
ABSTRACT
Macroautophagy/autophagy, a lysosome-dependent self-degradative process, is a critical mechanism for the clearance of misfolded proteins and dysfunctional organelles in neurons. In the peripheral nervous system, autophagic stress is associated with the development of peripheral neuropathy. However, the molecular mechanism of axonal neuropathy induced by autophagic stress due to dysfunction of autophagy in peripheral neurons in vivo is still unclear. We found that dorsal root ganglion (DRG) neuron-specific atg7 (autophagy related 7) knockout (atg7-cKO) mice employing two different Cre recombinase systems exhibited sensory neuropathy approximately 2 months after birth. In electron microscopy analysis, axon degeneration was clearly observed in the myelinated fibers of the sciatic nerve before the appearance of neuronal cell death. Dystrophic axons filled with abnormal vesicular accumulations and amorphous inclusions were specifically localized in the myelinated axons within the DRG in atg7-cKO mice, indicating the presence of autophagic stress in proximal axons. In line with the EM findings, the mutant mice showed preferential induction of axonal injury-associated genes, including ATF3 (activating transcription factor 3), in large-size DRG neurons that constitute myelinated fibers without axotomy. SARM1 (sterile alpha and HEAT/Armadillo motif containing 1), the central executioner of Wallerian degeneration, was activated in the sciatic nerves of atg7-cKO mice, and axonal degeneration and sensory neuropathy in atg7-cKO mice were prevented via expression of a dominant-negative Sarm1 transgene. Our findings demonstrate the importance of SARM1-dependent axon degeneration in the development of peripheral sensory neuropathy induced by impairment of autophagy.
Abbreviations
AAV: adeno-associated virus; ATF3: activating transcription factor 3; ATG7: autophagy related 7; AVIL: advillin; cADPR: cyclic ADP ribose; CALC: calcitonin/calcitonin-related polypeptide; CMT: Charcot-Marie-Tooth disease; cKO: conditional knockout; DEG: differentially expressed gene; DRG: dorsal root ganglion; FE-SEM: field emission scanning electron microscopy; IF: immunofluorescence; NCV: nerve conduction velocity; PVALB: parvalbumin; RAG: regeneration-associated gene; ROS: reactive oxygen species; SARM1: sterile alpha and HEAT/Armadillo motif containing 1; SYN1: synapsin I
Introduction
Repetitive electrical stimulation in neurons requires a high metabolic supply and is associated with the generation of oxidative stress and organelle damage. Autophagy is a lysosome-dependent defense mechanism by which neurons prevent the accumulation of dysfunctional cellular organelles such as aged mitochondria [Citation1]. Thus, the loss of autophagy in neurons results in the accumulation of toxic aggregates and damaged organelles that are involved in the neurodegeneration in the central nervous system (CNS) [Citation1]. Although the higher concentration of lysosomes in neuronal soma than in axons suggests that autophagy principally occurs in the cell body, recent studies have demonstrated axonal autolysosomal digestion via constitutive lysosomal transport through the axon [Citation2], suggesting a specific role of autophagy in the maintenance of axonal integrity. In accordance with this finding, in mice with neuronal autophagy deficiency in the CNS caused by the deletion of the Atg7 (autophagy related 7) gene, a critical gene for autophagosome formation in macroautophagy, there is widespread axonal dystrophy with an accumulation of phagophores and endolysosomal vesicular structures in the axons as well as neuronal death [Citation3–5]. Since axonal dystrophy and neuronal death are the pathologic hallmark of neurodegenerative diseases, such as Alzheimer disease [Citation4,Citation6,Citation7], these studies highlight the involvement of autophagic stress in the pathogenesis of the neurodegenerative diseases.
The association of genes involved in autophagy with inherited peripheral neuropathies further emphasizes the importance of autophagy in axonal maintenance [Citation8]. Charcot-Marie-Tooth disease (CMT) 2B is an inherited axonal peripheral neuropathy and mutations in RAB7A in CMT2B patients leads to a defect in basal and starvation-induced autophagy [Citation9,Citation10]. In addition, a mutation in RETREG1/Fam134B, a protein involved in reticulophagy, is involved in the development of the hereditary sensory axonal neuropathy-II, and knockout of Retreg1 in mice promotes the development of sensory neuropathy in aged animals [Citation11]. However, despite the emerging importance of autophagy in axonal maintenance in the peripheral nervous system, the mechanism of autophagy-dependent peripheral neuropathy has not been clearly determined [Citation12–14].
When an axon is severed, the distal portion of the injured axon undergoes catastrophic destruction known as Wallerian degeneration. The critical mechanism of Wallerian degeneration involves SARM1 (sterile alpha and HEAT/Armadillo motif containing 1)-dependent NAD+ and ATP depletion in injured axons [Citation15]. Loss of SARM1 in flies or mice resulted in marked protection of axons from degeneration after injury [Citation15,Citation16]. Interestingly, aberrant activation of SARM1 via damaged mitochondria or activating mutations in the Sarm1 gene can sufficiently induce axon degeneration, without any physical axon injury [Citation17,Citation18], suggesting that developing tools to disable SARM1 might be a promising method to protect axons from degeneration in various models of neurodegenerative diseases. Here we report that dorsal root ganglion (DRG) neuron-specific atg7 deletion is sufficient to induce SARM1-dependent Wallerian-like axon degeneration in mice and that SARM1 inhibition improves sensory axonal neuropathy in DRG-specific atg7-KO mice.
Results
Atg7-deficiency in DRG neurons induced postnatal sensory neuropathy
DRG neuron-specific atg7-deficient (Avil-atg7-cKO) mice were obtained by crossing Avil (advillin)-Cre mice, which express Cre recombinase specifically in peripheral sensory DRG neurons [Citation19], and Atg7f/f mice. The loss of autophagy in the DRG of Avil-atg7-cKO mice was confirmed by the reduction of ATG7 protein levels and the accumulation of SQSTM1/p62, an autophagy adaptor that is degraded by autolysosomes, in DRG extracts from adult mice (~2 months after birth) (). Massive accumulation of SQSTM1 in DRG neurons from Avil-atg7-cKO mice, but not WT mice, was also observed by immunofluorescence (IF) staining (). In addition, there was an increase of punctate SQSTM1 stainings in axons of the sciatic nerves in the mutant mice (Figure S1A). The gait of Avil-atg7-cKO mice was not distinguishable from the control mice until 3 weeks after birth, but the mice exhibited abnormal hindlimb extension during tail suspension around postnatal week 4, and this phenotype worsened with age. Abnormal sensory-motor coordination in the mutant mice was evident in the rotarod test in adults (), while grip strength was normal in the mutant mice (). The analysis of sensory nerve electrophysiology in adults revealed a reduced amplitude of sensory nerve action potential by 52.1% and a reduced nerve conduction velocity (NCV) by 38.76% in Avil-atg7-cKO mice compared to those of WT controls, whereas motor NCV and compound motor action potential were not altered in Avil-atg7-cKO mice (). Von Frey tests did not show a change in pain sensitivity in Avil-atg7-cKO mice compared to control mice (Figure S1B).
Figure 1. Atg7-deficiency in DRG neurons induced postnatal sensory neuropathy. (A) Demonstration of atg7 knockout (atg7-cKO)) by western blot analysis against ATG7 and SQSTM1/p62 or by immunofluorescence (IF) staining against SQSTM1 in the DRG from Avil-atg7-cKO and WT mice at 2 months. Scale bar: 50 μm. (B, C) The rotarod performance (B) and grip strength test (C) for WT and Avil-atg7-cKO mice. (D) Sensory and motor nerve conduction velocity (NCV) test. SNAP; sensory nerve action potential. CMAP; compound motor action potential. (E) Design of AAV-mediated Atg7 knockdown in mouse DRG. SYN1-Cre; SYN1-Cre recombinase. (F) Western blot analysis showing the levels of ATG7 and SQSTM1 in the DRG, cerebrum (Cbr) and cerebellum (Cbll) of SYN1-atg7-cKO and SYN1-tdTomato mice. (G) Representative IF staining against SQSTM1 in DRG from SYN1-tdTomato and SYN1-atg7-cKO (SYN1-Cre) mice. Scale bar: 50 μm. (H, I) The rotarod performance (H) and grip strength test (I) for SYN1-tdTomato and SYN1-atg7-Cko mice. (J) NCV test for SYN1-tdTomato and SYN1-atg7-Cko mice. *; p < 0.05, **; p < 0.01, ns; non-significant. Each dot in figures indicate the number of animals.
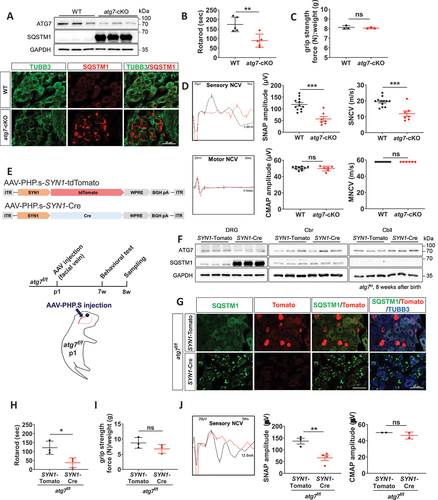
Avil-Cre mice are knockin mice [Citation19], and thus haploinsufficiency of the Avil gene in Avil-atg7-cKO mice may contribute to the development of peripheral neuropathy. We thus conditionally deleted Atg7 gene in DRG neurons by overexpressing Cre recombinase in Atg7f/f mice using AAV-SYN1 (synapsin I) promoter-Cre (SYN1-Cre). Serotype PHP.S AAV coats are known to be able to specifically infect peripheral neurons [Citation20] and thus, PHP.S-SYN1-Cre AAV or PHP.S-SYN1-tdTomato AAV was delivered intravenously via the facial vein of Atg7f/f mice at postnatal day 1 (). AAV-SYN1-Cre injection into Atg7f/f mice (SYN1-atg7-cKO mice) resulted in a reduction in ATG7 protein levels and massive accumulation of SQSTM1 in the DRG, but not in the cerebral cortex or cerebellum, after 2 months (). IF staining revealed high levels of SQSTM1 accumulation in DRG neurons in SYN1-atg7-cKO mice in adulthood (). The SYN1-atg7-cKO mice exhibited an abnormal gait around postnatal week 4, and sensory-motor coordination defects in the rotarod test were evident at postnatal week 8 (). However, hindlimb grip strength was normal in SYN1-atg7-cKO mice (). In addition, electrophysiological testing revealed a reduction in the amplitude of sensory nerve action potentials with normal compound motor action potential in SYN1-atg7-cKO mice compared to SYN1-tdTomato mice (). Together, these findings indicate that the loss of the Atg7 gene in DRG neurons during postnatal development was sufficient to induce autophagy impairment with the development of sensory neuropathy in adult mice.
Atg7-deficiency in DRG neurons induced peripheral axon degeneration
The development of sensory neuropathy in Avil-atg7-cKO mice and SYN1-atg7-cKO mice in adulthood suggests that axonal degeneration or sensory neuronal loss might occur when neuropathy phenotypes appear. To investigate this possibility, we first counted the number of DRG neurons after immunostaining three neuronal subtypes with cell type-specific markers. The numbers of NTRK2 (a low threshold mechanoreceptor), PVALB (parvalbumin; proprioceptive neurons), and CALC (calcitonin/calcitonin-related polypeptide; thermoreceptive and nociceptive neurons)-immunoreactive neurons in the DRG did not significantly decrease in Avil-atg7-cKO mice compared to WT mice in adults (Figure S1C), indicating that there was no significant neuronal loss, although the mutant mice exhibited sensory neuropathy at postnatal month 1 or 2. This is in agreement with a previous report showing late neuronal death in autophagy-defective DRG neurons approximately 9 months after birth in mice [Citation14].
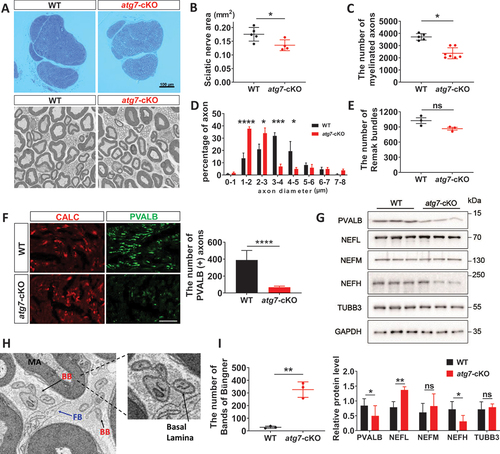
We next morphometrically examined the sciatic nerves of the mutant mice using EM at 2 months after birth. Although the general features of the sciatic nerves were similar between mutant and WT control mice (), the area of the sciatic nerve was reduced by 15.93% in Avil-atg7-cKO mice compared to control mice in adulthood (). The number of myelinated fibers in the sciatic nerves was significantly reduced in Avil-atg7-cKO mice compared to that in WT mice (). In addition, the distribution of axonal diameter in myelinated fibers shifted to the left, indicating a decrease in axonal diameter in Avil-atg7-cKO mice (). However, the development of unmyelinated fibers, as revealed by counting both the number of Remak bundles in the sciatic nerves () and the number of unmyelinated axons within each bundle (Figure S1D), was not significantly altered in Avil-atg7-cKO mice compared to WT mice. Thus, myelinated fibers appeared to be selectively compromised in Avil-atg7-cKO mice.
Figure 2. Atg7-deficiency in DRG neurons induced peripheral axon degeneration. (A) Semithin sections and FE-SEM of the sciatic nerves of WT and Avil-atg7-cKO mice in adults. Scale bar: 100 μm. (B) Measurement of the sciatic nerve areas in semithin sections from WT and Avil-atg7-cKO mice. (C, D) Counting the number and the measurement of axon diameter of myelinated fibers in FE-SEM images of the sciatic nerve. (E) the number of Remak bundles in the sciatic nerve. (F) IF staining against CALC and parvalbumin (PVALB) in the sciatic nerve of WT and Avil-atg7-cKO mice, and the number of Parvalbumin (+) axons in the sciatic nerve section in adults. Scale bar: 50 μm. (G) Western blot analysis showing the levels of Parvalbumin, neurofilament (NF) and beta-tubulin in the sciatic nerve of WT and Avil-atg7-cKO mice. The graph shows quantitative analysis on the western blot. n = 3. (H) The morphology of the Band of Büngner (BB) in the sciatic nerve of Avil-atg7-cKO mice. FB; fibroblast, MA; myelinated axon. (I) Counting the number of BB in the sciatic nerve. *; p < 0.05, **; p < 0.01, ***; p < 0.001, ****; p < 0.0001.
To substantiate sensory-prone axon degeneration in Avil-atg7-cKO mice, we examined the area and the number of myelinated axons in the sural nerve near trifurcation site of the sciatic nerve into tibial, peroneal and sural branches [Citation21]. The area of sural nerve was reduced by ~ 53.44% (p < 0.001) in Avil-atg7-cKO mice compared to control mice in adulthood (Figure S1E and F). In addition, the number of myelinated fibers in sural nerve was reduced by ~ 58.33% (p < 0.001) in Avil-atg7-cKO mice compared to control mice in adulthood (Figure S1G), indicating more apparent axon degeneration in mutant sural nerves than those of the sciatic nerves of the mutant mice.
To investigate the preferential axonal loss in large myelinated fibers in the peripheral nerves of Avil-atg7-cKO mice, we immunostained sciatic nerve sections with antibodies against specific markers of large myelinated fibers and thermoreceptive/nociceptive small fibers (PVALB and CALC, respectively). There was a selective loss of PVALB+, but not CALC+, axon staining in the sciatic nerve of Avil-atg7-cKO mice (). In addition, western blot analysis revealed a reduction in PVALB expression in the sciatic nerves of Avil-atg7-cKO mice compared to WT mice (). However, the expression of PVALB in DRG lysates was not decreased in Avil-atg7-cKO mice compared to WT mice (Figure S1H). Furthermore, we examined the expression levels of neurofilament 200, a marker of large myelinated axons, in the sciatic nerve of Avil-atg7-cKO mice, and found significant downregulation of neurofilament-200 in the sciatic nerves of Avil-atg7-cKO mice compared to control mice (). However, the expression levels of medium-size-NF and tubulin monomers were not altered in mutant mice compared control mice (). These data support a selective loss of large myelinated fibers in Avil-atg7-cKO, consistent with behavioral defects of the mutant mice.
The reduction in the number of myelinated fibers in Avil-atg7-cKO mice may result from postnatal axonal underdevelopment or axon degeneration of mature axons. To determine this, we tried to find an evidence of axon degeneration by FE-SEM. Interestingly, we found many “Bands of Büngner”, aligned Schwann cells enclosed within the basal lamina for repair, which are seen in the mature nerve after Wallerian degeneration, in the sciatic nerves of Avil-atg7-cKO mice in adults (). Morphometric analysis based on FE-SEM revealed that the number of Bands of Büngner in the sciatic nerves reached ~ 300 in the sciatic nerve in Avil-atg7-cKO, whereas only 5 Bands of Büngners were detected in WT mice at adulthood (). The appearance of a high number of Bands of Büngners of the sciatic nerves in Avil-atg7-cKO mice supports substantial degeneration of preexisting axons instead of axon development defects. Taken together, these findings indicate that selective axonal loss in myelinating fibers occurs in Avil-atg7-cKO mice and that peripheral axon degeneration might be related to the development of sensory neuropathy in atg7-cKO mice.
Axonal injury responses in DRG neurons of atg7-cKO mice without nerve injury
Following axonal injury to peripheral nerves, transcriptional regeneration-associated responses occur in the injured neurons. We thus examined whether there was a global change in injury-associated gene expression in uninjured DRG neurons of Avil-atg7-cKO mice using mRNA-Seq analysis. A comparison of the DRG transcript levels between WT and Avil-atg7-cKO mice in adults (Log2FC > 1, p < 0.05) showed that 934 genes were differentially expressed in the uncut Avil-atg7-cKO DRG compared to the uncut WT DRG (). Among the 934 differentially expressed genes (Atg7-DEGs), 463 genes were upregulated while 471 genes were downregulated. The expression changes of 5 arbitrarily selected Atg7-DEGs, including Ucn (urocortin) and Cckbr (cholecystokinin B receptor), were confirmed by q-PCR (). We also examined the expression of regeneration-associated genes (RAGs), whose expression was regulated by more than 2-fold (p < 0.05) after axotomy in WT DRG neurons, and found that 802 genes were upregulated and 449 genes were downregulated upon axotomy at 7 days post-injury (). In Gene Ontology analysis, we compared the enriched biological processes and cellular components between RAGs and Atg7-DEGs. The analysis showed that many significantly enriched biological processes and cellular components were shared by RAGs and Atg7-DEGs (Figure S2A and B, Table S1). In addition, among 934 Atg7-DEGs, 360 genes (39% of 934 Atg7-DEG) were found among RAGs, and conversely 29% of 1251 RAGs were detected among the Atg7-DEGs, indicating that axotomy and neuronal Atg7 KO induce shared transcriptional responses. The expression of representative “injury-induced genes”, such as Sprr1a (small proline-rich protein 1A), Npy (neuropeptide Y), and Atf3 (activating transcription factor 3) [Citation22] (Table S1), was increased in the uninjured Avil-atg7-cKO DRG compared to uninjured WT DRG. Finally, Pearson’s correlation coefficient analysis of the fold changes in the expression of each of whole transcriptomes from uncut Avil-atg7-cKO DRG:uncut WT DRG and injured WT DRG:uncut WT DRG revealed a correlation coefficient of 0.56 (p < 0.0001), indicating a strong positive correlation between them ().
Figure 3. Axonal injury responses in the DRG neurons of Avil-atg7-cKO mice without nerve injury. (A) mRNA-seq analysis of DRG. The volcano plots showing Atg7-DEG (DEG) and RAG. DEG; differentially expressed genes (2-fold, p < 0.05) in Avil-atg7-cKO DRG compared to WT DRG without nerve injury, RAG; regeneration-associated genes (2 fold, p < 0.05) identified at 7 days after nerve injury in the DRG of WT mice. (B) RT-qPCR results of 5 Atg7-DEGs those expression changes were compared with mRNA-seq results. (C) Pearson’s correlation coefficient analysis between the fold changes of whole transcripts in uncut Avil-atg7-cKO DRG/uncut WT DRG and injured WT DRG/uncut WT. r = 0.56, p < 0.0001. (D) Western blot analysis showing ATF3 induction in the DRG of WT, Avil-atg7-cKO and SYN1-atg7-cKO mice in adults. (E, F) IF staining against ATF3 in the DRG of WT, injured WT and uninjured Avil-atg7-cKO mice in adults. Scale bar: 50 μm. (F) Quantification of ATF3 (+) DRG neurons. (G) Western blot analysis of ATF3 in the DRG of WT and Avil-atg7-cKO mice at 1 week and 3 weeks after birth. (H) IF staining against parvalbumin in the sciatic nerves of WT and Avil-atg7-cKO mice at 1 week and 3 weeks after birth. (I) Quantification of the number of parvalbumin (+) axons in the sciatic nerve. ****; p < 0.0001. ns; non-significant. Scale bar: 50 μm.
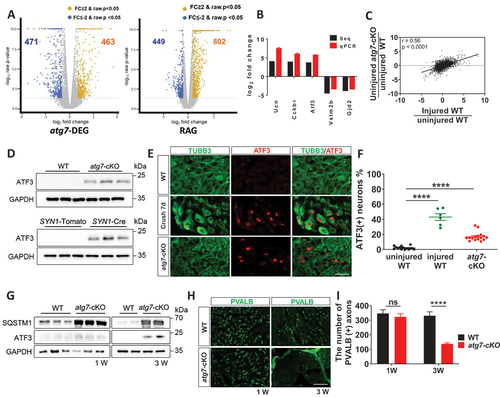
To substantiate potential axon damage in the DRG of uninjured Avil-atg7-cKO mice, we further examined the induction of ATF3, a marker of DRG neurons with axonal injury, in the DRG. We first examined induction of ATF3 in DRGs at protein levels using western blot and found a significant increase in the expression of ATF3 in the uncut DRG of Avil-atg7-cKO mice compared to WT mice in adulthood (). We also found a significant ATF3 induction in uninjured DRG in SYN1-atg7-cKO mice, but not in SYN1-tdTomato mice, in western blot (). We next examined ATF3 induction in DRGs using IF staining. In Avil-atg7-cKO mice, IF staining revealed that ~ 17% of DRG neurons exhibited nuclear ATF3 induction without injury, while only ~ 2% of DRG neurons showed nuclear ATF3 staining in uninjured WT mice in adults (). To determine parallel expression of ATF3 in WT DRG after nerve injury, we examined ATF3 induction in WT DRG at 7 days post-injury. Approximately, 43% of DRG neurons in WT mice exhibited nuclear ATF3 induction at 7 days post-injury (). Finally, we examined Atf3 mRNA expressions in DRGs of WT mice after injury and in DRGs of Avil-atg7-cKO mice, compared to respective controls using RT-qPCR. We found that induction of Atf3 mRNA expressions in both conditions, albeit nerve injury produced more pronounced effects than that of atg7-knockout on Atf3 mRNA expressions (Figure S2C).
We observed abnormal hindlimb reflexes approximately 1 month after birth in atg7-cKO mice, and thus we next examined when axonal injury responses occur in the DRG in atg7-cKO mice by analyzing ATF3 induction. The induction of ATF3 in the DRG of uninjured Avil-atg7-cKO mice was apparent at postnatal week 3, but not at postnatal week 1 in western blot analysis (). IF staining also revealed that ~ 8% of TuJ1-positive DRG neurons exhibited nuclear ATF3 staining in Avil-atg7-cKO mice at postnatal 3 weeks (Figure S2D). Consistent with the axonal injury response of DRG neurons at postnatal week 3 in Avil-atg7-cKO mice, we found that there was a loss of PVALB (+) axons in the sciatic nerves at postnatal week 3 (). Thus, these findings indicate that there was axon degeneration in the sciatic nerve approximately 3 weeks postnatally, which was temporally correlated with the first appearance of abnormal peripheral sensory-motor coordination, as revealed by the abnormal hindlimb extension reflex, in the mutant mice at postnatal week 4.
Selective vulnerability of the axons of mechanoreceptors and proprioceptors in atg7-cKO mice without nerve injury
Since axonal loss mainly occurred in the myelinated axons of Avil-atg7-cKO mice in EM analysis, we next examined the potential neuronal subtype-specific induction of ATF3 in the DRG of Avil-atg7-cKO mice without axotomy using double IF staining. Significant ATF3 induction in the uninjured DRG of Avil-atg7-cKO mice in adulthood was mainly observed in PVALB (+) and NTRK2 (+) neurons (). However, the accumulation of SQSTM1, an index of autophagy impairment, was similar among the three neuronal subtypes in Avil-atg7-cKO mice (), indicating a similar loss of ATG7-dependent autophagy in the three cell types.
Figure 4. Selective vulnerability of the axons of mechanoreceptive and proprioceptive DRG neurons in uninjured Avil-atg7-cKO mice. (A) IF staining against ATF3 with neuronal markers, parvalbumin, NTRK2 or CALC in the DRG of WT and Avil-atg7-cKO mice. Scale bar: 50 μm. (B) Quantification of the number of ATF3 (+) neurons in each subtype of DRG neurons. ***; p < 0.001, ****; p < 0.0001, ns; non-significant. (C) Double IF staining against SQSTM1 and NTRK2, PVALB or CALC in the DRG of WT and Avil-atg7-cKO mice. Scale bar: 50 μm. Quantification shows the percentage of SQSTM1 inclusion (+) neurons in the DRG of WT and Avil-atg7-cKO mice. N.D; non-detected. (D) Comparison of the number of neuronal subtype marker genes those expressions altered more than 2-fold (up and down) in Atg7-DEG and RAG.
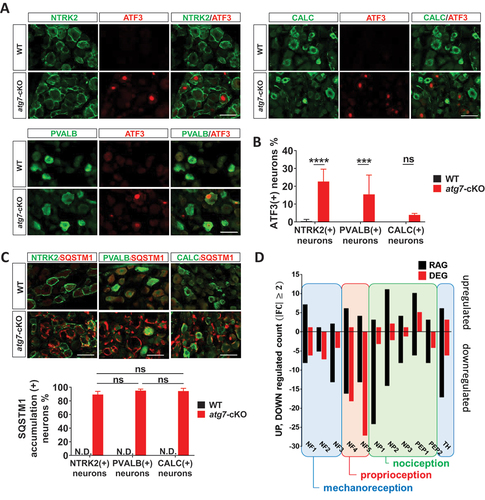
Previous single cell transcriptomic analyses revealed that there are 9 ~ 11 neuronal subtypes in the DRG and that axonal injury-induced transcriptional reprogramming results in the dysregulation of the expression of neuronal subtype marker genes [Citation22,Citation23]. To see injury-like transcriptional responses in each DRG neuronal subtype to autophagic dysfunction, we examined the expression changes of each of ~ 50 neuronal subtype marker genes for 11 cell subtypes in uncut Avil-atg7-cKO DRG, and analyzed Pearson’s correlation coefficients by comparing them to mRNA-Seq data from the injured WT DRG. This experiment revealed correlation coefficients of 0.379 ~ 0.676 (p < 0.01) in 11 neuronal subtypes, indicating moderate to strong positive correlation between them (Figure S3). In accordance with the previous report [Citation22], counting of the number of dysregulated marker genes (FC > 2, p < 0.05) in injured WT DRG showed that the expressions of many genes among subtype marker genes in each subtype neuron were up- or downregulated more than 2 folds after axotomy. In uncut Avil-atg7-cKO DRG, the expressions of marker genes of mechanoreceptive (NF1–3) and proprioceptive neurons (NF4, 5) showed comparable changes to injury-induced dysregulation of those marker genes in WT mice (). However, only a few marker genes of thermoreceptors and nociceptive neurons (NP1–3, PEP1,2) were dysregulated more than 2 folds in uncut Avil-atg7-cKO DRG compared to the injured WT DRG, again suggesting the selective vulnerability of large myelinated axons in Avil-atg7-cKO mice.
Dystrophic axons might be associated with distal axon degeneration of myelinated fibers in atg7-cKO mice
Autophagy defective neurons in the CNS exhibited dystrophic axons which may be related to distal axon degeneration. We tried to find dystrophic axons in the peripheral nerves of Avil-atg7-cKO mice at postnatal week 3 and in adults by EM. However, we could not find any dystrophic axons filled with vesicular accumulations in the sciatic nerves of Avil-atg7-cKO mice at postnatal week 3 () and in adults (Figure S4A).
Figure 5. Neuronal subtype-specific axon degeneration was associated with proximal dystrophic axons. (A) FE-SEM images of the sciatic nerve and DRG from WT and Avil-atg7-cKO mice at postnatal week 3. Scale bar: 5 μm. *; amorphous inclusion. HN; heminode. (B) Enlargement of amorphous inclusions (asterisks) and vesicular accumulation (middle panel) within sensory neuronal soma and proximal axon. DA; dystrophic axon, M; mitochondria (white arrowheads), Scale bar: 1 μm. (C) A dystrophic proximal axon within DRG. Scale bar: 1 μm. (D) Quantification of dystrophic axons within DRG. (E) Schematic drawing of a DRG neuron with a stem axon and two axonal branches (central and peripheral). Vesicular accumulation in the stem axon near neuronal soma may break normal distal axon transport. (F). FE-SEM images of L4 dorsal roots from WT and Avil-atg7-cKO mice at adults. Scale bar: 0.1 mm. (G) Measurement of the dorsal root areas in semithin sections from WT and Avil-atg7-cKO mice. (H) Counting the number of myelinated axons in the dorsal root of WT and Avil-atg7-cKO mice. ns; non-significant, *; p < 0.05, **; p < 0.01, ***; p < 0.001.
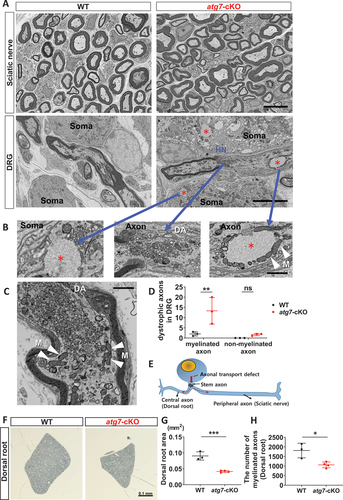
We next examined the DRG of Avil-atg7-cKO mice at 3 weeks postnatally by EM to find axonal changes in more proximal regions of the peripheral nerves. DRG of Avil-atg7-cKO mice, but not of WT mice, showed many amorphous inclusions in the neuronal soma and proximal axons within the DRG, which are typical features of SQSTM1/ubiquitin inclusions in autophagy-defective neurons [Citation14] (). Interestingly, we observed dystrophic axons filled with massive accumulations of small vesicles in proximal axons within the DRG of Avil-atg7-cKO mice, but not WT mice (). Many mitochondria were localized around amorphous inclusions and vesicular accumulations (). The proximal axon-specific accumulation of vesicular structures was also observed in the DRG of Avil-atg7-cKO mice in adult (Figure S4). We counted the number of dystrophic axons showing “vesicular accumulations” that occupied more than 50% of the cross sectional axoplasmic area in each axon within the adult DRG, and ~ 12 myelinated axons/ DRG section exhibited the vesicular accumulation in Avil-atg7-cKO mice, whereas ~ 2 myelinated axons/DRG section were dystrophic in WT DRG ( and Figure S4B). Vesicular accumulations were not apparent in un-myelinated axons within the DRG of Avil-atg7-cKO mice in adulthoods (), indicating that vesicular accumulations are specifically localized in the proximal axons of myelinated neurons within the DRG.
The proximal axons located within DRG may encompass a dendroaxon or a stem axon that is divided into a peripheral axon and a central axon to the spinal cord () [Citation24,Citation25]. Vesicular accumulations in a heminode is a definite evidence of axonal dystrophy of stem axons within DRG (). We hypothesized that if axon degeneration in the sciatic nerve is associated with a breakage of axonal transport in a dystrophic stem axon, it may lead to axon degeneration not only in peripheral axons (sciatic nerve) but also in central axons (). We thus examined a potential axon degeneration in central axons (dorsal roots) in Avil-atg7-cKO mice. The morphometric analysis of semi-thin sections of L4 dorsal roots from WT and Avil-atg7-cKO mice at adults revealed a significant decrease not only in the root area but also in the number of myelinated axons in Avil-atg7-cKO mice compared to WT mice (). In addition, there was a significant reduction in axonal diameter of myelinated fibers in Avil-atg7-cKO mice compared to WT mice (Figure S5A and B). We also examined the expression of PVALB in the dorsal roots using IF staining, and found a significant decrease of PVALB+ axons in the dorsal root of Avil-atg7-cKO mice compared to that in WT mice (Figure S5C and D). Taken together, our findings suggest that axonal dystrophy in the proximal axons of DRG neurons is associated with axonal degeneration of both peripheral and central branches in Avil-atg7-cKO mice.
Dominant-negative SARM1 ameliorated both axonal degeneration and sensory neuropathy in SYN1-atg7-cKO mice
The autophagic stress in dystrophic proximal axons of Avil-atg7-cKO mice may induce production of reactive oxygen species (ROS) which may be related to the induction of axon degeneration. Thus, we examined possible ROS production within the DRG by measuring CAT (catalase) activity in WT and Avil-atg7-cKO mice. This experiment revealed that CAT activity was increased by more than 2-fold in the DRG of Avil-atg7-cKO mice compared to the WT DRG (). Both autophagic stress including ROS generation and axonal transport defects by vesicular accumulations in dystrophic axons may induce Wallerian-like degeneration through SARM1 activation. We thus investigated the involvement of SARM1 in axonal degeneration in atg7-KO mice. We first examined the activation of the SARM1 NADase activity by measuring NAD+ levels in the sciatic nerves of WT and Avil-atg7-cKO mice (2-months-old), and this experiment revealed that there was a significant decrease of NAD levels in the sciatic nerve of Avil-atg7-cKO mice (46.1% of control value, p < 0.05) compared to that in WT mice (). We also examined the levels of cyclic ADP ribose (cADPR), which is a product of SARM1 NADase reaction and a potential biomarker of SARM1 activation [Citation26], in the sciatic nerves of WT and Avil-atg7-cKO mice at adults. However, we could not find a significant elevation of cADPR levels in the sciatic nerves of Avil-atg7-cKO mice compared to that in WT mice, even though its level was increased in WT mice after axotomy (Figure S6A-C). We suspected that chronic and continuous SARM1 activation in each individual nerve at different times in Avil-atg7-cKO mice might not be reflected in cADPR levels in our quantitative assay since cADPR is metabolically very unstable [Citation27].
Figure 6. Dominant-negative SARM1 reduced the phenotypes of sensory neuropathy in SYN1-atg7-cKO mice. (A) CAT activity in the DRG from WT and Avil-atg7-cKO mice. (B) NAD levels in the sciatic nerves of WT and Avil-atg7-cKO mice. (C) Production of SYN1-atg7-cKO mice expressing SARM1 dominant-negative mutant (SARM1-DN) or tdTomato using AAV. (D) Western blot analysis against SARM1-DN (Venus-Tag), ATG7, and SQSTM1 in the DRG. (E, F) The rotarod performance (E) and sensory nerve action potential (SNAP, F) in SYN1-atg7-cKO mice. (G) Quantification of PVALB-positive axons in the sciatic nerve from SYN1-atg7-cKO mice co-injected with AAV- SARM1-DN or AAV-tdTomato. (H) Western blot analysis against ATF3 in the DRG of the mutant mice. (I) Counting the number of ATF3 (+) neurons in the DRG of mutant mice after IF staining. (J) RT-qPCR results of Ucn and Cckbr mRNA in DRG of SYN1-atg7-cKO mice co-injected with AAV-tdTomato or AAV- SARM1-DN. *; p < 0.05, **; p < 0.01, ***; p < 0.001, ****; p < 0.0001. ns; non-significant.
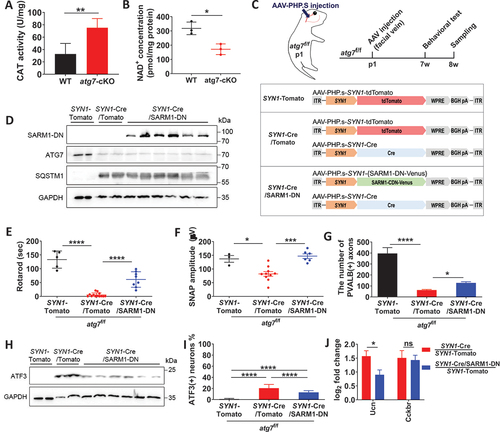
To know whether SARM1 inhibition suppresses the development of axonal neuropathy in atg7-KO mice, we intravenously coinjected AAV-dominant negative Sarm1 transgene (SARM1-DN), which has three mutations in ARM (K193, H194) and TIR domain (H685) of SARM1 and was very effective in inhibiting SARM1 in Wallerian degeneration [Citation28] or AAV-tdTomato with AAV-SYN1-Cre into Atg7f/f mice at postnatal day 1 (). Coinjection of AAV-SARM1-DN with AAV-SYN1-Cre into Atg7f/f mice resulted in reduction in ATG7 protein levels and massive accumulation of SQSTM1 in the DRG at levels comparable to those observed in AAV-tdTomato/AAV-SYN1-Cre coinjected Atg7f/f mice (), suggesting that autophagy impairment was similar in both groups of animals. Coinjection of AAV-SARM1-DN in SYN1-atg7-cKO mice resulted in significant recovery of not only sensory-motor coordination defects in the rotarod test () but also electrophysiological abnormalities () in adults.
We next examined whether SARM1 inhibition suppressed sciatic nerve axonal degeneration in SYN1-atg7-cKO mice. First, axonal downregulation of PVALB staining in the sciatic nerve of SYN1-atg7-cKO mice was partially recovered by coinjection of AAV-Sarm1-DN (). Second, it was found that the expression of ATF3 in the DRG was downregulated in SARM1-DN-injected SYN1-atg7-cKO mice compared to AAV-tdTomato injected SYN1-atg7-cKO mice in western blot analysis and IF staining (). Additionally, we examined the mRNA expression of Ucn and Cckbr in the DRG of SARM1-DN-injected SYN1-atg7-cKO mice to determine whether other injury-responsive genes in autophagy-deficient neurons are dependent on SARM1. RT-qPCR showed that the induction of mRNA expression of urocortin in SYN1-atg7-cKO mice was significantly rescued by injection of AAV-SARM1-DN (). Together, these findings indicate that loss of the Atg7 gene in DRG neurons during postnatal development leads to SARM1-dependent sensory axonal degeneration and sensory neuropathy.
Discussion
Although the involvement of autophagy dysfunction in the development of peripheral neuropathy has been reported, the mechanisms by which neuropathy develops are unclear. Here, we found that SARM1-dependent Wallerian-like axon degeneration participates in the development of axonal neuropathy in sensory neuron-specific autophagy-deficient mice. SARM1 is the critical executioner for axon degeneration following various axonal insults including axotomy. SARM1 is a NADase and its enzyme activity drives the degeneration of axons by energy failure due to NAD depletion in injured axons [Citation15]. In normal axons, SARM1 activity is restrained by NMNAT2 (nicotinamide nucleotide adenylyltransferase 2), a very labile NAD+ synthase, which level in axons is maintained through continuous supply from soma to axons [Citation29]. Thus, breaking axonal transport by axotomy results in the reduction of NMNAT2 levels in distal axons, consequently, NAD+ levels are decreased with an increase of a NAD+ precursor, NMN. An increase of NMN:NAD+ ratio leads to SARM1 activation by altering autoinhibitory conformation of SARM1 [Citation30,Citation31]. Our EM findings of vesicular accumulation in the proximal axons of DRG neurons of atg7-cKO mice suggest that there might be an axonal transport defect which would result in impaired delivery of axon survival factors, such as NMNAT2, that could eventually provoke SARM1 activation in distal axons.
SARM1-dependent axon degeneration has also been observed in chemotherapy-induced axon degeneration [Citation28], TNF (tumor necrosis factor)-induced optic nerve degeneration [Citation32] and TARDBP/TDP-43-linked axon destruction in an amyotrophic lateral sclerosis model [Citation33]. It is not entirely clear how various axonal stresses without physical axon severing induce SARM1 activation in axons. It was recently shown that impairment of mitochondrial function or overproduction of mitochondrial ROS could leads an axon to a “metastable condition” that was fated to degenerate via SARM1 activation [Citation34], underlining the importance of mitochondrial quality control in the prevention of aberrant SARM1 activation in normal axons [Citation18,Citation35,Citation36]. Moreover, it was recently shown that a mitochondrial-SARM1 feedback loop is implicated in MFN2 (mitofusin 2) mutation-induced axonal neuropathy, a CMT2A model [Citation37]. In that hypothesis, initial mitochondrial dysfunction by MFN2 mutation causes SARM1 activation that further disrupts mitochondrial function, potentially through interrupting mitochondrial NAD+ metabolism, which then additionally activates SARM1. Therefore, inhibiting SARM1 improved not only axonal neuropathy but also mitochondrial abnormalities caused by MFN2 mutation. Since autophagy is one of the main pathways to clear damaged mitochondria through mitophagy [Citation1] and because mitochondria are indeed accumulated in dystrophic axons of DRG neurons in atg7-cKO mice, it is likely that long-term autophagy loss in DRG neurons leads to the accumulation of dysfunctional mitochondria in axons which ultimately provokes axonal SARM1 activation. Taken together, the accumulation of dysfunctional mitochondria associated with axonal transport defects could be involved in SARM1 activation in atg7-cKO mice.
On the other hand, we found that behavioral phenotypes of atg7-cKO mice were not entirely attributed to SARM1-dependent axon degeneration since their rescue by SARM1-DN was modest in vivo. There may be some explanations to this. We used AAV-SARM1-DN to inhibit SARM1 in atg7-cKO mice. After injection of SARM1-DN into the mice at postnatal day 1, the phenotypes were analyzed at 6 ~ 8 weeks after AAV injection, and thus limited availability of SARM1-DN proteins in infected DRG neurons may affect the experimental outcomes. It would also be possible that autophagic stress or loss of Atg7 nonspecifically induces neuronal dysfunction. It was recently shown that atg5 KO neurons showed autophagy-independent alteration of axonal microtubule stability that hampers retrograde NTF (neurotrophin) signaling [Citation38], which may alter neuronal function. Therefore, SARM1-independent neuronal dysfunction or unidentified toxicity may contribute to phenotypes of atg7-cKO mice. Future studies using SARM1-KO-atg7-cKO mice would show more conclusive data on the extent of functional implication of SARM1-related axon degeneration in the development of sensory neuropathy in atg7-cKO mice.
Recent studies revealed that a defect in axonal lysosomal transport in Niemann-Pick disease type C resulted in the formation of dystrophic peripheral axons [Citation2], filled with autophagic vacuoles and multivesicular bodies, suggesting autolysosomal digestion within axons as a basal protection mechanism against axonal dystrophy under normal conditions [Citation3]. In the present study, we found dystrophic axons in the peripheral nerves of atg7-cKO mice. Considering previous reports showing that specific ablation of essential autophagy genes, such as Atg7 or Atg5, in cerebellar Purkinje neurons or cortical neurons resulted in axonal dystrophy [Citation4,Citation39,Citation40], our results indicate that autophagy dysfunction is related to the development of dystrophic axons not only in the CNS but also in the peripheral nerves. Interestingly, we found that dystrophic axons are primarily localized in proximal axons near the soma within the DRG. Neuronal soma and proximal axons are the major sites of autolysosome localization in neurons [Citation41], and recent studies have revealed that full maturation of axonal autophagic vesicles mainly occurs near neuronal soma [Citation38,Citation42]. Thus, our finding may indicate that the loss of autophagy results in the accumulation of both autophagy substrates and phagophore structures principally in these regions of DRG neurons.
Neuronal Atg5 or Atg7 KO causes early or late neurodegeneration depending on the types of neurons [Citation4,Citation5,Citation14,Citation39,Citation40]. Most neurodegeneration was associated with axonal dystrophy which precedes late neuronal death [Citation4,Citation5]. In DRG neurons, Atg7 KO induced neuronal death at 9 months after birth [Citation14]. Because long-term denervation after axotomy in the peripheral nerve induces delayed cell death of injured neurons [Citation43], it would be possible that axon degeneration in Atg7-deficient DRG neurons is causally associated with the late neuronal death of the autophagy-deficient mice. Further studies evaluating the role of SARM1 in delayed neuronal death in Atg7-deficient DRG neurons are necessary.
In this study, we have found that axonal vulnerability to autophagy deficiency was cell-type specific in sensory neurons. Several reports have shown neuronal subtype-specific vulnerability to nerve damage in peripheral nervous system. For example, loss of the class III phosphatidylinositol 3-kinase, an endocytotic regulator, in the DRG induces faster degeneration of large-diameter NTRK2 or PVALB neurons than small CALC+ neurons [Citation14]. In addition, neuronal adaptation to a harmful situation also seems to differ across subtypes of sensory neurons. It was recently reported that the loss of Schwann cells and demyelination specifically induced injury responses of low-threshold mechanoreceptors and proprioceptors in the DRG [Citation44]. Interestingly, ablation of non-myelinating Schwann cells did not provoke injury responses in unmyelinated C-fiber neurons [Citation44]. One speculative explanation is that those large-diameter neurons, proprioceptive neurons and low-threshold mechanoreceptors, undergo more active metabolic homeostasis than small nociceptor neurons due to their constant activity under normal conditions, and thus, they may be more sensitive to metabolic disturbance or harmful stresses than small neurons. It should also be mentioned that most CMT patients show abnormal signs of proprioception and vibration, and their symptoms slowly progress to decreased pain and temperature sensation [Citation45]. Similar findings were reported in an animal model of CMT2E expressing human neurofilament light chain mutant that exhibited selective loss of myelinated axons in the peripheral nerves [Citation46]. Therefore, selective axon degeneration in large myelinated axons in Atg7-deficient sensory neurons might be related to the differential vulnerability of sensory neurons following various neuroglial insults in the peripheral nervous system.
In conclusion, we show that autophagy impairment induced by deletion of the Atg7 gene in the peripheral sensory neurons leads to axonal dystrophy and SARM1-dependent Wallerian-like degeneration. This study provides mechanistic insights into how autophagy dysfunction leads to sensory neuropathy. Because many genes related to autophagy/mitophagy quality control are causative genes for axonal CMT2 [Citation47,Citation48], preventing SARM1 activation is a promising strategy against axonal neuropathy characterized by autophagic dysfunction.
Materials and Methods
Materials
Antibodies against ATG7 (A2856), NEFH/neurofilament-200 (N5389) were purchased from Sigma Aldrich. Antibodies against NEFM/neurofilament-M (AB1987) was purchased form Millipore. Antibodies against SQSTM1/p62 (ab109012), GAPDH (ab181603), and CALC (ab22560) were obtained from Abcam. TUBB3/tubulin β3 antibody (clone TUJ1, MMS-435P) was purchased from BioLegend. Antibodies against ATF3 (NBP1–85816) and PVALB/parvalbumin (NB120-11427SS) were purchased from Novus Biologicals. NEFL/neurofilament-L (2837), horseradish peroxidase-linked anti-rabbit IgG (7074) and anti-mouse IgG (7076) were obtained from Cell Signaling Technology. Antibodies against NTRK2/TRKB (AF1494-SP) and NTRK3/TRKC (AF-1404-SP) were purchased from R&D system.
Animals
C57BL6 mice were obtained from Samtako Bio Korea (Korea), and most experiments were performed with 2- to 3-month-old mice. Avil-Cre mice were kindly provided by Dr. Jung Eun Shin [Citation19,Citation49]. Atg7f/f mice were obtained from Dr. Masaki Komatsu [Citation39]. DRG-specific autophagy knockout mice were produced through the crossbreeding of Atg7f/f mice and Avil-Cre mice. All animal experiments were performed according to protocols approved by the Dong-A University Committee on Animal Research which follows the guidelines for animal experiments that were established by the Korean Academy of Medical Sciences.
The genotype of the mice was confirmed by polymerase chain reaction (PCR). The following PCR primers were used: Avil_Cre forward primer (5’-GCCTGGGGGTAACTAAACTGGTCGAGCGAT-3’) and reverse primer (5’-GCACTGTGTCCAGACCAGGCCAGGTATCTC-3’); Atg7 forward primer (5’-TGGCTGCTACTTCTGCAATG-3’) and reverse primer (5’-CAGGACAGAGACCATCAGCTCCAC-3’).
For a sciatic nerve injury, wild type mice were anesthetized with 10% ketamine hydrochloride (Yuhan, 0.2 ml/100 g body weight), and the left sciatic nerves of adult mice (7 ~ 8 weeks) were axotomized as previously reported [Citation49].
Adeno-associated virus (AAV) production and infection
Gene delivery into mice was performed using AAV vectors expressing tdTomato, Cre, and SARM1-DN under the control of the human SYN1 (synapsin I) promoter, a neuron-specific promoter (Addgene, 50465; deposited by Dr. Bryan Roth); the vectors were packaged using the AAV-PHP.s serotype. AAV-PHP.s-SYN1-tdTomato (Vector ID: VB210328–1088utk), AAV-PHP.s-SYN1-Cre (Vector ID: VB210324–1043×en), and AAV-PHP.s-SYN1-SARM1-DN (Vector ID: VB210512–1261mtk) were constructed by VectorBuilder (Guangzhou, China). SARM1-DN, which has three mutations (K193R, H194A and H685) in SARM1, was kindly provided by Dr. A. DiAntonio (Washington University, MO) [Citation28]. Atg7f/f mice at postnatal day 1 were anesthetized with ice for 5 min. The mice were then placed under a stereomicroscope (Olympus, SZ51, Japan), and AAV virus particles were injected into the superior temporal vein at a dose of 1 × 1012 CG/10 µl using a Hamilton syringe with a 33-gauge needle [Citation50,Citation51]. Then, the mice were returned to a warm cage, and housed until sacrifice.
Immunofluorescence (IF) staining
The sciatic nerve and DRG were harvested and postfixed in 4% paraformaldehyde (Biosolution, BP031a) for overnight. The tissues were transferred to 30% sucrose for cryoprotection and then were embedded using OCT compound (Scigen, 4583). The tissue was sectioned with 10 μm thickness using Cryostat (Feigocut, Leica) and sections were attached to the Superfrost-slide (VWR, 48311–703). Slide was blocked with PBS (Hyclone, SH30258.02) containing 5% fetal bovine serum (Gibco, 16000044) and 5% bovine serum albumin (Bioshop, ALB001.100) for 1 h. The slide was incubated with primary antibodies for overnight at 4°C, and then washed with PBS. The slide was incubated with secondary antibodies at room temperature for 2 h, then washed with PBS. The nuclei were counterstained with Hoechst (ThermoFisher Scientific, 62249) for 15 min at 37°C. The sections were then observed using by Axiolmager 2 research microscope (Carl Zeiss Meditec, AG, Germany) or laser confocal microscope (ImageXpress, Molecular Devices (San Jose, CA, USA) For counting the immunopositive neurons or axons, 4 captured images (×20 objective) from L4/L5 DRGs or sciatic nerve from a mouse (n = 3 for each genotype) were used.
Field Emission Scanning electron microscopy (FE-SEM)
The sciatic nerves and DRG were immersion-fixed with a mixture of 4% paraformaldehyde and 3% glutaraldehyde (Sigma Aldrich, G7776) for overnight. Afterward, the tissues were transferred to 0.05 M sodium cacodylate buffer (EMS, 11650) and were post-fixed in 2% osmium tetroxide (OSO4; Ted pella, 18463) in cacodylate buffer for 90 min. The solution was replaced with 1.5% potassium ferrocyanide (Sigma Aldrich, P3289) in cacodylate buffer, and samples were sequentially incubated in filtered 1% thiocarbohydrazide (Sigma Aldrich, 223220) for 45 min at 40°C, 2% OSO4 aqueous solution for 90 min, and 1% uranyl acetate solution for overnight at 4°C. On the next day, the sample (still in the uranyl acetate solution) was warmed in 50°C oven for 120 min, then solution was replaced by lead aspartate solution for 120 min at 50°C. After washing with PBS, sample were sequentially dehydrated and embedded with a graded ethanol series, acetone (2 times, 15 min), acetone: Spurr’s mixture and final pure Spurr’s resin (DDSA; 00563–450, Epon812; 08791–500, MNA; 00886–500, DMP30; 00553–100, Polyscience) overnight at room temperature. The samples were incubated in fresh pure resin for another 6 h and polymerized for 48 h at 60°C. The embedded samples were sectioned using an ultramicrotome (Leica, Germany) with 100-µm thickness and then the sections were attached to a silicon wafer (20 mm diameter). Large-area high resolution imaging (15,000×, pixel size 3072 × 2048) was performed using Apreo 2S Lovac scanning electron microscopy (ThermoFisher Scientific, USA) at the Neuroscience Translational Research Solution Center.
For the morphometric analysis of FE-SEM images, three sciatic nerves, L4 dorsal roots and L4 dorsal root ganglions from each genotype were used. The whole number of myelinated axons, the Band of Büngner, and Remak bundles were counted using the mosaic image of the sciatic nerve. The number of dystrophic axons in DRG was counted using DRG images from wild type (n = 3) and Atg7 KO (n = 3) mice.
Western blot analysis
The tissues immersed in RIPA buffer was homogenized by TissueLyser LT (Qiagen, Hilden, Germany). The lysates were centrifuged for 15 min (17000 g) at 4°C, then the separated supernatant was collected. Proteins were separated by SDS-PAGE, and then were transferred to the nitrocellulose membrane (Amersham, 10600003) using a semi-dry transfer system by BioRad (Hercules, CA, USA). The membrane was blocked for 1 h with TBST (iNtRON Biotechnology, IBS-BT008A) and 5% skim milk (BD DIFCO, 232100). The membrane was incubated with primary antibody that diluted in TBST containing skim milk for overnight at 4°C. Then, the membrane was washed three times with TBST. The horseradish peroxidase-conjugated second antibody (1; 2000) was applied to the membrane for 2 h at room temperature. Thereafter, Enhanced Chemiluminescence reaction was performed and image was taken using Lumino GraphIII (Atto Corporation, Tokyo, Japan). The intensity of western band was analyzed using CSANALYZER4 software (Atto Corporation, Tokyo, Japan).
mRNA-Sequencing analysis
The total RNA from uninjured or injured L4–5 DRG was purified by using RNAqueous-Micro Kit (Invitrogen, AM1931). Two independent sets of experiments were performed for each genotype. After purifying mRNA from the total RNA using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, 20020595), the cDNA libraries were prepared and then mRNA-Sequencing was performed using the NovaSeq 6000 S4 Reagent Kit (Illumina, 20028312). The whole processes of mRNA-Seq were performed by Macrogen (Seoul, Korea). Functional analysis of the differentially expressed genes was conducted by using g:Profiler tool and DAVID version 6.8. Enrichment of biological process terms was tested for statistical significance (adjusted P value < 0.05). The gene lists of each analysis were displayed in Table S1.
RT-qPCR
The cDNA was synthesized using M-MulV Reverse Transcriptase (New England BioLabs, M0253). qRT-PCR was performed by using Maxima SYBR Green/ROX qPCR kit (Applied Biosystems, K0221) with QuantStudio 5 (Thermo Fisher Scientific, MA, USA) at the Neuroscience Translational Research Solution Center. The primers for Ucn; 5`-GGCACCATGATACAGAGGGG-3` and 5`-CTGTGCCAAGAGCAGCAAC-3`, for Cccbr; 5`-CCAACAAATGTGGTCCGTGC-3` and 5`-GGTAGAGTTCGCGGGAGATG-3`, for ATF3; 5`-GAGGCGGCGAGAAAGAAATA-3` and 5`-CTGTCTTCTCCTTTTTCTTGTTT-3`; for Vstm2b; 5`-CGTTTCGGACTACAGCGATG-3` and 5`-AGGTGGGTTCTTGTCATCGT-3`, for Gjb2; 5`-ATCTTGGAGAGGCTGCTGGAA-3` and 5`-CCACAATGAGTATCCGGAAGATC-3`, for Gapdh; 5`-GGGAAGCCCATCAAAAGTGGC-3` and 5`-GACAAGTCTCTGGCATCTGAG-3`.
Behavioral tests
For Rotarod test, mice were placed on gradually accelerating (1 RPM per every 8.3 second) the circular rod ROTA-ROD (Harvard Apparatus, USA), and the length of time that a given animal stays on the rotating rod was measured three times for a test.
The muscle strength of mice was measured using a grip strength-meter (Bioseb, Bio-GT3, France). The mouse was placed on the grid, and then the mouse’s tail was pulled back to measure the hindlimb grip strength. This procedure was conducted three times for a test.
Electrophysiology
Mice were anesthetized with 2.5% isoflurane (Vspharm, Gyeonggi, South Korea) using an animal anesthesia system (Terrell, Minrad Inc., PA, USA), and nerve conduction velocity (NCV) and the amplitude of action potential were measured using a Viking Quest System from Nicolet Biomedical (Madison, WI, USA). Measurement of motor NCV was performed in the sciatic nerves, and sensory NCV was measured in the tail.
Measurement of NAD+ and cADPR
For tissue metabolites extraction, the sciatic nerves were homogenized by sonication (Sonics VibraCell VCX130, pulse on 2 s, pulse off 2 s, 50% Amplitude, total 20 s) in 50% methanol (Burdick&Jackson, RP230–1) in water (100 μl/nerve). Homogenates were centrifuged (17,000 g, 10 min, 4°C) and cleared supernatants were transferred to new tubes containing one third volume of chloroform (Burdick&Jackson, 100199), mixed, and centrifuged (17,000 g, 10 min, 4°C). The aqueous phase (100 μl) was transferred to a new tube and lyophilized and stored at − 70°C until analysis. For metabolites extraction from eDRG neurons, at DIV7, we first removed cell bodies in cultures, and then immediately the medium was replaced with 160 μl ice cold 50% metabolites in water. The neurites were incubated for 5 min on ice with the 50% methanol solution and then the solution was transferred to tubes containing 50 μl chloroform on ice, vortexed, and centrifuged at 20,000 g for 15 min at 4°C. The clear aqueous phase collected and lyophilized under vacuum.
Lyophilized samples were reconstituted with 5 mM ammonium formate and centrifuged (17,000 g, 10 min, 4°C), and 10 μl supernatant was loaded on LC-MS/MS (Agilent 6410LC-QQQ/MS) with internal standard (2-chloroadenosine at 2 ng/μl) at Metabolomics Research Center for Functional Materials. To increase the sensitivity and specificity of the analysis, we worked in multiple reaction monitoring and followed the MS/MS transitions: NAD+ MH+, 664.1–136.1. The spray chamber settings were as follows: heated capillary, 325°C; vaporizer temperature, 40°C; Capillary voltage, 4000 V. For quantitative analysis, the precursor ion and product ion settings are as follows: cADPR precursor ion [MH]+ m/z 542, product ion m/z 428, m/z 136. Serial dilutions of standard for NAD and cADPR in 5 mM ammonium formate were used for calibration. Metabolites were quantified by MassHunter quantitative analysis tool (Agilent, version B.03.01) with standard curves.
Statistical analysis
All Statistical analysis was performed using GraphPad Prism software, after the normality test. The single comparisons were done using two-tailed unpaired Student’s t-test between two groups. The one-way ANOVA was performed after post hoc multiple comparisons by using Tukey’s HSD test. The results were given as mean ± SEM. P-value <0.05 was considered significant.
Supplemental Material
Download Zip (4.1 MB)Acknowledgements
We thank to Dr. A. DiAntonio and Yo Sasaki for providing SARM1-DN cDNA and information for the measurement of NAD and cADPR.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2244861
Additional information
Funding
References
- Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011 Nov 11;147(4):728–741. PubMed PMID: 22078875. 10.1016/j.cell.2011.10.026
- Roney JC, Li S, Farfel-Becker T, et al. Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev Cell. 2021 May 17;56(10):1452–1468.e8. PubMed PMID: 33878344. 10.1016/j.devcel.2021.03.032.
- Tamim-Yecheskel C, Fraiberg M, Kokabi K, et al. A tecpr2 knockout mouse exhibits age-dependent neuroaxonal dystrophy associated with autophagosome accumulation. Autophagy. 2021 Oct;17(10):3082–3095. doi: 10.1080/15548627.2020.1852724. PubMed PMID: 33218264.
- Komatsu M, Qing JW, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007 Sep 4;104(36):14489–14494. PubMed PMID: 17726112. 10.1073/pnas.0701311104.
- Nishiyama J, Miura E, Mizushima N, et al. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007 Nov-Dec;3(6):591–596. doi: 10.4161/auto.4964. PubMed PMID: 17912025.
- Yong Y, Hunter-Chang S, Stepanova E, et al. Axonal spheroids in neurodegeneration. Mol Cell Neurosci. 2021 Dec;117:103679. PubMed PMID: 34678457. doi: 10.1016/j.mcn.2021.103679
- Gowrishankar S, Yuan P, Wu Y, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A. 2015 Jul 14;112(28):E3699–708. PubMed PMID: 26124111. 10.1073/pnas.1510329112.
- Haidar M, Timmerman V. Autophagy as an Emerging Common Pathomechanism in Inherited Peripheral Neuropathies. Front Mol Neurosci. 2017 May 11;10:143. PubMed PMID: 28553203. 10.3389/fnmol.2017.00143
- Romano R, Stefania V, Fiore D, et al. Carrying the RAB7 K126R Mutation. Cells. 2022 Jan 31;11(3):496. PubMed PMID: 35159308. 10.3390/cells11030496.
- Romano R, Del Fiore VS, Saveri P, et al. Autophagy and Lysosomal Functionality in CMT2B Fibroblasts Carrying the RAB7K126R Mutation. Cells. 2022 Jan 31;11(3):496. PubMed PMID: 35159308. 10.3390/cells11030496.
- Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy FAM134B-HA. Nature. 2015 Jun 18;522(7556):354–358. PubMed PMID: 26040720. 10.1038/nature14498.
- Rudnick ND, Griffey CJ, Guarnieri P, et al. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci U S A. 2017 Sep 26;114(39):E8294–E8303. PubMed PMID: 28904095. 10.1073/pnas.1704294114.
- Tashiro Y, Urushitani M, Inoue H, et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012 Dec 14;287(51):42984–42994. PubMed PMID: 23095749. 10.1074/jbc.M112.417600.
- Zhou X, Wang L, Hasegawa H, et al. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc Natl Acad Sci U S A. 2010 May 18;107(20):9424–9429. PubMed PMID: 20439739. 10.1073/pnas.0914725107.
- Figley MD, DiAntonio A. The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease. Curr Opin Neurobiol. 2020 Aug;63:59–66. PubMed PMID: 32311648. doi: 10.1016/j.conb.2020.02.012
- Osterloh JM, Yang J, Rooney TM, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012 Jul 27;337(6093):481–484. PubMed PMID: 22678360. 10.1126/science.1223899.
- Bloom AJ, Mao X, Strickland A, et al. Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients. Mol Neurodegener. 2022 Jan 6;17(1):1–15. PubMed PMID: 34991663. 10.1186/s13024-021-00511-x.
- Loreto A, Hill CS, Hewitt VL, et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol Dis. 2020 Feb;134:104678. PubMed PMID: 31740269. doi: 10.1016/j.nbd.2019.104678
- Hasegawa H, Abbott S, Han B-X. Analyzing Somatosensory Axon Projections with the Sensory Neuron-Specific Advillin Gene. J Neurosci. 2007 Dec 26;27(52):14404–14414. PubMed PMID: 18160648. 10.1523/JNEUROSCI.4908-07.2007
- Chan KY, Jang MJ, Yoo BB, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017 Aug;20(8):1172–1179. doi: 10.1038/nn.4593. PubMed PMID: 28671695.
- Badia J, Pascual‐Font A, Vivó M, et al. Topographical distribution of motor fascicles in the sciatic‐tibial nerve of the rat. Muscle Nerve. 2010 Aug;42(2):192–201. doi: 10.1002/mus.21652. PMID: 20544926.
- Renthal W, Tochitsky I, Yang L, et al. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron. 2020 Oct 14;108(1):128–144.e9. PubMed PMID: 32810432. 10.1016/j.neuron.2020.07.026.
- Usoskin D, Furlan A, Islam S, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015 Jan;18(1):145–153. doi: 10.1038/nn.3881. PubMed PMID: 25420068.
- Devor M. Unexplained peculiarities of the dorsal root ganglion. Pain. 1999 Aug;82(6):S27–S35. Pubmed PMID: 10491970. doi: 10.1016/S0304-3959(99)00135-9
- Zenker W, Högl E. The prebifurcation section of the axon of the rat spinal ganglion cell. Cell Tissue Res. 1976 Jan 27;165(3):345–363. Pubmed PMID: 1248030. 10.1007/BF00222438
- Sasaki Y, Engber TM, Hughed RO, et al. cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp Neurol. 2020 Jul;329:113252. Pubmed PMID: 32087251. doi: 10.1016/j.expneurol.2020.113252
- Lee HC, Aarhus R. Wide distribution of an enzyme that catalyzes the hydrolysis of cyclic ADP-ribose. Biochim Biophys Acta. 1993 Jun 24;1164(1):68–74. Pubmed PMID: 8518298. 10.1016/0167-4838(93)90113-6
- Geisler S, Huang SX, Strickland A, et al. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med. 2019 Feb 4;216(2):294–303. PubMed PMID: 30642945. 10.1084/jem.20181040.
- Milde S, Fox AN, Freeman MR, et al. Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Sci Rep. 2013;3(1):2567. Pubmed PMID: 23995269. doi: 10.1038/srep02567
- Figley MD, Gu W, Nanson JD, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron. 2021 Apr 7;109(7):1118–1136.e11. Pubmed PMID: 33657413. 10.1016/j.neuron.2021.02.009.
- Shen C, Vohra M, Zhang P, et al. Multiple domain interfaces mediate SARM1 autoinhibition. Proc Natl Acad Sci U S A. 2021 Jan 26;118(4):e2023151118. Pubmed PMID: 33468661. 10.1073/pnas.2023151118.
- Ko KW, Milbrandt J, DiAntonio A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J Cell Bio. 2020 Aug 3;219(8):e201912047. PubMed PMID: 32609299. 10.1083/jcb.201912047
- White MA, Lin Z, Kim E, et al. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol Commun. 2019 Oct 28;7(1):1–66. PubMed PMID: 31661035. 10.1186/s40478-019-0800-9.
- Hughes RO, Bosanac T, Mao X, et al. Small molecule SARM1 inhibitors recapitulate the SARM1−/− phenotype and allow recovery of a metastable pool of axons fated to degenerate. Cell Rep. 2021 Jan 5;34(1):108588. Pubmed PMID: 33406435. 10.1016/j.celrep.2020.108588.
- Merlini E, Coleman MP, Loreto A. Mitochondrial dysfunction as a trigger of programmed axon death. Trends Neurosci. 2022 Jan;45(1):53–63. Pubmed PMID: 34852932. doi: 10.1016/j.tins.2021.10.014
- Summers DW, Frey E, Walker LJ, et al. DLK activation synergizes with mitochondrial dysfunction to downregulate axon survival factors and promote SARM1-dependent axon degeneration. Mol Neurobiol. 2020 Feb;57(2):1146–1158. doi: 10.1007/s12035-019-01796-2. Pubmed PMID: 31696428.
- Sato-Yamada Y, Strickland A, Sasaki Y, et al. A SARM1-mitochondrial feedback loop drives neuropathogenesis in a charcot-marie-tooth disease type 2A rat model. J Clin Invest. 2022 Dec 1;132(23):e161566. Pubmed PMID: 36287202. 10.1172/JCI161566.
- Negrete-Hurtado A, Overhoff M, Bera S, et al. Autophagy lipidation machinery regulates axonal microtubule dynamics but is dispensable for survival of mammalian neurons. Nat Commun. 2020 Mar 24;11(1):1535. PubMed PMID: 32210230. 10.1038/s41467-020-15287-9.
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006 Jun 15;441(7095):880–884. PubMed PMID: 16625205. 10.1038/nature04723.
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006 Jun 15;441(7095):885–889. PubMed PMID: 16625204. 10.1038/nature04724.
- Gowrishankar S, Wu Y, Ferguson SM. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J Cell Bio. 2017 Oct 2;216(10):3291–3305. PubMed PMID: 28784610. 10.1083/jcb.201612148
- Cason SE, Mogre SS, Holzbaur ELF, et al. Spatiotemporal analysis of axonal autophagosome–lysosome dynamics reveals limited fusion events and slow maturation. Mol Biol Cell. 2022 Nov 1;33(13):ar123. Pubmed PMID: 36044338. 10.1091/mbc.E22-03-0111.
- Tandrup T, Woolf CJ, Coggeshall RE. Delayed loss of small dorsal root ganglion cells after transection of the rat sciatic nerve. J Comp Neurol. 2000 Jun 26;422(2):172–180. PubMed PMID: 10842225. 10.1002/(SICI)1096-9861(20000626)422:2<172:AID-CNE2>3.0.CO;2-H
- Elbaz B, Yang L, Vardy M, et al. Sensory neurons display cell-type-specific vulnerability to loss of neuron-glia interactions. Cell Rep. 2022 Jul 19;40(3):111130. PubMed PMID: 35858549. 10.1016/j.celrep.2022.111130.
- Szigeti K, Lupski JR. Charcot-Marie-Tooth disease. Eur J Hum Genet. 2009 Jun;17(6):703–710. PubMed PMID: 19277060. doi: 10.1038/ejhg.2009.31
- Dale JM, Villalon E, Shannon SG, et al. Expressing Hnf-L E397K results in abnormal gaiting in a transgenic model of CMT2E. Genes Brain Behav. 2012 Apr;11(3):360–365. doi: 10.1111/j.1601-183X.2012.00771.x. PubMed PMID: 22288874.
- Zhou Y, Carmona S, Muhammad AKMG, et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest. 2019 Mar 18;129(4):1756–1771. PubMed PMID: 30882371. 10.1172/JCI124194.
- Guardia CM, Nakagawa M, Polishchuk R, et al. Impaired mitochondrial mobility in charcot-marie-tooth disease. Front Cell Dev Biol PubMed PMID: 33598463 2021 Feb 1;9:624823. doi: 10.3389/fcell.2021.624823
- Shin JE, Ha H, Kim YK, et al. DLK regulates a distinctive transcriptional regeneration program after peripheral nerve injury HHS Public Access. Neurobiol Dis. 2019 Jul;127:178–192. PubMed PMID: 30735704. doi: 10.1016/j.nbd.2019.02.001
- Gessler DJ, Tai PWL, Li J, et al. Intravenous infusion of AAV for widespread gene delivery to the nervous system. Methods Mol Biol. 2019;1950:143–163. Pubmed PMID: 30783972.
- Lee J, Shin JE, Lee B, et al. The stem cell marker Prom1 promotes axon regeneration by down-regulating cholesterol synthesis via Smad signaling. Proc Natl Acad Sci U S A. 2020 Jul 7;117(27):15955–15966. Pubmed PMID: 32554499. 10.1073/pnas.1920829117.