
- Chen C, Wang D, Yu Y, et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021;12:65.
- Sun Y, Berleth N, Wu W, et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 2021;12:1028.
- Yu S, Li Z, Zhang Q, et al. GPX4 degradation via chaperone-mediated autophagy contributes to antimony-triggered neuronal ferroptosis. Ecotoxicol Environ Saf. 2022;234:113413.
- Liu J, Liu Y, Wang Y, et al. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy. 2023;19:945–956.
- Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77:2064–2077.
- Han L, Bai L, Fang X, et al. SMG9 drives ferroptosis by directly inhibiting GPX4 degradation. Biochem Biophys Res Commun. 2021;567:92–98.
- Dong K, Wei R, Jin T, et al. HOIP modulates the stability of GPx4 by linear ubiquitination. Proc Natl Acad Sci U S A. 2022;119:e2214227119.
- Vuckovic AM, Bosello Travain V, Bordin L, et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3epsilon. FEBS Lett. 2020;594:611–624.
- Liu H, Forouhar F, Lin AJ, et al. Small-molecule allosteric inhibitors of GPX4. Cell Chem Biol. 2022;29:1680–1693.e9.
- Hochstein P, Ernster L. Adp-activated lipid peroxidation coupled to the Tpnh oxidase system of microsomes. Biochem Biophys Res Commun. 1963;12:388–394.
- Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438.
- Wu G, Fang YZ, Yang S, et al. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492.
- Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331.
- Imai H, Koumura T, Nakajima R, et al. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J. 2003;371:799–809.
- Zhang Y, Swanda RV, Nie L, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12:1589.
- Liu Y, Wang Y, Liu J, et al. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2021;28:55–63.
- Buczynski MW, Dumlao DS, Dennis EA. Thematic review series: proteomics. An integrated omics analysis of eicosanoid biology. J Lipid Res. 2009;50:1015–1038.
- Wang T, Fu X, Chen Q, et al. Arachidonic acid metabolism and kidney inflammation. Int J Mol Sci. 2019;20:20.
- Li C, Deng X, Xie X, et al. Activation of glutathione peroxidase 4 as a novel anti-inflammatory strategy. Front Pharmacol. 2018;9:1120.
- Schwarzler J, Mayr L, Vich Vila A, et al. PUFA-Induced metabolic enteritis as a fuel for Crohn’s disease. Gastroenterology. 2022;162:1690–1704.
- Mayr L, Grabherr F, Schwarzler J, et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat Commun. 2020;11:1775.
- Kryukov GV, Castellano S, Novoselov SV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443.
- Daniels LA. Selenium metabolism and bioavailability. Biol Trace Elem Res. 1996;54:185–199.
- Li Z, Ferguson L, Deol KK, et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol. 2022;18:751–761.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516.
- Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
- Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–193.
- Nomura K, Imai H, Koumura T, et al. Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J Biol Chem. 1999;274:29294–29302.
- Brielmeier M, Bechet JM, Suppmann S, et al. Cloning of phospholipid hydroperoxide glutathione peroxidase (PHGPx) as an anti-apoptotic and growth promoting gene of Burkitt lymphoma cells. BioFactors. 2001;14:179–190.
- Ran Q, Liang H, Gu M, et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem. 2004;279:55137–55146.
- Hurst R, Korytowski W, Kriska T, et al. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic Biol Med. 2001;31:1051–1065.
- Huang HS, Chang WC, Chen CJ. Involvement of reactive oxygen species in arsenite-induced downregulation of phospholipid hydroperoxide glutathione peroxidase in human epidermoid carcinoma A431 cells. Free Radic Biol Med. 2002;33:864–873.
- Kriska T, Korytowski W, Girotti AW. Hyperresistance to photosensitized lipid peroxidation and apoptotic killing in 5-aminolevulinate-treated tumor cells overexpressing mitochondrial GPX4. Free Radic Biol Med. 2002;33:1389–1402.
- Nomura K, Imai H, Koumura T, et al. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193.
- Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698.
- Ufer C, Wang CC, Fahling M, et al. Translational regulation of glutathione peroxidase 4 expression through guanine-rich sequence-binding factor 1 is essential for embryonic brain development. Genes Dev. 2008;22:1838–1850.
- Liu P, Zhu J, Yuan G, et al. The effects of selenium on GPX4-mediated lipid peroxidation and apoptosis in germ cells. J Appl Toxicol. 2022;42:1016–1028.
- Weinlich R, Oberst A, Beere HM, et al. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–136.
- Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119.
- Xie Y, Zhu S, Zhong M, et al. Inhibition of aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153:1429–43 e5.
- Vandenabeele P, Galluzzi L, Vanden Berghe T, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714.
- Zhang Y, Su SS, Zhao S, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329.
- Basit F, van Oppen LM, Schockel L, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716.
- Xue Y, Enosi Tuipulotu D, Tan WH, et al. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40:1035–1052.
- Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671.
- Zou J, Zheng Y, Huang Y, et al. The versatile gasdermin family: their function and roles in diseases. Front Immunol. 2021;12:751533.
- Wu R, Wang N, Comish PB, et al. Inflammasome-dependent coagulation activation in sepsis. Front Immunol. 2021;12:641750.
- Gu X, Wang Y, He Y, et al. MiR-1656 targets GPX4 to trigger pyroptosis in broilers kidney tissues by activating NLRP3 inflammasome under Se deficiency. J Nutr Biochem. 2022;105:109001.
- Gong Z, Li Q, Yang J, et al. Identification of a pyroptosis-related gene signature for predicting the immune status and prognosis in lung adenocarcinoma. Front Bioeng Biotechnol. 2022;10:852734.
- Chen Y, Liao Y, Du Q, et al. Roles of pyroptosis-related gene signature in prediction of endometrial cancer outcomes. Front Med. 2022;9:822806.
- Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072.
- Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–282.
- Chen X, Huang J, Yu C, et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun. 2022;13:6318.
- Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–1343.
- Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
- Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
- Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022;289:7038–7050.
- Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379.
- Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–2081.
- Gaschler MM, Andia AA, Liu H, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–515.
- Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–125.
- Tang D, Kroemer G, Kang R. Ferroptosis in hepatocellular carcinoma: from bench to bedside. Hepatology. 2023;Publish Ahead of Print. DOI: 10.1097/HEP.0000000000000390
- Chen X, Kang R, Kroemer G, et al. Targeting ferroptosis in pancreatic cancer: a double-edged sword. Trends Cancer. 2021;7:891–901.
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658.
- Andrabi SA, Kim NS, Yu SW, et al. Poly(adp-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313.
- Liu L, Liu B, Guan G, et al. Cyclophosphamide-induced GPX4 degradation triggers parthanatos by activating AIFM1. Biochem Biophys Res Commun. 2022;606:68–74.
- Xie Y, Li J, Kang R, et al. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. 2020;8:431.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy. 2021;17:1–382.
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28–45.
- Vernon PJ, Tang D. Eat-me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid Redox Signal. 2013;18:677–691.
- Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428.
- Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032.
- Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238.
- Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003.
- Song X, Zhu S, Chen P, et al. AMPK-Mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc– activity. Curr Biol. 2018;28(15):2388–2399.e5. DOI:10.1016/j.cub.2018.05.094
- Kang R, Zhu S, Zeh HJ, et al. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14:2173–2175.
- Chen X, Song X, Li J, et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy. 2023;19:54–74.
- Xue Q, Kang R, Klionsky DJ, et al. Copper metabolism in cell death and autophagy. Autophagy. 2023;1–21. DOI:10.1080/15548627.2023.2200554.
- Wu K, Yan M, Liu T, et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat Cell Biol. 2023;25:714–725.
- Rubio N, Coupienne I, Di Valentin E, et al. Spatiotemporal autophagic degradation of oxidatively damaged organelles after photodynamic stress is amplified by mitochondrial reactive oxygen species. Autophagy. 2012;8:1312–1324.
- Jiang W, Li Y, Zhao Y, et al. L-carnitine supplementation during in vitro culture regulates oxidative stress in embryos from bovine aged oocytes. Theriogenology. 2020;143:64–73.
- Morgan AH, Hammond VJ, Sakoh-Nakatogawa M, et al. A novel role for 12/15-lipoxygenase in regulating autophagy. Redox Biol. 2015;4:40–47.
- Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27:420–435.
- Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285.
- Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–296.
- Yang C, Zhang Y, Lin S, et al. Correction for: suppressing the KIF20A/NUAK1/Nrf2/GPX4 signaling pathway induces ferroptosis and enhances the sensitivity of colorectal cancer to oxaliplatin. Aging. 2021;13:19077.
- Ni J, Chen K, Zhang J, et al. Inhibition of GPX4 or mTOR overcomes resistance to Lapatinib via promoting ferroptosis in NSCLC cells. Biochem Biophys Res Commun. 2021;567:154–160.
- Wang Q, Bin C, Xue Q, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426.
- Yuan L, Li S, Chen Q, et al. EBV infection-induced GPX4 promotes chemoresistance and tumor progression in nasopharyngeal carcinoma. Cell Death Differ. 2022;29:1513–1527.
- Zheng J, Sato M, Mishima E, et al. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021;12:698.
- Wiernicki B, Maschalidi S, Pinney J, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676.
- Hangauer MJ, Viswanathan VS, Ryan MJ, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–250.
- Liu S, Zhao X, Shui S, et al. PDTAC: targeted photodegradation of GPX4 triggers ferroptosis and potent antitumor immunity. J Med Chem. 2022;65:12176–12187.
- Luo T, Zheng Q, Shao L, et al. Intracellular delivery of glutathione peroxidase degrader induces ferroptosis in vivo. Angew Chem Int Ed Engl. 2022;61:e202206277.
- Han L, Bai L, Qu C, et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun. 2021;576:33–39.
- Kim R, Hashimoto A, Markosyan N, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338–346.
- Xu C, Sun S, Johnson T, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35:109235.
- Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069–2083.
- Liu J, Zhu S, Zeng L, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy. 2022;18:2036–2049.
- Wirth EK, Conrad M, Winterer J, et al. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. Faseb J. 2010;24:844–852.
- Wirth EK, Bharathi BS, Hatfield D, et al. Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol Trace Elem Res. 2014;158:203–210.
- Jakaria M, Belaidi AA, Bush AI, et al. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J Neurochem. 2021;159:804–825.
- Ko CJ, Gao SL, Lin TK, et al. Ferroptosis as a major factor and therapeutic target for neuroinflammation in Parkinson’s disease. Biomedicines. 2021;9:1679.
- Banning A, Schnurr K, Bol GF, et al. Inhibition of basal and interleukin-1-induced VCAM-1 expression by phospholipid hydroperoxide glutathione peroxidase and 15-lipoxygenase in rabbit aortic smooth muscle cells. Free Radic Biol Med. 2004;36:135–144.
- Rayman MP, Taylor EW, Zhang J. The relevance of selenium to viral disease with special reference to SARS-CoV-2 and COVID-19. Proc Nutr Soc. 2023;2022:1–12.
- Bedford J, Enria D, Giesecke J, et al. COVID-19: towards controlling of a pandemic. Lancet. 2020;395:1015–1018.
- Khoso PA, Yang Z, Liu C, et al. Selenium deficiency downregulates selenoproteins and suppresses immune function in chicken thymus. Biol Trace Elem Res. 2015;167:48–55.
- Drijvers JM, Gillis JE, Muijlwijk T, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T cells. Cancer Immunol Res. 2021;9:184–199.
- Zhang RX, Kang R, Tang DL. STING1 in sepsis: mechanisms, functions, and implications. Chin J Traumatol. 2022;25:1–10.
- Zhang R, Kang R, Tang D. The STING1 network regulates autophagy and cell death. Signal Transduct Target Ther. 2021;6:208.
- Jia M, Qin D, Zhao C, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol. 2020;21:727–735.
- Li C, Liu J, Hou W, et al. STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front Cell Dev Biol. 2021;9:698679.
- Foresta C, Flohe L, Garolla A, et al. Male fertility is linked to the selenoprotein phospholipid hydroperoxide glutathione peroxidase. Biol Reprod. 2002;67:967–971.
- Chen X, Kang R, Kroemer G, et al. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218. DOI:10.1084/jem.20210518
- Dabkowski ER, Williamson CL, Hollander JM. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic Biol Med. 2008;45:855–865.
- Sun W, Shi R, Guo J, et al. MiR-135b-3p promotes cardiomyocyte ferroptosis by targeting GPX4 and aggravates myocardial Ischemia/reperfusion injury. Front Cardiovasc Med. 2021;8:663832.
- Kromer A, Moosmann B. Statin-induced liver injury involves cross-talk between cholesterol and selenoprotein biosynthetic pathways. Mol Pharmacol. 2009;75:1421–1429.
- Luo Y, Chen H, Liu H, et al. Protective effects of ferroptosis inhibition on high fat diet-induced liver and renal injury in mice. Int J Clin Exp Pathol. 2020;13:2041–2049.
- Yang Y, Chen J, Gao Q, et al. Study on the attenuated effect of Ginkgolide B on ferroptosis in high fat diet induced nonalcoholic fatty liver disease. Toxicology. 2020;445:152599.
- Zhao T, Yu Z, Zhou L, et al. Regulating Nrf2-GPx4 axis by bicyclol can prevent ferroptosis in carbon tetrachloride-induced acute liver injury in mice. Cell Death Discov. 2022;8:380.
- Zhu K, Zhu X, Liu S, et al. Glycyrrhizin attenuates hypoxic-ischemic brain damage by inhibiting ferroptosis and neuroinflammation in neonatal rats via the HMGB1/GPX4 pathway. Oxid Med Cell Longev. 2022;2022:1–18.
- Fukai M, Hayashi T, Yokota R, et al. Lipid peroxidation during ischemia depends on ischemia time in warm ischemia and reperfusion of rat liver. Free Radic Biol Med. 2005;38:1372–1381.
- Weiwer M, Bittker JA, Lewis TA, et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett. 2012;22:1822–1826.
- Eaton JK, Furst L, Ruberto RA, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497–506.
- Cheff DM, Huang C, Scholzen KC, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023;14:9401–9418.
- Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
- Muller T, Dewitz C, Schmitz J, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74:3631–3645.
- Su LJ, Zhang JH, Gomez H, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843.
- Blaner WS, Shmarakov IO, Traber MG. Vitamin a and vitamin E: will the real antioxidant please stand up? Annu Rev Nutr. 2021;41:105–131.
- Maiorino M, Coassin M, Roveri A, et al. Microsomal lipid peroxidation: effect of vitamin E and its functional interaction with phospholipid hydroperoxide glutathione peroxidase. Lipids. 1989;24:721–726.
- Zhang R, Kang R, Tang D. Reductive cell death: the other side of the coin. Cancer Gene Ther. 2023. DOI:10.1038/s41417-023-00612-3