
ABSTRACT
Colitis-associated colon cancer (CAC) is a distinct form of colorectal cancer that emerges from prolonged colitis in individuals with inflammatory bowel disease (IBD). Chronic inflammation is considered as the most critical contributor to the development and progression of CAC, with the degree and persistence of the inflammatory response being the primary drivers of CAC progression in individuals with IBD. Macroautophagy/autophagy, which is a cellular process that aids in preserving the intestinal mucosal barrier and regulating intestinal inflammation, is effective in reducing IBD progression. However, the precise contribution of impaired autophagy to CAC pathogenesis has yet to be fully elucidated. Nevertheless, it is thought that autophagy may reduce the risk of CAC by regulating intestinal inflammation and other factors. This review summarizes recent research on CAC development and investigates the possible functions of autophagy in reducing CAC initiation. Furthermore, we explore the functional transformation of autophagy after CAC transformation and propose that promoting autophagy as a means of attenuating CAC initiation may serve as a novel strategy for preventing IBD-related CAC.
Abbreviations
A. muciniphila: Akkermansia muciniphila; AIEC: adherent invasive Escherichia coli; AOM/DSS: azoxymethane-dextran sodium sulfate; ATG: autophagy related; BECN1: beclin1, autophagy related; CAC: colitis-associated colon cancer; CCDC50: coiled-coil domain containing 50; CLDN2: claudin 2; CoPEC: colibactin-producing Escherichia coli; CRC: colorectal cancer; DAMPs: danger/damage-associated molecular patterns; DC: dendritic cell; DSS: dextran sulfate sodium; DTP: drug-resistant persistent; ER: endoplasmic reticulum; ERN1/IRE1α: endoplasmic reticulum to nucleus signaling 1; IBD: inflammatory bowel disease; IECs: intestinal epithelial cells; IKK: IkappaB kinase; IL: interleukin; IRGM1: immunity-related GTPase family M member 1; ISC: intestinal stem cell; LPS: lipopolysaccharide; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MAPK: mitogen-activated protein kinase; MDP: muramyl dipeptide; MELK: maternal embryonic leucine zipper kinase; MHC: major histocompatibility complex; miRNA: microRNA; MTOR: mechanistic target of rapamycin kinase; NLRP3: NLR family, pyrin domain containing 3; NOD2: nucleotide-binding oligomerization domain containing 2; NRBF2: nuclear receptor binding factor 2; PAMPs: pathogen-associated molecular patterns; PI3K: class I phosphoinositide 3-kinase; PtdIns3K: class III phosphatidylinositol 3-kinase; PYCARD/ASC: PYD and CARD domain containing; RALGAPA2/RalGAPα2: Ral GTPase activating protein protein, alpha subunit 2 (catalytic); RIPK2/CARD3: receptor (TNFRSF)-interacting serine-threonine kinase 2; RIPK3: receptor-interacting serine-threonine kinase 3; ROS: reactive oxygen species; sCRC: sporadic colorectal cancer; SMARCA4/BRG1: SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4; SQSTM1: sequestosome 1; STAT3: signal transducer and activator of transcription 3; TNF/TNFA: tumor necrosis factor; ULK1: unc-51 like autophagy activating kinase 1; UPR: unfolded protein response; WT: wild-type
Introduction
Autophagy is a cellular process that facilitates cellular adaptation to various forms of stress, such as nutritional insufficiency, infection, and hypoxia. This process plays a vital role in preserving cell homeostasis by providing cells with nutrients during times of stress and removing damaged mitochondria and mitochondrial DNA. Autophagy is associated with a variety of physiological functions, such as the removal of bacteria from cells [Citation1], inflammatory cytokine secretion [Citation2], controlled inflammation, antigen presentation [Citation3], and lymphocyte growth [Citation4]. Autophagy impairment can result in increased inflammation and an increased risk of malignancy. Autophagy initiation is triggered by the ULK1/Atg1 functional complex, which comprises ULK1, ATG13, RB1CC1/FIP200, and ATG101. This complex stimulates a series of ATG (autophagy related) genes and forms a phagophore, that matures into an autophagosome with a double-membrane structure. Phagophores enclose and, via autophagosomes, transport cellular components, pathogens, and damaged organelles to lysosomes, where they are digested into usable macromolecules [Citation5]. Autophagy impairment is associated with several human pathologies, including inflammatory bowel disease (IBD) [Citation6].
A significant compromise in the intestinal barrier is a distinguishing characteristic of IBD. A vital part of the intestinal mucosal barrier is the intestinal epithelial cells. These cells regulate the permeability of the intestines and act as a defense against pathogenic invasion [Citation7]. The constant renewal of these cells is enabled by LGR5 intestinal stem cells (ISC) located in the basal region of the crypt [Citation8]. Conditions such as IBD often involve disruption of the epithelial barrier, abnormal microbial interactions, and dysautonomia. Significant abnormalities have been observed during the microscopic assessment of intestinal tissues obtained from individuals with IBD. These include a diminished number of goblet cells, altered mucus composition, and mucus barrier disruptions [Citation9]. Malfunctions in Paneth cells, which leads to the decreased secretion of defensins, are considered significant contributors to the pathogenesis of IBD [Citation10,Citation11]. Moreover, IBD is primarily attributed to immune deficiencies within the intestinal immune system. Mucosal immune cells like macrophages, T cells, dendritic cells, and innate lymphoid cells (ILCs) have a critical role in this dysfunction [Citation12]. Dysregulation of the cytokine balance, specifically the disproportional expression of pro-inflammatory versus anti-inflammatory cytokines, has been identified as a contributing factor to the suppressed resolution of inflammation in IBD. Instead, it may perpetuate disease and accelerate tissue destruction [Citation13]. Among the physiological mechanisms of IBD development described above, autophagy has been found to play a fundamental role in the regulation of all of these factors, and defects in autophagy-related genes are directly associated with IBD development [Citation14].
Several retrospective investigations and meta-analyses have demonstrated that the existence of an extended inflammatory response in IBD is an independent factor for the emergence of colitis-associated colon cancer (CAC) [Citation15]. It has been reported that IBD elevates the risk of colorectal cancer (CRC) development, and every 10-unit increase on the inflammation index is associated with a 2.1% increase in CRC risk [Citation16]. This suggests that controlling inflammation in IBD may be an effective strategy for preventing the development of CAC. Recently, several mechanisms other than inflammation have been identified as contributors to the development of CAC. These mechanisms include reactive oxygen species (ROS) [Citation17], inflammasomes [Citation18], and specific types of cell death, such as pyroptosis and necrosis [Citation19]. Furthermore, studies in animal models of colitis have revealed that commensal microbiota or specific bacteria are necessary to trigger inflammation and promote CAC development [Citation20].
However, it is worth noting that the factors involved in the onset of CAC can be regulated by autophagy. The objective of this review was to examine the involvement of autophagy in IBD progression and CAC genesis and to examine the literature on the potential role of autophagy modulators.
Role of autophagy in CAC development
Autophagy defects exacerbate the severity of IBD
CAC is often considered as a prototype of inflammation-induced cancer and presents differently to sporadic CRC (sCRC), which usually follows a sequence of “normal mucosa-adenoma-dysplasia-carcinoma”, while CAC generally appears as an “inflamed mucosa-dysplasia-carcinoma” sequence [Citation21]. The escalation of inflammation in IBD increases the likelihood of malignancy by generating cytokines that promote tumor growth, such as TNF/TNFA (tumor necrosis factor), IL1 (interleukin 1), IL6, IL17A, and IL23, and by NFKB/NF-κB (nuclear factor of kappa light polypeptide gene enhancer in B cells) and STAT3 (signal transducer and activator of transcription 3) [Citation22]. Immune cells are initially activated by factors such as IL1, TNF, and injury- or pathogen-related molecular pathways. The process of activation results in the generation of various pro-inflammatory chemokines, cytokines, and growth factors, such as VEGF (vascular endothelial growth factor), IL1, IL6, and TNF. The activation of NFKB and STAT3 activators by pro-inflammatory cytokines in both cancerous cells and the tumor microenvironment has been found to result in various oncogenic processes, such as cancer cell survival, growth, epithelial-mesenchymal transition/EMT, angiogenesis, invasion, and metastasis [Citation23] ().
Figure 1. Molecular patterns from intestinal inflammation to CAC development. The initial phase of intestinal inflammation activates the NFKB (nuclear factor kappa B) in immune cells via TNF (tumor necrosis factor), IL1, and danger-associated molecular patterns (DAMPs), which leads to an influx of pro-inflammatory factors, including TNF, IL1, IL6, IL11, IL22, VEGF (vascular endothelial growth factor), ROS, among others. TNF plays a pivotal role by activating fibroclasts to produce VEGF, which provides essential nutritional support for tumor growth, and concurrently stimulates Th17 cells to produce IL17 and IL22. In the presence of ROS and RNS, further DNA damage is induced within cells, a crucial event in the progression of CAC. This DNA damage, along with the concerted activation of STAT and NFKB within tumor cells by cytokines, ultimately drives tumor proliferation.
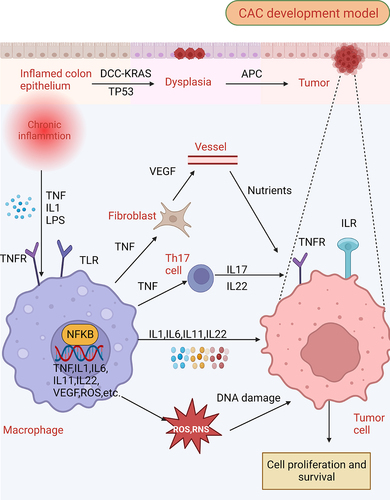
Since the degree of inflammation in IBD is strongly associated with CAC development and progression, we discuss the crucial function of impaired autophagy in facilitating inflammatory mechanisms in IBD. Genome-wide correlation investigations have revealed a strong relationship between susceptibility to IBD and ATG, particularly IRGM1 (immunity related GTPase M) and ATG16L1 [Citation6,Citation24]. Autophagy plays a significant role in preserving homeostasis within various cells in the intestine, including intestinal epithelial cells, intestinal stem cells, Paneth cells, and diverse immune cells. By effectively managing intestinal immunity, autophagy serves a protective function by preventing the onset of IBD in healthy individuals and mitigating the exacerbation of inflammation in patients with IBD.
Autophagy maintains intestinal epithelial cell homeostasis
The apical membrane of intestinal epithelial cells contains tight junctions (TJ), which consist of transmembrane proteins, such as OCLN (occludin), claudins, and junctional adhesion molecules (JAM) [Citation25]. These proteins render the intestine permeable to water and nutrients, while preventing the entry of pathogens [Citation26]. Claudin dysregulation has been linked to increased intestinal permeability, persistent inflammatory activation, and epithelial-mesenchymal transition, which may lead to the progression of IBD and subsequent CAC development [Citation27]. Autophagy plays a vital role in regulating proteins associated with intestinal tight junctions, such as claudins and OCLN. Increased levels of CLDN2 (claudin2), a pore-forming protein that selectively allows cations to pass through the intestinal membrane, can damage the intestinal barrier by increasing paracellular permeability. Autophagy can modulate the intestinal barrier by promoting the lysosomal degradation of CLDN2, thereby reducing epithelial permeability [Citation28]. Recently, through co-immunoprecipitation trials, it was discovered that CLDN2 exhibits an increased connection with clathrin and the adaptor protein AP2 (comprising the AP2A1 and AP2M1 subunits), along with LC3 and lysosomes, under starvation conditions. This demonstrates the integral function of endocytosis, which is mediated by the autophagy-stimulated degradation of CLDN2. Understanding the autophagy-modulated molecular pathways that link AP2M1, CLDN2, and LC3 May pave the way for innovative therapeutic strategies aimed at combating intestinal inflammation [Citation29] (). Sinecetine, which is an autophagy activator, has been shown to effectively mitigate the impairment of the intestinal barrier in colitis by stimulating autophagy in epithelial cells. This process inhibits apoptosis and facilitates the degradation of CLDN2 [Citation30]. Numerous in vitro and in vivo investigations have demonstrated that OCLN functions as a barrier-forming protein in TJ, whereby a reduction in OCLN levels results in an increase in the permeability of macromolecular TJ [Citation31]. Recent studies have also established that autophagy reduces OCLN degradation and promotes membrane localization, thereby decreasing paracellular permeability [Citation32,Citation33] (). In vivo studies have shown that rapamycin, which induces autophagy, increases OCLN expression in the intestines of wild-type (WT) mice. This effect provides protection against the loss of the TJ barrier induced by lipopolysaccharides (LPS) and TNF. In contrast, the acute knockout of Atg7 in adult mice hinders autophagy, resulting in reduced OCLN expression in the intestines, an increase in the standard permeability of colonic TJ, and a worsening impact of experimental colitis [Citation32]. Moreover, one study demonstrated that the activation of BECN1 reduced the total OCLN protein levels through a constitutional, autophagy-independent role, while autophagy blocked constitutional BECN1 function and upregulated OCLN levels [Citation34]. Rapamycin has also been found to enhance autophagy during sepsis-induced intestinal barrier dysfunction. This effect is associated with an increase in TJP1/ZO-1 (tight junction protein 1) and OCLN expression, which presents a promising therapeutic approach for preventing intestinal barrier dysfunction [Citation35].
Figure 2. Defective autophagy leads to an increase in intestinal inflammation. (A) autophagy promotes the membrane localization of OCLN (occludin), a barrier-forming protein, by reducing its endocytosis in an MAPK1/ERK2-MAPK3/ERK1-dependent manner. In contrast, defective autophagy fails to degrade the pore-forming protein CLDN2, leading to increased intestinal permeability and bacterial translocation. (B) autophagy-deficient intestinal epithelium cells exhibit increased apoptosis and an increased generation of danger/damage-associated molecular patterns (DAMPs) and promotes the generation of inflammation. (C) during an invasive pathogen infection, lysozyme is secreted by the secretory autophagy pathway. This process requires endoplasmic reticulum (ER) stress-mediated EIF2AK3-EIF2A activation in Paneth cells. Contrastingly, impaired autophagy leads to the impaired ability of Paneth cells to secrete lysozyme, further increasing pathogen invasion. (D) damage of ATG16L1 in intestinal stem cells can lead to failed cell protection induced by muramyl dipeptide (MDP) and damage to the homeostasis of intestinal stem cells. Moreover, autophagy defects lead to the increased sensitivity of differentiated cells to reactive oxygen species (ROS) produced by commensal bacteria and may inactivate the hpo-yki pathway to promote the excessive proliferation of stem cells.
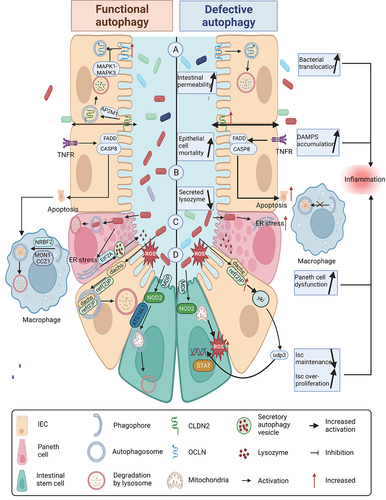
Several intestinal diseases, including IBD and colitis, are characterized by extensive epithelial erosion caused by an abnormal increase in intestinal epithelial cell (IEC) mortality. CAC development is facilitated by a vicious cycle of events involving the disruption of the intestinal barrier, dysregulation of cell death, and consequent inflammation [Citation30]. In a dextran sulfate sodium (DSS)-induced colitis model, the exposure of atg5 KO mouse IECs to oxidative stress resulted in higher susceptibility to apoptosis when compared to normal IECs. Additionally, stimulation of the MAPK (mitogen-activated protein kinase) signaling mechanism has been observed [Citation36]. Numerous studies have shown that autophagy prevents apoptosis and necrosis during colonic inflammation. In chronic colitis caused by the opportunistic pathogen Helicobacter hepatica in mouse intestines, pro-inflammatory cytokine secretion and IEC apoptosis are increased when specific Atg16l1 is deleted from IECs. These findings indicate that ATG16L1 is associated with susceptibility to IBD and that intraepithelial autophagy plays a crucial role in regulating inflammation-triggered apoptosis and maintaining barrier integrity [Citation37]. Autophagy also plays a role in eliminating apoptotic cells. The ATG14-BECN1/Beclin1-PIK3C3/VPS34 complex is regulated by NRBF2, which is a subunit with regulatory functions that enhance macroautophagy. Administration of DSS to mice deficient in NRBF2 resulted in notable intestinal inflammation and the accumulation of apoptotic cells. This could be attributed to the reduced activity of RAB7 and blocked phagosome maturation caused by a NRBF2 deficiency, ultimately resulting in the accumulation of apoptotic cells (). A correlation between the disease severity among individuals with ulcerative colitis (UC) and the number of apoptotic cells present in colon tissue has been established [Citation38].
In summary, autophagy plays a critical role in preserving epithelial cell stability by regulating CLDN2 and other tight junction proteins. It helps regulate permeability and resist pathogens, thereby ensuring the stability of the epithelial barrier. Moreover, autophagy in IECs plays a crucial function in averting apoptosis caused by inflammation and in facilitating the elimination of apoptotic cells.
Autophagy maintains Paneth cell and intestinal stem cell homeostasis
Intestinal inflammation may arise from the dysfunction of Paneth cells [Citation39]. However, autophagy may also affect the antibacterial effectiveness of Paneth cells by influencing their functional capacity. The limitation of bacterial invasion by intestinal Paneth cells has been documented through the secretion of antimicrobial proteins such as lysozyme, which are routed through secretory autophagy (an alternative secretory pathway based on autophagy) during bacterial infection (). The autophagic process responsible for secretion was found to be impaired in the Paneth cells of mice with an autophagy gene mutation [Citation40]. Changes in Paneth cell morphology have been reported for different autophagy gene deletions, such as ATG16L1 [Citation40], TFEB [Citation41], Irgm1 [Citation42], ATG4B [Citation43], ATG5 and ATG7 [Citation44].
Autophagy is particularly important in protecting intestinal stem cells (ISCs) from ROS-mediated oxidative stress damage. Autophagy protein deficiency in ISCs has been found to increase intracellular ROS levels, thereby reducing the number of ISCs and impairing their ability to regenerate in vitro, which is similar to the effects observed in epithelial cells and macrophages [Citation45]. The peptidoglycan motif muramyl dipeptide (MDP) participates in the survival of LGR5+ (leucine-rich repeat-containing G protein-coupled receptor 5) ISCs through NOD2 activation. It was previously reported that the gut of atg16l1 KO mice did not benefit from cytoprotection induced by MDP, suggesting that MDP stimulates mitochondrial clearance through ATG16L1 to counteract ISC mitochondrial ROS [Citation46]. Along with its function in protecting ISCs from oxidative stress, recent studies have indicated that autophagy has the potential to mitigate the susceptibility of differentiated intestinal cells to ROS generated in response to symbiotic bacteria. The ref(2)P/SQSTM1/p62 protein, which is a substrate for autophagy, directly interacts with dachs, a regulator of hpo (hippo)-yki signaling. In the absence of autophagy, ref(2)P/SQSTM1 tends to accumulate, resulting in the activation of yki and the increased production of upd3, which leads to excessive stem cell proliferation (). Consequently, insufficient autophagy leads to an overabundance of ROS-dependent regeneration, resulting in epithelial dysplasia and impaired barrier function [Citation47].
Autophagy regulates intestinal immunity
Autophagy plays a role in the regulation of intestinal immune homeostasis, thereby facilitating the development of macrophages induced by anti-TNF throughout innate immune responses. The presence of the ATG16L1T300A risk allele in donors has been associated with the impaired induction of these macrophages [Citation48]. Several studies have found that a lack of ATG16L1 or ATG7 in macrophages results in the progression of colitis [Citation49,Citation50]. Furthermore, when autophagy in macrophages was repressed, the activation of NFKB and the release of pro-inflammatory cytokines in immune cells, specifically dendritic cells (DC) and B cells, was increased [Citation51,Citation52]. Autophagy disruption in ITGAX/CD11c+ DCs increases the generation of IL1B/IL-1β and TNF, which exacerbates DSS-induced colitis [Citation53]. Furthermore, autophagy promotes the polarization of anti-inflammatory M2 in macrophages and inhibits the polarization of M1, leading to the increased secretion of anti-inflammatory cytokines, thereby enhancing inflammation resolution, tissue healing, and remodeling [Citation54].
Autophagy also plays a fundamental role in adaptive immunity. The maintenance of T cell homeostasis is dependent on basal autophagy, and the absence of pro-autophagy mediators, such as ATG3, ATG5, ATG7, BECN1 and the class III phosphatidylinositol 3-kinase (PtdIns3K), results in impaired autophagy, which in turn, hinders T cell survival, activation, growth, and differentiation [Citation55]. Furthermore, autophagy facilitates the presentation of major MHC-II [Citation3] and the cross-presentation of MHC-I [Citation56] during adaptive immune responses.
Autophagy defects may enhance CAC initiation
As inflammation in IBD worsens, various cytokines, including IL6, IL1, IL17A, TNF, and IL23, are released, which enhances NFKB signaling. Several studies have shown that NFKB signaling forms a critical link between inflammation and malignant transformation [Citation22,Citation23]. An elevated degree of LPS-induced NFKB signaling was directly observed in atg7 KO intestinal epithelial cells [Citation57]. Autophagy is known to modulate IKK (IκB kinase) activity, an important kinase in the NFKB signaling pathway, either by eliminating damaged or abnormal IKK complexes or by inhibiting their activity [Citation58]. Autophagy can also downregulate other factors that promote the activation of the NFKB signaling mechanism and the malignant transformation of CAC, such as ROS and inflammasomes, as described below.
Autophagy attenuates ROS production and endoplasmic reticulum stress
ROS have been implicated in the development of dysplasia in IBD, and play crucial roles in cancer initiation, progression, angiogenesis, and metastasis through various mechanisms [Citation59,Citation60]. Excessive accumulation of ROS can induce protein dysfunction and DNA damage, which in turn, may result in gene mutations and cellular apoptosis. ROS can also act as chemical messengers to activate signaling pathways, such as the NFKB and p38 MAPK pathways, and affect cell proliferation [Citation17]. The shortening of telomeres, which is induced by ROS, causes chromosomal instability, resulting in the loss, amplification, and translocation of chromosomes, thereby fostering CAC development [Citation17]. Autophagy, particularly mitophagy, reduces ROS production by removing damaged mitochondria and reducing NADPH oxidase complexes [Citation53]. However, ATG5-deficient epithelial cells exhibit enhanced RLR (RIG-I-like receptor) signaling pathway and massive amplification of ROS [Citation61]. Macrophages from mice lacking Atg16l1 exhibit elevated levels of ROS upon exposure to LPS compared to their WT counterparts [Citation2]. Excessive ROS production in response to defective autophagy promotes CAC. Transcription of ATG16L1, Ambra1, ATG7, and Wipi2, which are crucial for autophagosome biogenesis, is directly regulated by SMARCA4/BRG1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4). It has been reported that a deficiency of SMARCA4/BRG1 in IECs results in impaired autophagy, causing ROS overproduction. This phenomenon may contribute to the malignant progression of CAC in the presence of inflammation-induced DNA damage. Furthermore, an overabundance of SMARCA4/BRG1 has been found to mitigate colonic inflammation and tumorigenesis via autophagy-dependent oxidative stress sequestration, while the loss of SMARCA4/BRG1 results in compensatory regeneration [Citation62].
The endoplasmic reticulum (ER) is a highly dynamic organelle that performs a multitude of essential cellular functions. Pathological conditions can lead to insufficient nutrients in the ER, resulting in uncontrolled protein production. Activation of the unfolded protein response (UPR) can occur due to the accumulation of misfolded or unfolded proteins within the ER lumen [Citation63]. The UPR is an essential mechanism in ER stress and consists of three branches: EIF2AK3/PERK, ERN1/IRE1α, and ATF6. These branches facilitate an inflammatory response signaling mechanism through interactions with transcription factors, such as NFKB and AP-1 (activator protein 1), via crosstalk [Citation64]. The UPR signaling pathway may result in a complex inflammatory process referred to as the acute-phase response, in addition to individual transcription factor-based pro-inflammatory approaches [Citation63]. The activation of these mechanisms can contribute to the development of severe intestinal inflammation and the onset of CAC. Genetic ablation of ERN1, as an ER stress sensor, has been found to prevent the development of CAC in mice by reducing CTNNB1/β-catenin levels [Citation65]. A recent investigation discovered that PDIA2 (protein disulfide isomerase-associated 2), which is located in the ER, acts as a molecular link between ER stress and energy reprogramming. The protein PDIA2 undergoes translocation from the ER to the mitochondria, where it interacts with multiple elements of complexes I and II. This results in the inhibition of oxygen phosphorylation, while simultaneously promoting glycolysis. The DSS-induced inhibition of PDIA2 in colon cancer cells resulted in the restoration of metabolic balance and a significant reduction in CAC growth in a xenograft animal model [Citation66]. Autophagy has been shown to reduce ER stress through multiple mechanisms, including the recycling and elimination of damaged organelles, protein aggregates, and worn-out proteins [Citation67], and through the removal of ERN1 via ATG16L1-dependent autophagy [Citation68]. Recent studies have also suggested that the genetic inhibition of BECN1-induced autophagy impairs mucus excretion by reducing ER stress and prevents chemical- and microbial-driven intestinal inflammation [Citation69].
Furthermore, a robust reciprocal enhancement exists between ER stress and ROS production. In the ER lumen, disulfide bond formation is particularly susceptible to shifts in redox homeostasis. Exogenous oxidants, including peroxides and ROS generators, can infringe upon protein folding and consequently induce ER stress [Citation70]. The phenomenon of ER stress has been found to enhance the production of ROS, which occurs as a result of the transfer of ER Ca2+ to the mitochondria, thereby intensifying metabolic activity and increasing the generation of ROS signaling within the mitochondria. This signaling then reciprocates back to the ER, resulting in a further release of Ca2+. The ensuing discharge of Ca2+ from the ER, combined with the generation of mitochondrial ROS, induces a self-perpetuating cycle of stress response [Citation71]. ER stress has also been shown to mediate ROS production in a colitis model, and one study reported that Bifidobacterium dentium effectively reduced the severity of colitis by reducing the ER stress-mediated ROS response [Citation72]. Overall, a close association has been observed between ER stress, increased ROS production, and CAC onset (). Notably, a mutual and amplifying feedback mechanism links ER stress and ROS, potentially accelerating the initiation of CAC. However, several autophagy-related genes have demonstrated the capacity to attenuate both ROS and ER stress, thereby presenting promising strategies to curb CAC progression.
Figure 3. Autophagy deficiency promotes CAC initiation. (A) defects in autophagy lead to elevated levels of reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress. The increased ROS can induce DNA damage in epithelial cells, while ERN1/IRE1α (endoplasmic reticulum to nucleus signaling 1) elevation promotes the production of CTNNB1. PDIA2 (protein disulfide isomerase family a member 2) further facilitates metabolic reprogramming. (B) during the impairment of autophagy, obstacles to the activation of inflammasomes are eliminated, which promotes the cleavage of GSDMD (gasdermin D) to form N-GSDMD (N-terminal GSDMD). This process enables the conversion of the IL1B precursor into its mature form, and stimulates the production of matrix metalloproteinases (MMPs). (C) defects in autophagy contribute to an increase in invasive pathogenic microorganisms such as adherent-invasive E. coli (AIEC) and colibactin-producing E. coli (CoPEC), leading to the production of inflammatory mediators such as TNF and IL6. CoPEC can directly induce DNA damage in epithelial cells. Moreover, autophagy defects increase ER stress and decrease akkermansia muciniphila abundance, thereby reducing the proportion of cytotoxic T lymphocytes (CTL) in the intestine and weakening the anti-tumor immunity of CAC. (D) necrosis and pyroptosis escalate when autophagy is deficient. N-GSDMD, RIPK3 (receptor interacting serine/threonine kinase 3), and MLKL (mixed lineage kinase domain like pseudokinase) can activate the MAPK/JNK pathway, contributing to the malignant transformation in CAC.
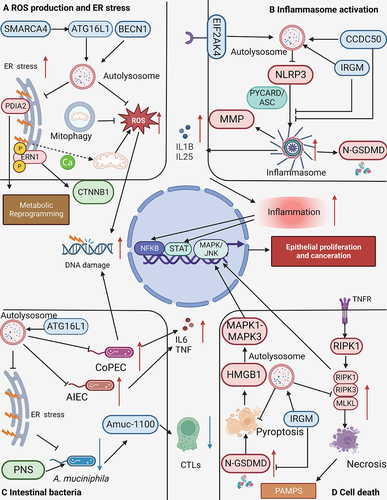
Autophagy reduces inflammasome activation
Recently, there is growing evidence suggesting that an overactivation of inflammasomes and elevated IL1B expression may cause CAC occurrence and development. Chen et al. revealed that the regulation of intestinal homeostasis and antimicrobial immunity by inflammasomes may be involved in promoting inflammatory diseases and CAC [Citation18]. The regulation of CAC is significantly influenced by RALGAPA2 (Ral GTPase activating protein, alpha subunit 2 (catalytic)), which serves as the primary catalytic subunit responsible for negatively regulating the small guanosine triphosphatase Ral. Mice lacking RALGAPA2 exhibited a greater incidence and magnitude of tumorigenesis, as well as a higher ratio of tumors infiltrating the submucosal layer when compared to WT mice. Moreover, the upregulation of IL1B through NLRP3 inflammasome activation and the subsequent promotion of CAC were observed upon the downregulation of RALGAPA2. The RAL-activated NLRP3 inflammasome also plays a vital role in matrix metalloproteinase (MMP) upregulation in CAC, and promotes tumor invasion and metastasis [Citation73]. A recent report indicated that the NLRP3 inflammasome also plays a role in the maturation of IL1B and increased 5-HT biosynthesis in CRC cells. This is achieved through the induction of TPH1 (tryptophan hydroxylase 1) transcription. It has been reported that activation of the NLRP3 inflammasome in macrophages is enhanced by the excessive production of 5-HT via the ion channel receptor HTR3A, resulting in the establishment of a positive feedback cycle between 5-HT and NLRP3. NLRP3 knockdown, and pCPA (TPH1 suppressor) and TPS (HTR3A antagonist) therapy were found to substantially prevent AOM/DSS-induced CRC development, suggesting that the 5-HT-NLRP3 positive feedback loop in the CRC microenvironment may promote cancer development and progression. However, repression of tumor progression can be achieved by blocking this positive feedback loop [Citation74]. Therefore, several studies have shown that the inhibition of NLRP3 activation reduces CAC progression, and the administration of small molecule andrographolide (Andro) significantly reduces colitis development and cancer burden, and protects mice against DSS-induced colitis and CAC by suppressing the NLRP3 inflammasome [Citation75]. A study also reported that Arctigenin could effectively prevent CAC in mice by inhibiting fatty acid oxidation, disrupting NLRP3 inflammasome assembly, and downregulating IL1B expression [Citation76]. In addition, it is also thought that bacteria can either promote or limit CAC via their inflammasomes. Shao et al. reported that Bacteroides fragilis restricts CAC by negatively regulating the NLRP3 axis [Citation77]. Another study of APCMin/+ mice reported that Porphyromonas gingivalis promotes the production of protumorigenic IL1B by triggering the NLRP3 inflammasome in recruited tumor-infiltrating myeloid cells, thus facilitating CAC development [Citation78].
Autophagy plays an essential role in limiting inflammasome activation. The elimination of damaged mitochondria suppresses the activation of the NLRP3 inflammasome, which effectively prevents the overproduction of ROS and mtDNA [Citation79]. As a consequence of nutrient deprivation, IL25/IL17E (secreted by IL1) destroys the intestinal epithelium. Autophagy stimulation maintains the intestinal barrier by suppressing inflammasome activation and the release of pro-inflammatory cytokines, including IL1B and IL25 [Citation80]. In addition, the amino acid sensor EIF2AK4/GCN2 has been found to control inflammatory processes in the intestine and reduce colitis by inducing autophagy and suppressing inflammasome activation. This was confirmed in EIF2AK4/GCN2-deficient mice, which exhibited decreased autophagy and increased ROS levels, resulting in inflammasome activation and the worsening of colitis [Citation81]. Autophagic pathways that inhibit NLRP3 inflammasome activation have also been investigated. Studies have shown that IRGM/IRGM1 can inhibit NLRP3 inflammasome activation through at least two mechanisms: (1) binding to the oligomerization domain (NACHT) of NLRP3, which blocks NLRP3 oligomerization and apoptosis-associated speck-like protein containing a CARD (ASC) protein polymerization, thus interfering with inflammasome assembly; and (2) directly interacting with NLRP3 and PYCARD/ASC, and mediating its degradation via SQSTM1/p62-dependent autophagy [Citation82]. In an experimental mouse model of IBD, irgm1−/− mice demonstrated increased inflammation following DSS administration, indicating that IRGM/IRGM1 inhibits colitis by suppressing the NLRP3 inflammasome [Citation82]. Furthermore, the autophagy receptor CCDC50 has been found to have a vital function in the activation and inhibition of the inflammasome. It serves as a receptor for macroautophagy/autophagy cargo, specifically identifying K63-polyubiquitinated NLRP3, and transporting it to phagophores conjugated with MAP1LC3/LC3 for degradation. CCDC50 can also inhibit NLRP3 inflammasome activity by targeting K63-polyubiquitinated NLRP3 for selective autophagic degradation and inhibiting NLRP3 polymerization and PYCARD/ASC recruitment, thereby inhibiting inflammasome assembly (). Mice with ccdc50 KO were found to be more susceptible to DSS-induced colitis, and exhibited increased NLRP3 inflammasome activity and severe intestinal inflammation [Citation83].
Autophagy regulates intestinal bacteria and reduces oncogenic bacterial invasion
The gut microbiota has been associated with CRC progression [Citation20], and specific changes in the microbial composition have been linked to CAC development compared to sporadic cancer. For example, CAC is characterized by a reduction in Fusobacterium and Ruminococcus, and an increase in bacteria from the Enterobacteriaceae family and Sphingomonas [Citation84]. Autophagy plays a significant role in the maintenance of intestinal microbiota, with evidence suggesting that it can regulate microbial composition by stabilizing goblet cell function [Citation85], maintaining the normal secretion of Paneth cells [Citation86], and reducing the overproduction of inflammatory factors [Citation2]. Gram-negative anaerobes connected with mucin, such as Akkermansia muciniphila, are selectively reduced in the fecal microbiota of IBD patients, and it has been reported that A. muciniphila plays a role in CAC progression [Citation87]. Research shows that the administration of A. muciniphila or a particular outer membrane protein (Amuc_1100) may result in a substantial increase in the percentage of cytotoxic lymphocytes (CTLs) in the colon. Additionally, the procedure may upregulate TNF expression in CTLs, which subsequently intensifies apoptosis in cancer cells [Citation87]. Furthermore, Panax notoginseng saponins (PNS) have been shown to prevent CAC progression by increasing the abundance of A. muciniphila [Citation88]. However, a reduced abundance of A. muciniphila has also been observed in multiple autophagy-associated gene defects [Citation85,Citation89]. Researchers noted marked changes and a reduced variety in the intestinal microbiota of Atg5-deficient mice compared to their WT counterparts using an atg5 KO mouse model. This study also observed a decrease in the abundance of certain organisms, such as A. muciniphila and members of the Lachnospiraceae family, which are linked to the control of inflammation. Conversely, there was a significant increase in the abundance of pro-inflammatory bacteria, such as Candidatus Arthromitus, as well as potential pathogens, such as those from the Pasteurellaceae family, in atg5 KO mice [Citation89]. Similarly, gut bacterial analysis of Atg16l1T300A mice revealed that they had a different microbiota in the terminal ileum and colon, in contrast to co-resident WT mice, with a substantial reduction in A. muciniphila abundance [Citation85]. Notably, a recent study suggested that autophagy directly promotes the expansion of A. muciniphila by reducing ER stress to promote goblet cell mucus secretion [Citation69]. In addition to anti-inflammatory genera, such as A. muciniphila, autophagy was also found to regulate Ruminococcus for the downregulation of CAC. A study demonstrated that polymethoxyflavones (PMFs) enhance autophagy by encouraging autophagosomes to merge with lysosomes, and the population of Ruminococcus was found to increase to 9.4% in the 1.0% PMF-treated group, which was a significant increase when compared to the benzo[a]pyrene/dextran sulfate sodium (BaP/DSS) group, where Ruminococcus only increased to 2.7% [Citation90].
Autophagy also plays a fundamental role in mitigating the invasion of pathogenic microorganisms and reducing the oncogenic effects of microorganisms. Defects in autophagy can lead to increased susceptibility to various pathogens. For instance, autophagy-deficient cells promote the replication of pathogenic adherent invasive Escherichia coli (AIEC) [Citation91]. Additionally, when ATG16L1, IRGM, or NOD2 expression is reduced, macrophages demonstrate heightened intracellular AIEC as well as augmented production of IL6 and TNF in reaction to AIEC infection [Citation92]. Research has shown that the levels of Mir30c (microRNA 30c) and Mir130a are upregulated in mouse enterocytes upon the activation of NFKB due to AIEC infection. The increased expression of these miRNAs results in a decrease in ATG5 and ATG16L1 levels, thereby impeding autophagy. This, in turn, leads to an increase in the quantity of intracellular AIEC and an escalation in the inflammatory response [Citation93]. The abnormal colonization of the colonic mucosa of individuals with CRC is facilitated by colibactin-producing Escherichia coli (CoPEC), which have been found to foster colorectal carcinogenesis in mouse models of CRC. Intestinal epithelial autophagy inhibits CoPEC-induced CRC in ApcMin/+ mice and can also induce invasive cancer formation in DSS-induced colitis models [Citation94]. Furthermore, autophagy is inhibited in HCT-116 cells after transfection with ATG16L1- and ATG5-specific siRNA, and transfected cells exhibited an increase in intracellular 11G5 (a CoPEC strain isolated from the patient) and secreted IL6 and IL8. These results suggested that autophagy, specifically the ATG5- and ATG16L1-dependent pathways, is essential for eliminating intracellular 11G5 bacteria and mitigating inflammation induced by 11G5 [Citation95]. Moreover, it was also demonstrated that autophagy is necessary to limit CoPEC-induced colonic DNA damage in a chronic DSS-treated mouse model, thus preventing the development of CAC [Citation94].
Hence, defects in autophagy can cause severe dysregulation of intestinal bacteria (), especially a decrease in the abundances of bacteria that reduce inflammation, which may accelerate the onset of CAC.
The function of autophagy in specific types of cell death
The significance of autophagy in mitigating necrosis has been demonstrated in animal models of murine norovirus (MNV)-induced IBD and allogeneic hematopoietic stem cell transplantation. The models under consideration indicate that intestinal organoids lacking ATG16L1 demonstrate a depletion of Paneth cells and an increase in TNF-mediated necrosis, accompanied by elevated levels of phosphorylated-RIPK3 and phosphorylated-MLKL [Citation96]. Specifically, the impairment of autophagy was found to enhance necroptosis mediated by TNF and TLR (toll like receptor) ligands, while macrophages lacking the Atg16l1 accumulated highly insoluble variants of RIPK1, RIPK3, TRIF, and ZBP1 [Citation97]. Immunoprecipitation assays indicated that SQSTM1/p62, which identifies ubiquitinated cargo proteins for degradation via autophagy, can form complexes with RIPK3 and MLKL, which was consistent with the function of SQSTM1/p62 in recognizing autophagy [Citation98]. Recent studies have indicated that RIPK3 and MLKL are crucial for CAC development. MTOR regulates ubiquitin-dependent RIPK3 degradation via autophagy, and the MTOR-mediated blockade of autophagy can result in RIPK3 accumulation and an increased susceptibility to necroptosis [Citation98]. A study examining live mice observed significant colonic tumorigenesis in Tsc1-deficient mice, which had twice as many tumors per colon as WT mice [Citation98]. Furthermore, another study showed that mice lacking RIPK3 exhibited substantially reduced tumorigenesis related to colitis, indicating that RIPK3 May promote CAC progression by supporting epithelial proliferation and tumor growth through MAPK/JNK signaling, and by mediating CXCL1-induced immunosuppression [Citation99].
According to a recent study, the progression of CAC is facilitated by pyroptosis, which is a type of programmed cell death regulated by pro-inflammatory gasdermin proteins. This study utilized a mouse model of CAC and found that the gsdme-/- mice resulted in reduced weight loss, attenuated rectal prolapse, colon shortening, and minimized tumor numbers and sizes in contrast to the WT littermates. The study also revealed that HMGB1 (high-mobility group box protein 1) released during pyroptosis induces cancer cell growth and the expression of PCNA (proliferating cell nuclear antigen) through the MAPK1/ERK2-MAPK3/ERK1 mechanism [Citation19]. As described above, autophagy effectively attenuates inflammasome activation and reduces caspase activation, thereby preventing pyroptosis. In addition, IRGM/Irgm1 has also been found to prevent pyroptosis in an experimental mouse model of Crohn disease [Citation82].
In summary, various specific types of cell death, including necrosis and pyroptosis (), produce pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), which generate large amounts of inflammatory mediators and have been shown to promote CAC development. Autophagy has a significant function in preventing the activation of these pathways, thus preventing CAC progression.
The role of autophagy after tumor transformation
Following CAC transformation, transcriptome-based tumor subtype analysis indicated that, in contrast to sCRC, the typical epithelial tumor subtype related to WNT signaling is entirely absent in IBD-CRC. Instead, a mesenchymal stroma-rich subtype prevails [Citation100], suggesting that autophagy may play different roles in conventional CRC and CAC models. Various CAC animal models have been established in rodents, including mice, rats, and hamsters. The most extensively researched chemically inducible model requires the administration of a procarcinogen, AOM, through a single injection, which is then metabolized to methylazoxymethanol (MAM), a hepatic active agent. This is succeeded by an inflammatory insult using DSS [Citation101]. The AOM/DSS model results in a pathology marked by acute colitis, substantial weight loss, and bloody diarrhea, which eventually leads to the development of multiple colonic cancers. This progression mirrors the sequence observed in human CAC formation, moving from inflammation to heterogeneous proliferation, and eventually culminating in cancer [Citation102]. Moreover, in the AOM/DSS model, the molecular disruptions that regulate CAC development and progression are similar to those detected in human subjects. The molecular biomarkers of CAC following AOM/DSS treatment include NFKB, the JAK-STAT3 pathway, pro-inflammatory cytokines (TNF and IL6), CTNNB1, etc [Citation103], which have been described as processes that can be inhibited by autophagy. However, the elucidation of autophagy in this model remains limited, as most studies of autophagy in colon cancer have been conducted using the CRC model. Recent studies investigating autophagy in AOM/DSS-induced colon cancer models have proposed the potentially cytoprotective role of autophagy following CAC transformation as well as the possibility of autophagy-mediated tumor cell death.
Cytoprotective role of autophagy in CAC
CAC progresses through multiple stages and develops in different microenvironments, with autophagy playing a dual role. Deletion of ATG10, ATG7, ATG5, and BECN1 has been associated with an unfavorable prognosis and may cause cytoprotective autophagy in CRC cells [Citation104–106]. Autophagy is an essential mechanism for the progression of chemoresistance and has a cytoprotective role in CRC cells. The ability of CRC cells to enter a reversible drug-resistant persistent (DTP) state to evade cell death induced by chemotherapy and targeted drugs, is a major contributor to treatment failure and the recurrence of tumors. A recent study revealed that key autophagy genes are upregulated and the MTOR module is downregulated in the DTP-state in CRC. Furthermore, combining autophagy suppressors with CPT-11 significantly inhibited the acquisition of the DTP state and led to robust cell death, highlighting the significance of autophagy in maintaining the DTP state of tumors [Citation107]. Although there are differences in the molecular composition of tumor cells in CAC and sCRC, recent studies using AOM/DSS-induced cancer models have found that autophagy may have similar cytoprotective effects on tumor cells. One study revealed that ripk2/card3 KO mice had fewer colorectal tumors than control mice after AOM/DSS administration. Tumors from ripk2/card3 KO mice exhibited reduced LC3-II and BECN1 levels and elevated SQSTM1/p62 levels when compared to control mice, suggesting that Fusobacterium nucleatum activates autophagic signaling by upregulating Ripk2/Card3 expression and promoting CRC metastasis [Citation108]. Additionally, in mouse models of CAC, a study observed high LAMP2A (a chaperone-mediated autophagy marker) expression in CAC tissues, whereas it was nearly imperceptible in tissues adjacent to the tumor. This suggests that chaperone-mediated inhibition of autophagy enhances apoptosis and blocks CAC growth [Citation109].
In summary, autophagy has a cytoprotective function in the CAC model. Nevertheless, further investigation is required to reveal differences in the potential roles of autophagy between conventional CRC and CAC models, particularly in the context of chemoresistance.
Autophagy-related cell death in CAC
Autophagy, which is cytoprotective in nature, can prevent the tumor apoptosis induced by anticancer drugs. Conversely, autophagy, which is also lethal in nature, can facilitate tumor cell apoptosis in collaboration with anticancer drugs. Autophagy-related cell death is vital in CAC-CRC, and its induction attenuates CAC-CRC progression. Autophagy-related pathways, such as PI3K-AKT-MTOR and AMPK-MTOR, regulate cell death associated with autophagy in CAC-CRC cells. Overactivation of the PI3K-AKT-MTOR or AMPK-MTOR axis plays a fundamental role in CAC-CRC development and is related to apoptosis and the induction of drug resistance in CAC-CRC cells [Citation110,Citation111]. Furthermore, several studies have demonstrated the ability of LC3B, an important autophagy marker, to induce apoptosis in colon cancer cells [Citation112–115]. Research has shown that apoptosis induced by ATG7 knockdown is nuclear-LC3 dependent and is augmented by chemotherapy in CRC cells [Citation104]. Recent studies have also shown that the mitophagy protein, PINK1, inhibits colonic tumor growth through TP53 activation and reduces the metabolic reprogramming of acetyl-CoA [Citation116]. Moreover, PINK1-PRKN/Parkin-dependent mitophagy has also been found to be associated with cell apoptosis; Aloe gel glucomannan and dietary-valerobetaine induced apoptosis in colon cancer cells via a mitochondrial damage-driven PINK1-PRKN mitophagy mechanism; thus, driving the PINK1-PRKN mitophagy pathway may be an effective anti-tumor strategy [Citation117,Citation118].
Several drugs have been found to promote autophagy-related cell death in CAC models. For example, the induction of apoptosis associated with autophagy and cell cycle arrest in AOM-induced mouse colon carcinogenesis was demonstrated through the use of hesperidin [Citation119]. However, research on the role of novel pathways, such as the PINK1/Parkin mitophagy pathway, in CAC models remains limited. Additional studies are required to explore these pathways and their possible roles in CAC.
Autophagy regulators in CAC
Molecular Regulatory Mechanisms of Autophagy in CAC
Autophagy is regulated by various molecules in inflamed intestinal epithelial cells and functions to reduce stress and inflammation. In CAC cells, autophagy can be regulated to promote nutritional support or induce tumor cell death. Molecular regulators, such as AMPK, PI3K, MTOR, and microRNAs, have crucial functions in autophagy modulation during CAC initiation and tumor transformation. Understanding these molecular mechanisms is essential for developing targeted therapies.
MTOR: The protein kinase MTOR plays an essential role in cellular proliferation and growth. The MTORC1 complex is reliant on this particular component, which functions as a highly effective suppressor of autophagy [Citation120]. In mammals, MTORC1 directly hinders the activation of ULK1-ATG13, a critical factor in initiating autophagy through phosphorylation [Citation121,Citation122]. Additionally, MTORC1 effectively regulates the later stages of autophagy. It has been reported that MTORC1 negatively regulates autophagosome and endosome maturation by UVRAG (UV radiation resistance associated) [Citation123]. Furthermore, the MTORC1 pathway suppresses autophagy when nutrients are abundant, while the activity of the MTORC1 pathway decreases under nutrient-deficient conditions, resulting in the reactivation of autophagy [Citation124]. Studies have revealed that glutamine attenuates CAC initiation by inhibiting MTOR and inducing autophagy [Citation125]. However, in established cancer, reduced MTOR activity in DTP-state CRC plays a crucial role in promoting autophagy resistance in tumor cells [Citation107].
AMPK: AMPK is a key energy sensor that depends on intracellular energy levels to function [Citation126]. When cells are nutrient-deficient, AMPK can directly activate ULK1 [Citation127] to initiate autophagy and acts upstream of MTOR to inhibit MTORC1 activity [Citation121], thus exerting a powerful autophagy-promoting effect. AMPK activators, such as GL-V9, have been found to significantly enhance autophagy in macrophages by activating the AMPK-ULK1 pathway. Moreover, it was found that GL-V9 treatment effectively prevented CAC by enhancing autophagy-mediated inflammasome degradation in a CAC model [Citation128]. Similarly, Ginsenoside Rd activates the AMPK-ULK1 pathway to promote autophagy-mediated inflammasome degradation, thereby alleviating colitis [Citation129]. In an established CRC model, the AMPK-MTOR and AMPK-ULK1 mechanism has been shown to induce apoptosis related to autophagy in CRC cells [Citation130–135], and therefore, AMPK-mediated autophagy-associated apoptosis may be a potential target in the CAC model.
PtdIns3K: PtdIns3K has been found to be upregulated in CAC and plays an important role in the development of CAC [Citation136]. Although the class III PtdIns3K subunit PIK3C3/VPS34 binds to BECN1 and PIK3R4, and generates phosphatidylinositol-3-phosphate, which is a lipid necessary for autophagy initiation, the activation of class I PI3K inhibits autophagy via the well-established PI3K-AKT-MTORC1 pathway [Citation137] and this pathway has been extensively studied. A study involving Andro suggested that autophagy effectively prevents CAC production by inhibiting the NLRP3 inflammasome, and that the mechanism responsible for autophagy upregulation after Andro administration involves the inhibition of the PI3K-AKT-MTORC1 pathway [Citation75]. Inhibition of the PI3K-AKT-MTORC1 pathway, in turn, has been shown to play a fundamental role in the induction of autophagy-related apoptosis in established cancer models, with a large number of small molecule complexes exerting antitumor effects by inhibiting this pathway to trigger autophagy-associated apoptosis in conventional CRC models [Citation138–142]. Similar results have also been observed in specific CAC models [Citation119].
MicroRNAs: MicroRNAs are involved in the regulation of autophagy via multiple pathways [Citation143]. Mir142-3p has been found to reduce autophagy by targeting ATG16L1 in IBD and CRC [Citation144], and the high expression of Mir93 and Mir106b has also been found to target ATG16L1 in order to reduce autophagosome formation and increase inflammation in the gut [Citation145]. Moreover, aberrant activation of the NFKB pathway in CRC cell lines with Mir143 overexpression or ATG2B depletion suggests that Mir143 may inhibit autophagy in Crohn disease by targeting ATG2B and increasing the inflammatory response of the NFKB pathway [Citation146]. miRNAs have also been found to inhibit autophagy, effectively attenuate protective autophagy in CRC cells, and promote the sensitivity of CRC cells to chemotherapy and radiation therapy, which is a phenomenon observed in a variety of miRNAs, including Mir22 [Citation147], Mir409-3p [Citation148], Mir34a [Citation149], Mir27b-3p [Citation150] and Mir502 [Citation151].
Therapeutic potential of autophagy regulators in CAC
Autophagy plays a crucial role in the prevention of CAC progression during the initial stages of malignant transformation by alleviating intestinal inflammation and reducing the levels of certain tumorigenic factors. In support of this, numerous autophagy regulators have recently emerged in CAC models, effectively reducing tumorigenesis by promoting the attenuation of inflammatory responses. Moreover, some autophagy regulators that promote autophagy-related cell death have been identified in AOM/DSS-induced colon cancer models. However, unlike conventional CRC models, no autophagy modulators that attenuate CAC development by enhancing cytoprotective autophagy and promoting sensitivity to chemotherapeutic agents have been identified in CAC models. We list a series of studies of autophagy modulators found to attenuate intestinal pathology scores as well as CAC development by promoting autophagy in CAC models. ()
Table 1. Examples of autophagy modulators that inhibit colitis-associated colon cancer (CAC) development by promoting autophagy.
Discussion
The development of CAC in patients with IBD is significantly associated with chronic inflammation, which is a prominent risk factor. The etiology of CAC differs significantly from that of sCRC, and there has been growing recognition of non-conventional forms of dysplasia. Additionally, the concept of cumulative inflammatory burden highlights the importance of considering the presence of histological inflammation over time as a crucial risk factor for CAC [Citation21]. A growing body of evidence indicates that chronic inflammation can augment the factors associated with CAC development through diverse processes. Tumor initiation and progression are facilitated by various factors, including oxidative stress-induced DNA damage, aberrant immune responses, and gut dysbiosis. Therefore, identifying novel therapeutic targets to reduce inflammation and promote homeostasis in the intestinal environment is crucial.
Our review suggests that defective autophagy plays a pivotal role in creating a pro-inflammatory environment in the intestine. Autophagy-deficient epithelial cells produce ROS, inflammasomes, and tumorigenic microorganisms that are closely related to CAC development in animal studies. These products can upregulate signaling pathways, such as the MAPK/JNK and MMP pathways, which accelerate the aggressive and malignant transformation of epithelial cells. Additionally, the inflammasome-induced production of IL1B, as well as the production of PAMPs and DAMPs due to epithelial cell death, contribute to both inflammatory deterioration and CAC development.
Drugs such as 5-ASA and anti-TNF have exhibited a reduction in CAC incidence in numerous clinical and preclinical trials by decreasing intestinal inflammation [Citation158–160]. Therefore, we present a series of preclinical studies that demonstrate the considerable potential of autophagy modulators in reducing colitis and CAC development. Many autophagy promoters, such as andrographolide, are safer and have fewer side effects than drugs such as 5-ASA, indicating promising prospects for future CAC treatment.
A recent study on the genetics and epigenetics of CAC suggested that its molecular composition differs from that of sCRC, which could lead to variations in the role of autophagy after CAC and sCRC formation. Similar to sCRC, we observed cytoprotective autophagy in cancer cells in the CAC model and the existence of autophagy-related tumor cell death, which are two distinct aspects of tumor growth. However, unlike in sCRC, chloroquine/CQ and hydroxychloroquine/HCQ have been found to be effective in reducing tumor cell-protective autophagy in sCRC and promotes therapeutic efficacy when combined with chemotherapeutic agents [Citation105,Citation161]. In contrast, few studies have explored the efficacy of agents such as chloroquine, especially in combination with chemotherapy, in CAC models. Considering the molecular differences between CAC and conventional sCRC models, it is crucial to conduct further studies on the effects of these autophagy regulators in CAC models. This would help to determine any potential differences in the conventional model and further our understanding of the specific roles of autophagy regulators in CAC.
This review integrates current research on the possible involvement of autophagy and its associated protein deletions in the development of CAC; however, there are still very few studies on the prevention of CAC using autophagy regulators. The potential therapeutic benefits of targeting autophagy to prevent or reduce the incidence of CAC have been explored in preclinical studies. However, additional investigations are required to fully elucidate the underlying pathways. The main limitations associated with current autophagy regulators are poor selectivity and a lack of specificity for certain cell types. Targeting autophagy regulators to prevent or reduce the incidence of CAC is a promising strategy, and further research is needed to identify more specific and effective therapeutic targets.
Disclosure statement
The authors do not have any potential conflicts of interests to declare.
Additional information
Funding
References
- Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014 Apr 24;54(2):224–233. doi: 10.1016/j.molcel.2014.03.009
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008 Nov 13;456(7219):264–268. doi: 10.1038/nature07383
- Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Nat Acad Sci. 2005;102(22):7922–7927. doi: 10.1073/pnas.0501190102
- Wei J, Long L, Yang K, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016 Mar;17(3):277–285. doi: 10.1038/ni.3365
- Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020 Jan 22;19(1):12. doi: 10.1186/s12943-020-1138-4
- Nguyen HT, Lapaquette P, Bringer MA, et al. Autophagy and Crohn’s disease. J Innate Immun. 2013;5(5):434–443. doi: 10.1159/000345129
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014 Mar;14(3):141–153. doi: 10.1038/nri3608
- Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007 Oct 25;449(7165):1003–1007. doi: 10.1038/nature06196
- Martini E, Krug SM, Siegmund B, et al. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017 Jul;4(1):33–46. doi: 10.1016/j.jcmgh.2017.03.007
- Shimizu Y, Nakamura K, Yoshii A, et al. Paneth cell α-defensin misfolding correlates with dysbiosis and ileitis in Crohn’s disease model mice. Life Sci Alliance. 2020 Jun;3(6):e201900592. doi: 10.26508/lsa.201900592
- Aldhous MC, Noble CL, Satsangi J. Dysregulation of human beta-defensin-2 protein in inflammatory bowel disease. PLoS One. 2009 Jul 20;4(7):e6285. doi: 10.1371/journal.pone.0006285
- Hou Q, Huang J, Ayansola H, et al. Intestinal stem cells and immune cell relationships: potential therapeutic targets for inflammatory bowel diseases. Front Immunol. 2020;11:623691. doi: 10.3389/fimmu.2020.623691
- Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014 May;14(5):329–342. doi: 10.1038/nri3661
- Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020 Jan;16(1):38–51. doi: 10.1080/15548627.2019.1635384
- Beaugerie L, Itzkowitz SH, Longo DL. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015 Apr 9;372(15):1441–1452. doi: 10.1056/NEJMra1403718
- Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013 Mar;35(2):229–244. doi: 10.1007/s00281-012-0352-6
- Wang Z, Li S, Cao Y, et al. Oxidative stress and Carbonyl Lesions in ulcerative colitis and associated colorectal cancer. Oxid Med Cell Longev. 2016;2016:9875298. doi: 10.1155/2016/9875298
- Chen GY, Núñez G. Inflammasomes in intestinal inflammation and cancer. Gastroenterology. 2011 Dec;141(6):1986–1999. doi: 10.1053/j.gastro.2011.10.002
- Tan G, Huang C, Chen J, et al. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J Hematol Oncol. 2020 Nov 7;13(1):149. doi: 10.1186/s13045-020-00985-0
- Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006 Jul;3(7):390–407. doi: 10.1038/ncpgasthep0528
- Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022 Mar;162(3):715–730.e3. doi: 10.1053/j.gastro.2021.10.035
- Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol. 2016 Mar;17(3):230–240. doi: 10.1038/ni.3384
- Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018 May;18(5):309–324. doi: 10.1038/nri.2017.142
- Kim S, Eun HS, Jo EK. Roles of autophagy-related genes in the pathogenesis of inflammatory bowel disease. Cells. 2019 Jan 21;8(1):77. doi: 10.3390/cells8010077
- Neunlist M, Van Landeghem L, Mahé MM, et al. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nat Rev Gastroenterol Hepatol. 2013 Feb;10(2):90–100. doi: 10.1038/nrgastro.2012.221
- Buckley A, Turner JR. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb Perspect Biol. 2018 Jan 2;10(1):a029314. doi: 10.1101/cshperspect.a029314
- Zhu L, Han J, Li L, et al. Claudin family participates in the pathogenesis of inflammatory bowel diseases and colitis-associated colorectal cancer. Front Immunol. 2019;10:1441. doi: 10.3389/fimmu.2019.01441
- Hu CA, Hou Y, Yi D, et al. Autophagy and tight junction proteins in the intestine and intestinal diseases. Anim Nutr. 2015 Sep;1(3):123–127. doi: 10.1016/j.aninu.2015.08.014
- Ganapathy AS, Saha K, Suchanec E, et al. AP2M1 mediates autophagy-induced CLDN2 (claudin 2) degradation through endocytosis and interaction with LC3 and reduces intestinal epithelial tight junction permeability. Autophagy. 2022 Sep;18(9):2086–2103. doi: 10.1080/15548627.2021.2016233
- Xiong YJ, Deng ZB, Liu JN, et al. Enhancement of epithelial cell autophagy induced by sinensetin alleviates epithelial barrier dysfunction in colitis. Pharmacol Res. 2019 Oct;148:104461.
- Cummins PM. Occludin: one protein, many forms. Mol Cell Biol. 2012 Jan;32(2):242–250. doi: 10.1128/MCB.06029-11
- Saha K, Ganapathy AS, Wang A, et al. Autophagy reduces the degradation and promotes membrane localization of occludin to enhance the intestinal epithelial tight junction barrier against Paracellular Macromolecule Flux. J Crohns Colitis. 2022 Oct 11;17(3):433–449. doi: 10.1093/ecco-jcc/jjac148
- Ureshino RP, Rocha KK, Lopes GS, et al. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid Redox Signal. 2014 Jul 1;21(1):123–137. doi: 10.1089/ars.2013.5777
- Wong M, Ganapathy AS, Suchanec E, et al. Intestinal epithelial tight junction barrier regulation by autophagy-related protein ATG6/beclin 1. Am J Physiol Cell Physiol. 2019 May 1;316(5):C753–c765. doi: 10.1152/ajpcell.00246.2018
- Cao YY, Qiao Y, Wang ZH, et al. The polo-like kinase 1–mammalian target of rapamycin axis regulates autophagy to prevent intestinal barrier dysfunction during sepsis. Am J Pathol. 2022 Dec 9;193(3):296–312. doi: 10.1016/j.ajpath.2022.11.008
- Kubota M, Kakimoto K, Nakagawa T, et al. Autophagy deficiency exacerbates colitis through excessive oxidative stress and MAPK signaling pathway activation. PLoS One. 2019;14(11):e0225066. doi: 10.1371/journal.pone.0225066
- Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-Induced apoptosis during chronic colitis in mice. Cell Host Microbe. 2018 Feb 14;23(2):191–202.e4. doi: 10.1016/j.chom.2017.12.017
- Wu MY, Liu L, Wang EJ, et al. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy. 2021 May;17(5):1096–1111. doi: 10.1080/15548627.2020.1741332
- Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503(7475):272–276. doi: 10.1038/nature12599
- Bel S, Pendse M, Wang Y, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017 Sep 8;357(6355):1047–1052. doi: 10.1126/science.aal4677
- Murano T, Najibi M, Paulus GLC, et al. Transcription factor TFEB cell-autonomously modulates susceptibility to intestinal epithelial cell injury in vivo. Sci Rep. 2017 Oct 24;7(1):13938. doi: 10.1038/s41598-017-14370-4
- Liu B, Gulati AS, Cantillana V, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2013 Oct 15;305(8):G573–84. doi: 10.1152/ajpgi.00071.2013
- Cabrera S, Fernández AF, Mariño G, et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy. 2013 Aug;9(8):1188–1200. doi: 10.4161/auto.24797
- Cadwell K, Patel KK, Komatsu M, et al. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy. 2009 Feb;5(2):250–252. doi: 10.4161/auto.5.2.7560
- Asano J, Sato T, Ichinose S, et al. Intrinsic autophagy is required for the maintenance of intestinal stem cells and for Irradiation-induced intestinal regeneration. Cell Rep. 2017 Aug 1;20(5):1050–1060. doi: 10.1016/j.celrep.2017.07.019
- Levy A, Stedman A, Deutsch E, et al. Innate immune receptor NOD2 mediates LGR5(+) intestinal stem cell protection against ROS cytotoxicity via mitophagy stimulation. Proc Natl Acad Sci U S A. 2020 Jan 28;117(4):1994–2003. doi: 10.1073/pnas.1902788117
- Nagai H, Tatara H, Tanaka-Furuhashi K, et al. Homeostatic regulation of ROS-Triggered Hippo-Yki pathway via autophagic clearance of Ref(2)P/p62 in the drosophila intestine. Dev Cell. 2021 Jan 11;56(1):81–94.e10. doi: 10.1016/j.devcel.2020.12.007
- Levin AD, Koelink PJ, Bloemendaal FM, et al. Autophagy contributes to the induction of anti-TNF induced macrophages. J Crohns Colitis. 2016 Mar;10(3):323–329. doi: 10.1093/ecco-jcc/jjv174
- Zhang H, Zheng L, McGovern DP, et al. Myeloid ATG16L1 facilitates Host-bacteria interactions in maintaining intestinal homeostasis. J Immunol. 2017 Mar 1;198(5):2133–2146. doi: 10.4049/jimmunol.1601293
- Lee HY, Kim J, Quan W, et al. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy. 2016 Aug 2;12(8):1390–1403. doi: 10.1080/15548627.2016.1184799
- Takagawa T, Kitani A, Fuss I, et al. An increase in LRRK2 suppresses autophagy and enhances dectin-1-induced immunity in a mouse model of colitis. Sci Transl Med. 2018 Jun 6;10(444). doi: 10.1126/scitranslmed.aan8162
- Lahiri A, Hedl M, Abraham C. MTMR3 risk allele enhances innate receptor-induced signaling and cytokines by decreasing autophagy and increasing caspase-1 activation. Proc Natl Acad Sci U S A. 2015 Aug 18;112(33):10461–10466. doi: 10.1073/pnas.1501752112
- Zhang H, Zheng L, Chen J, et al. The protection role of Atg16l1 in CD11c(+)dendritic cells in murine colitis. Immunobiology. 2017 Jul;222(7):831–841. doi: 10.1016/j.imbio.2017.03.002
- Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11(2):271–284. doi: 10.1080/15548627.2015.1009787
- Pan H, Chen L, Xu Y, et al. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget. 2016 Apr 19;7(16):21235–21246. doi: 10.18632/oncotarget.6908
- Fiegl D, Kägebein D, Liebler-Tenorio EM, et al. Amphisomal route of MHC class I cross-presentation in bacteria-infected dendritic cells. J Immunol. 2013 Mar 15;190(6):2791–2806. doi: 10.4049/jimmunol.1202741
- Fujishima Y, Nishiumi S, Masuda A, et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-κB activation. Arch Biochem Biophys. 2011 Feb 15;506(2):223–235. doi: 10.1016/j.abb.2010.12.009
- Li D. Selective degradation of the IkappaB kinase (IKK) by autophagy. Cell Res. 2006 Nov;16(11):855–856. doi: 10.1038/sj.cr.7310110
- Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014 Apr 10;345(2):235–241. doi: 10.1016/j.canlet.2013.07.032
- Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci. 2020 Nov;77(22):4459–4483. doi: 10.1007/s00018-020-03536-5
- Tal MC, Sasai M, Lee HK, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2770–2775. doi: 10.1073/pnas.0807694106
- Liu M, Sun T, Li N, et al. BRG1 attenuates colonic inflammation and tumorigenesis through autophagy-dependent oxidative stress sequestration. Nat Commun. 2019 Oct 10;10(1):4614. doi: 10.1038/s41467-019-12573-z
- Lin Y, Jiang M, Chen W, et al. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019 Oct;118:109249.
- Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013 May 28;332(2):249–264. doi: 10.1016/j.canlet.2010.07.016
- Li XX, Zhang HS, Xu YM, et al. Knockdown of IRE1α inhibits colonic tumorigenesis through decreasing β-catenin and IRE1α targeting suppresses colon cancer cells. Oncogene. 2017 Nov 30;36(48):6738–6746. doi: 10.1038/onc.2017.284
- Tao J, Yin L, Wu A, et al. PDIA2 bridges endoplasmic reticulum stress and metabolic reprogramming during malignant transformation of chronic colitis. Front Oncol. 2022;12:836087. doi: 10.3389/fonc.2022.836087
- Rashid HO, Yadav RK, Kim HR, et al. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015 Nov 2;11(11):1956–1977. doi: 10.1080/15548627.2015.1091141
- Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis. J Exp Med. 2017 Feb;214(2):401–422. doi: 10.1084/jem.20160791
- Naama M, Telpaz S, Awad A, et al. Autophagy controls mucus secretion from intestinal goblet cells by alleviating ER stress. Cell Host Microbe. 2023 Jan 31;31(3):433–446.e4. doi: 10.1016/j.chom.2023.01.006
- Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014 Jul 20;21(3):396–413. doi: 10.1089/ars.2014.5851
- Wan Y, Yang L, Jiang S, et al. Excessive apoptosis in ulcerative colitis: crosstalk between apoptosis, ROS, ER stress, and intestinal homeostasis. Inflamm Bowel Dis. 2022 Mar 30;28(4):639–648. doi: 10.1093/ibd/izab277
- Engevik MA, Herrmann B, Ruan W, et al. Bifidobacterium dentium-derived y-glutamylcysteine suppresses ER-mediated goblet cell stress and reduces TNBS-driven colonic inflammation. Gut Microbes. 2021 Jan-Dec;13(1):1–21. doi: 10.1080/19490976.2021.1902717
- Iida T, Hirayama D, Minami N, et al. Down-regulation of RalGTPase-activating protein promotes colitis-associated cancer via NLRP3 inflammasome activation. Cell Mol Gastroenterol Hepatol. 2020;9(2):277–293. doi: 10.1016/j.jcmgh.2019.10.003
- Li T, Fu B, Zhang X, et al. Overproduction of gastrointestinal 5-HT promotes colitis-associated colorectal cancer progression via enhancing NLRP3 inflammasome activation. Cancer Immunol Res. 2021 Sep;9(9):1008–1023. doi: 10.1158/2326-6066.CIR-20-1043
- Guo W, Sun Y, Liu W, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014 Jun;10(6):972–985. doi: 10.4161/auto.28374
- Qiao S, Lv C, Tao Y, et al. Arctigenin disrupts NLRP3 inflammasome assembly in colonic macrophages via downregulating fatty acid oxidation to prevent colitis-associated cancer. Cancer Lett. 2020 Oct 28;491:162–179. doi: 10.1016/j.canlet.2020.08.033
- Shao X, Sun S, Zhou Y, et al. Bacteroides fragilis restricts colitis-associated cancer via negative regulation of the NLRP3 axis. Cancer Lett. 2021 Dec 28;523:170–181. doi: 10.1016/j.canlet.2021.10.002
- Wang X, Jia Y, Wen L, et al. Porphyromonas gingivalis promotes colorectal carcinoma by activating the Hematopoietic NLRP3 inflammasome. Cancer Res. 2021 May 15;81(10):2745–2759. doi: 10.1158/0008-5472.CAN-20-3827
- Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011 Mar;12(3):222–230. doi: 10.1038/ni.1980
- Yun Y, Baek A, Kim DE. Autophagy down-regulates NLRP3-dependent inflammatory response of intestinal epithelial cells under nutrient deprivation. BMB Rep. 2021 May;54(5):260–265. doi: 10.5483/BMBRep.2021.54.5.211
- Ravindran R, Loebbermann J, Nakaya HI, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. 2016 Mar 24;531(7595):523–527. doi: 10.1038/nature17186
- Mehto S, Jena KK, Nath P, et al. The Crohn’s disease risk factor IRGM Limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell. 2019 Feb 7;73(3):429–445.e7. doi: 10.1016/j.molcel.2018.11.018
- Hou P, Tian T, Jia P, et al. The regulation of NLRP3 inflammasome activation by CCDC50-mediated autophagy. Autophagy. 2022 May 27;19(1):1–2. doi: 10.1080/15548627.2022.2080957
- Richard ML, Liguori G, Lamas B, et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes. 2018 Mar 4;9(2):131–142. doi: 10.1080/19490976.2017.1379637
- Liu H, Gao P, Jia B, et al. IBD-Associated Atg16L1T300A polymorphism regulates commensal microbiota of the intestine. Front Immunol. 2021;12:772189. doi: 10.3389/fimmu.2021.772189
- Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008 Nov 13;456(7219):259–263. doi: 10.1038/nature07416
- Wang L, Tang L, Feng Y, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8(+) T cells in mice. Gut. 2020 Nov;69(11):1988–1997. doi: 10.1136/gutjnl-2019-320105
- Chen L, Chen MY, Shao L, et al. Panax notoginseng saponins prevent colitis-associated colorectal cancer development: the role of gut microbiota. Chin J Nat Med. 2020 Jul;18(7):500–507. doi: 10.1016/S1875-5364(20)30060-1
- Yang L, Liu C, Zhao W, et al. Impaired autophagy in intestinal epithelial cells alters gut microbiota and Host immune responses. Appl Environ Microbiol. 2018 Sep 15;84(18). doi: 10.1128/AEM.00880-18
- Wu JC, Tsai ML, Lai CS, et al. Polymethoxyflavones prevent benzo[a]pyrene/dextran sodium sulfate-induced colorectal carcinogenesis through modulating xenobiotic metabolism and ameliorate autophagic defect in ICR mice. Int J Cancer. 2018 Apr 15;142(8):1689–1701. doi: 10.1002/ijc.31190
- Lapaquette P, Glasser AL, Huett A, et al. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010 Jan;12(1):99–113. doi: 10.1111/j.1462-5822.2009.01381.x
- Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012 Jun;14(6):791–807. doi: 10.1111/j.1462-5822.2012.01768.x
- Nguyen HT, Dalmasso G, Müller S, et al. Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology. 2014 Feb;146(2):508–519. doi: 10.1053/j.gastro.2013.10.021
- Salesse L, Lucas C, Hoang MHT, et al. Colibactin-producing Escherichia coli induce the formation of invasive carcinomas in a chronic inflammation-associated mouse model. Cancers (Basel). 2021 Apr 24;13(9):2060. doi: 10.3390/cancers13092060
- Lucas C, Salesse L, Hoang MHT, et al. Autophagy of intestinal epithelial cells inhibits colorectal carcinogenesis induced by colibactin-producing Escherichia coli in Apc(Min/+) mice. Gastroenterology. 2020 Apr;158(5):1373–1388. doi: 10.1053/j.gastro.2019.12.026
- Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. 2017 Dec 4;214(12):3687–3705. doi: 10.1084/jem.20170558
- Lim J, Park H, Heisler J, et al. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. Elife. 2019 Jul 9;8. doi: 10.7554/eLife.44452.
- Xie Y, Zhao Y, Shi L, et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J Clin Invest. 2020 Apr 1;130(4):2111–2128. doi: 10.1172/JCI133264
- Liu ZY, Zheng M, Li YM, et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics. 2019;9(12):3659–3673. doi: 10.7150/thno.32126
- Rajamäki K, Taira A, Katainen R, et al. Genetic and Epigenetic Characteristics of inflammatory bowel disease-associated colorectal cancer. Gastroenterology. 2021 Aug;161(2):592–607. doi: 10.1053/j.gastro.2021.04.042
- Tanaka T. Development of an inflammation-associated colorectal cancer model and its application for research on carcinogenesis and chemoprevention. Int J Inflam. 2012;2012:658786. doi: 10.1155/2012/658786
- De Robertis M, Massi E, Poeta ML, et al. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. 2011 Mar 24;10:9. doi: 10.4103/1477-3163.78279
- Modesto R, Estarreja J, Silva I, et al. Chemically induced colitis-associated cancer models in rodents for pharmacological modulation: a systematic review. J Clin Med. 2022 May 12;11(10):2739. doi: 10.3390/jcm11102739
- Scherr AL, Jassowicz A, Pató A, et al. Knockdown of Atg7 induces nuclear-LC3 dependent apoptosis and augments chemotherapy in colorectal cancer cells. Int J Mol Sci. 2020 Feb 7;21(3):1099. doi: 10.3390/ijms21031099
- Selvakumaran M, Amaravadi RK, Vasilevskaya IA, et al. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res. 2013 Jun 1;19(11):2995–3007. doi: 10.1158/1078-0432.CCR-12-1542
- Jo YK, Kim SC, Park IJ, et al. Increased expression of ATG10 in colorectal cancer is associated with lymphovascular invasion and lymph node metastasis. PLoS One. 2012;7(12):e52705. doi: 10.1371/journal.pone.0052705
- Rehman SK, Haynes J, Collignon E, et al. Colorectal cancer cells enter a diapause-like DTP state to survive chemotherapy. Cell. 2021 Jan 7;184(1):226–242.e21. doi: 10.1016/j.cell.2020.11.018
- Chen Y, Chen Y, Zhang J, et al. Fusobacterium nucleatum promotes metastasis in colorectal cancer by activating autophagy signaling via the upregulation of CARD3 expression. Theranostics. 2020;10(1):323–339. doi: 10.7150/thno.38870
- Peng JQ, Han SM, Chen ZH, et al. Chaperone-mediated autophagy regulates apoptosis and the proliferation of colon carcinoma cells. Biochem Biophys Res Commun. 2020 Feb 5;522(2):348–354. doi: 10.1016/j.bbrc.2019.11.081
- Zou Z, Tao T, Li H, et al. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10(1):31. doi: 10.1186/s13578-020-00396-1
- Sanaei MJ, Baghery Saghchy Khorasani A, Pourbagheri-Sigaroodi A, et al. The PI3K/Akt/mTOR axis in colorectal cancer: oncogenic alterations, non-coding RNAs, therapeutic opportunities, and the emerging role of nanoparticles. J Cell Physiol. 2022 Mar;237(3):1720–1752. doi: 10.1002/jcp.30655
- Grasso S, Pereira GJS, Palmeira-Dos-Santos C, et al. Autophagy regulates selumetinib (AZD6244) induced-apoptosis in colorectal cancer cells. Eur J Med Chem. 2016 Oct 21;122:611–618. doi: 10.1016/j.ejmech.2016.06.043
- Jiang K, Liu M, Lin G, et al. Tumor suppressor Spred2 interaction with LC3 promotes autophagosome maturation and induces autophagy-dependent cell death. Oncotarget. 2016 May 3;7(18):25652–25667. doi: 10.18632/oncotarget.8357
- Gao GY, Ma J, Lu P, et al. RETRACTED: ophiopogonin B induces the autophagy and apoptosis of colon cancer cells by activating JNK/c-Jun signaling pathway. Biomed Pharmacother. 2018 Dec;108:1208–1215.
- Wang L, Yu Z, Ren S, et al. Metabolic reprogramming in colon cancer reversed by DHTS through regulating PTEN/AKT/HIF1α mediated signal pathway. Biochim Biophys Acta Gen Subj. 2018 Oct;1862(10):2281–2292. doi: 10.1016/j.bbagen.2018.07.017
- Yin K, Lee J, Liu Z, et al. Mitophagy protein PINK1 suppresses colon tumor growth by metabolic reprogramming via p53 activation and reducing acetyl-CoA production. Cell Death Differ. 2021 Aug;28(8):2421–2435. doi: 10.1038/s41418-021-00760-9
- D’Onofrio N, Martino E, Mele L, et al. Colorectal cancer apoptosis induced by dietary δ-valerobetaine involves PINK1/Parkin dependent-mitophagy and SIRT3. Int J Mol Sci. 2021 Jul 29;22(15):8117. doi: 10.3390/ijms22158117
- Zhang K, Zhang D, Wang J, et al. Aloe gel glucomannan induced colon cancer cell death via mitochondrial damage-driven PINK1/Parkin mitophagy pathway. Carbohydr Polym. 2022 Nov 1;295:119841. doi: 10.1016/j.carbpol.2022.119841
- Saiprasad G, Chitra P, Manikandan R, et al. Hesperidin induces apoptosis and triggers autophagic markers through inhibition of aurora-A mediated phosphoinositide-3-kinase/Akt/mammalian target of rapamycin and glycogen synthase kinase-3 beta signalling cascades in experimental colon carcinogenesis. Eur J Cancer. 2014 Sep;50(14):2489–2507. doi: 10.1016/j.ejca.2014.06.013
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017 Mar 9;168(6):960–976. doi: 10.1016/j.cell.2017.02.004
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011 Feb;13(2):132–141. doi: 10.1038/ncb2152
- Ganley IG, Lam du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009 May 1;284(18):12297–12305. doi: 10.1074/jbc.M900573200
- Kim YM, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015 Jan 22;57(2):207–218. doi: 10.1016/j.molcel.2014.11.013
- Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010 Jun 17;465(7300):942–946. doi: 10.1038/nature09076
- Tian Y, Wang K, Fan Y, et al. Chemopreventive effect of dietary glutamineon colitis-associated colorectal cancer is associated with modulation of the DEPTOR/mTOR Signaling pathway. Nutrients. 2016 May 2;8(5):261. doi: 10.3390/nu8050261
- Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018 Feb 6;27(2):299–313. doi: 10.1016/j.cmet.2017.10.009
- Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011 Jan 28;331(6016):456–461. doi: 10.1126/science.1196371
- Zhao Y, Guo Q, Zhao K, et al. Small molecule GL-V9 protects against colitis-associated colorectal cancer by limiting NLRP3 inflammasome through autophagy. Oncoimmunology. 2017;7(1):e1375640. doi: 10.1080/2162402X.2017.1375640
- Liu C, Wang J, Yang Y, et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem Pharmacol. 2018 Sep;155:366–379.
- Sun D, Tao W, Zhang F, et al. Trifolirhizin induces autophagy-dependent apoptosis in colon cancer via AMPK/mTOR signaling. Signal Transduct Target Ther. 2020 Aug 27;5(1):174. doi: 10.1038/s41392-020-00281-w
- Hu C, Cao Y, Li P, et al. Oleanolic acid induces autophagy and apoptosis via the AMPK-mTOR signaling pathway in colon cancer. J Oncol. 2021;2021:8281718. doi: 10.1155/2021/8281718
- Kong W, Zhu H, Zheng S, et al. Larotrectinib induces autophagic cell death through AMPK/mTOR signalling in colon cancer. J Cell Mol Med. 2022 Nov;26(21):5539–5550. doi: 10.1111/jcmm.17530
- Bu H, Liu D, Zhang G, et al. AMPK/mTOR/ULK1 axis-mediated pathway participates in apoptosis and autophagy induction by oridonin in colon cancer DLD-1 cells. Onco Targets Ther. 2020;13:8533–8545. doi: 10.2147/OTT.S262022
- Wang GY, Zhang L, Geng YD, et al. β-Elemene induces apoptosis and autophagy in colorectal cancer cells through regulating the ROS/AMPK/mTOR pathway. Chin J Nat Med. 2022 Jan;20(1):9–21. doi: 10.1016/S1875-5364(21)60118-8
- Ni X, Shang FS, Wang TF, et al. Ellagic acid induces apoptosis and autophagy in colon cancer through the AMPK/mTOR pathway. Tissue Cell. 2023 Apr;81:102032.
- Setia S, Nehru B, Sanyal SN. Upregulation of MAPK/Erk and PI3K/Akt pathways in ulcerative colitis-associated colon cancer. Biomed Pharmacother. 2014 Oct;68(8):1023–1029. doi: 10.1016/j.biopha.2014.09.006
- Yu X, Long YC, Shen HM. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy. 2015;11(10):1711–1728. doi: 10.1080/15548627.2015.1043076
- You P, Wu H, Deng M, et al. Brevilin a induces apoptosis and autophagy of colon adenocarcinoma cell CT26 via mitochondrial pathway and PI3K/AKT/mTOR inactivation. Biomed Pharmacother. 2018 Feb;98:619–625.
- Ha HA, Chiang JH, Tsai FJ, et al. Novel quinazolinone MJ‑33 induces AKT/mTOR‑mediated autophagy‑associated apoptosis in 5FU‑resistant colorectal cancer cells. Oncol Rep. 2021 Feb;45(2):680–692. doi: 10.3892/or.2020.7882
- Wang J, Liang D, Zhang XP, et al. Novel PI3K/Akt/mTOR signaling inhibitor, W922, prevents colorectal cancer growth via the regulation of autophagy. Int J Oncol. 2021 Jan;58(1):70–82. doi: 10.3892/ijo.2020.5151
- Cao ZX, Yang YT, Yu S, et al. Pogostone induces autophagy and apoptosis involving PI3K/Akt/mTOR axis in human colorectal carcinoma HCT116 cells. J Ethnopharmacol. 2017 Apr 18;202:20–27. doi: 10.1016/j.jep.2016.07.028
- Liu M, Zhao G, Zhang D, et al. Active fraction of clove induces apoptosis via PI3K/Akt/mTOR-mediated autophagy in human colorectal cancer HCT-116 cells. Int J Oncol. 2018 Sep;53(3):1363–1373. doi: 10.3892/ijo.2018.4465
- Long J, He Q, Yin Y, et al. The effect of miRNA and autophagy on colorectal cancer. Cell Prolif. 2020 Oct;53(10):e12900. doi: 10.1111/cpr.12900
- Zhai Z, Wu F, Dong F, et al. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy. 2014 Mar;10(3):468–479. doi: 10.4161/auto.27553
- Lu C, Chen J, Xu HG, et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology. 2014 Jan;146(1):188–199. doi: 10.1053/j.gastro.2013.09.006
- Lin XT, Zheng XB, Fan DJ, et al. MicroRNA-143 targets ATG2B to inhibit autophagy and increase inflammatory responses in Crohn’s disease. Inflamm Bowel Dis. 2018 Mar 19;24(4):781–791. doi: 10.1093/ibd/izx075
- Zhang H, Tang J, Li C, et al. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett. 2015 Jan 28;356(2 Pt B):781–790. doi: 10.1016/j.canlet.2014.10.029
- Tan S, Shi H, Ba M, et al. miR-409-3p sensitizes colon cancer cells to oxaliplatin by inhibiting beclin-1-mediated autophagy. Int J Mol Med. 2016 Apr;37(4):1030–1038. doi: 10.3892/ijmm.2016.2492
- Li Y, Li C, Li D, et al. lncRNA KCNQ1OT1 enhances the chemoresistance of oxaliplatin in colon cancer by targeting the miR-34a/ATG4B pathway. Onco Targets Ther. 2019;12:2649–2660. doi: 10.2147/OTT.S188054
- Sun W, Li J, Zhou L, et al. The c-Myc/miR-27b-3p/ATG10 regulatory axis regulates chemoresistance in colorectal cancer. Theranostics. 2020;10(5):1981–1996. doi: 10.7150/thno.37621
- Zhai H, Song B, Xu X, et al. Inhibition of autophagy and tumor growth in colon cancer by miR-502. Oncogene. 2013 Mar 21;32(12):1570–1579. doi: 10.1038/onc.2012.167
- Wu H, Lu XX, Wang JR, et al. TRAF6 inhibits colorectal cancer metastasis through regulating selective autophagic CTNNB1/β-catenin degradation and is targeted for GSK3B/GSK3β-mediated phosphorylation and degradation. Autophagy. 2019 Sep;15(9):1506–1522. doi: 10.1080/15548627.2019.1586250
- Liu J, Cai J, Fan P, et al. The abilities of Salidroside on ameliorating inflammation, skewing the Imbalanced Nucleotide oligomerization domain-like receptor family Pyrin domain containing 3/Autophagy, and maintaining intestinal barrier are profitable in colitis. Front Pharmacol. 2019;10:1385. doi: 10.3389/fphar.2019.01385
- Xuan H, Ou A, Hao S, et al. Galangin protects against symptoms of dextran sodium sulfate-induced acute colitis by activating autophagy and modulating the gut microbiota. Nutrients. 2020 Jan 29;12(2):347. doi: 10.3390/nu12020347
- Yang HL, Liu HW, Shrestha S, et al. Antrodia salmonea induces apoptosis and enhances cytoprotective autophagy in colon cancer cells. Aging. 2021 May 24;13(12):15964–15989. doi: 10.18632/aging.203019
- Trivedi PP, Jena GB, Tikoo KB, et al. Melatonin modulated autophagy and Nrf2 signaling pathways in mice with colitis-associated colon carcinogenesis. Mol Carcinog. 2016 Mar;55(3):255–267. doi: 10.1002/mc.22274
- Yang PM, Lin YT, Shun CT, et al. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci Rep. 2013 Nov 14;3(1):3219. doi: 10.1038/srep03219
- Zhao LN, Li JY, Yu T, et al. 5-aminosalicylates reduce the risk of colorectal neoplasia in patients with ulcerative colitis: an updated meta-analysis. PLoS One. 2014;9(4):e94208. doi: 10.1371/journal.pone.0094208
- Velayos FS, Terdiman JP, Walsh JM. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies. Am J Gastroenterol. 2005 Jun;100(6):1345–1353. doi: 10.1111/j.1572-0241.2005.41442.x
- Biancone L, Petruzziello C, Calabrese E, et al. Long-term safety of Infliximab for the treatment of inflammatory bowel disease: does blocking TNFalpha reduce colitis-associated colorectal carcinogenesis? Gut. 2009 Dec;58(12):1703. doi: 10.1136/gut.2008.176461
- Sasaki K, Tsuno NH, Sunami E, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010 Jul 15;10(1):370. doi: 10.1186/1471-2407-10-370