
ABSTRACT
Streptococcus pneumoniae (S. pneumoniae) represents a major human bacterial pathogen leading to high morbidity and mortality in children and the elderly. Recent research emphasizes the role of extracellular vesicles (EVs) in bacterial pathogenicity. However, the contribution of S. pneumoniae EVs (pEVs) to host-microbe interactions has remained unclear. Here, we observed that S. pneumoniae infections in mice led to severe lung injuries and alveolar epithelial barrier (AEB) dysfunction. Infections of S. pneumoniae reduced the protein expression of tight junction protein OCLN (occludin) and activated macroautophagy/autophagy in lung tissues of mice and A549 cells. Mechanically, S. pneumoniae induced autophagosomal degradation of OCLN leading to AEB impairment in the A549 monolayer. S. pneumoniae released the pEVs that could be internalized by alveolar epithelial cells. Through proteomics, we profiled the cargo proteins inside pEVs and found that these pEVs contained many virulence factors, among which we identified a eukaryotic-like serine-threonine kinase protein StkP. The internalized StkP could induce the phosphorylation of BECN1 (beclin 1) at Ser93 and Ser96 sites, initiating autophagy and resulting in autophagy-dependent OCLN degradation and AEB dysfunction. Finally, the deletion of stkP in S. pneumoniae completely protected infected mice from death, significantly alleviated OCLN degradation in vivo, and largely abolished the AEB disruption caused by pEVs in vitro. Overall, our results suggested that pEVs played a crucial role in the spread of S. pneumoniae virulence factors. The cargo protein StkP in pEVs could communicate with host target proteins and even hijack the BECN1 autophagy initiation pathway, contributing to AEB disruption and bacterial pathogenicity.
Abbreviations: AEB: alveolarepithelial barrier; AECs: alveolar epithelial cells; ATG16L1: autophagy related 16 like 1; ATP:adenosine 5’-triphosphate; BafA1: bafilomycin A1; BBB: blood-brain barrier; CFU: colony-forming unit; co-IP: co-immunoprecipitation; CQ:chloroquine; CTRL: control; DiO: 3,3′-dioctadecylox-acarbocyanineperchlorate; DOX: doxycycline; DTT: dithiothreitol; ECIS: electricalcell-substrate impedance sensing; eGFP: enhanced green fluorescentprotein; ermR: erythromycin-resistance expression cassette; Ery: erythromycin; eSTKs: eukaryotic-like serine-threoninekinases; EVs: extracellular vesicles; HA: hemagglutinin; H&E: hematoxylin and eosin; HsLC3B: human LC3B; hpi: hours post-infection; IP: immunoprecipitation; KD: knockdown; KO: knockout; LAMP1: lysosomal associated membrane protein 1; LC/MS: liquid chromatography-mass spectrometry; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MVs: membranevesicles; NC:negative control; NETs:neutrophil extracellular traps; OD: optical density; OMVs: outer membrane vesicles; PBS: phosphate-buffered saline; pEVs: S.pneumoniaeextracellular vesicles; protK: proteinase K; Rapa: rapamycin; RNAi: RNA interference; S.aureus: Staphylococcusaureus; SNF:supernatant fluid; sgRNA: single guide RNA; S.pneumoniae: Streptococcuspneumoniae; S.suis: Streptococcussuis; TEER: trans-epithelium electrical resistance; moi: multiplicity ofinfection; TEM:transmission electron microscope; TJproteins: tight junction proteins; TJP1/ZO-1: tight junction protein1; TSA: tryptic soy agar; WB: western blot; WT: wild-type
Introduction
Streptococcus pneumoniae (S. pneumoniae) is a common opportunistic human pathogen and is the primary cause of bacterial pneumonia worldwide [Citation1,Citation2]. This bacterium is responsible for millions of deaths each year due to invasive pneumococcal diseases, such as community-acquired pneumonia, sepsis, and meningitis [Citation3,Citation4]. S. pneumoniae typically colonizes the human nasopharynx and upper airways but can cause severe illnesses when it spreads to the lower respiratory tract [Citation5]. Bacteria can enter the bloodstream from the air space by passing through the alveolar epithelial barrier (AEB) and the vascular endothelial barrier, thus increasing pulmonary permeability [Citation6,Citation7]. Generally, the development of lobar pneumonia is caused by the bacteria’s adherence to the alveolar epithelium, followed by replication and the initiation of host damage responses. These processes are driven by the interactions between bacterial-secreted products and the alveolar epithelium as well as subsequent innate immune defenses [Citation1,Citation5]. Nevertheless, the precise mechanisms by which S. pneumoniae induces dysfunction of the alveolar epithelial barrier are still not fully understood.
Bacterial membrane vesicles (MVs) have been identified as a unique bacterial secretion pathway for transporting specific cargo [Citation8]. These MVs, which are also known as outer membrane vesicles (OMVs), have been extensively studied and characterized in gram-negative bacteria but less extensively in gram-positive bacteria [Citation9]. Recent studies have provided evidence that gram-positive bacteria can also release spherical membrane particles from the cell surface, referred to as extracellular vesicles (EVs) [Citation10]. These EVs have been found to play a role in host-pathogen interactions by delivering bacterial components into host cells, which causes cytotoxic effects, and thus increases bacterial pathogenicity [Citation11–16]. For example, EVs derived from Staphylococcus aureus (S. aureus) have been shown to activate toll-like receptors and induce pro-inflammatory cytokine responses in epithelial cells [Citation12] and macrophages [Citation17]. Although the internalization of EVs into epithelial and endothelial cells has been observed to increase barrier permeability [Citation11], there is still a lack of evidence on how EVs disrupt AEB integrity.
Macroautophagy/autophagy is a conserved lysosomal degradation pathway that eliminates damaged organelles and proteins, thereby avoiding excessive injury and cellular dysfunction in multiple cells and organs [Citation18]. On the one hand, autophagy can act as a protective response to external stimuli, such as hypoxia, under which it helps to prevent endothelial CLDN5 redistribution and abnormal accumulation, thus alleviating BBB dysfunction [Citation19,Citation20]. However, on the other hand, excessive autophagy can lead to autophagic cell death, resulting in tissue damage. Research has shown that autophagy can degrade tight junction proteins (TJ proteins) and increase epithelial barrier permeability in the intestines [Citation21,Citation22] and respiratory tracts [Citation23]. It has also been reported that S. pneumoniae infection can induce autophagy activation in human alveolar epithelial cells (AECs) [Citation24], human corneal epithelial cells [Citation25], and mouse bone-marrow-derived neutrophils [Citation26]. Intracellular S. pneumoniae can be recognized by bactericidal autophagy, and certain S. pneumoniae-derived proteins have been considered autophagy activators, such as surface-exposed choline-binding protein CbpC [Citation27], pore-forming toxin pneumolysin PLY [Citation28,Citation29], and S. pneumoniae endopeptidase O PepO [Citation30]. However, the direct evidence and underlying mechanisms of AEB disruption related to autophagy under S. pneumoniae infections are still unknown, and the manipulation of these autophagic processes by S. pneumoniae EVs (pEVs) remains unexplored.
In this study, we demonstrated that pEVs could induce autophagy in alveolar epithelial cells, leading to the disruption of AEB integrity. We also found that the internalization of pEVs in A549 cells led to an increase of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-II, a marker of autophagy activation and autophagosome formation, as well as the degradation of OCLN (occludin) through the process of autophagy. Furthermore, we identified that the eukaryotic-like serine-threonine kinases (eSTKs) protein StkP was included in pEVs, and intracellular StkP was found to induce autophagy activation by phosphorylating BECN1 (beclin 1). Collectively, our findings suggested that the bacterial protein StkP, as a cargo of pEVs, could induce host autophagy during S. pneumoniae infections and indicated a previously unknown role of EVs from gram-positive bacteria in causing the disruption of the epithelial barrier through the autophagy-mediated cellular degradation pathway. Overall, these results improved our understanding of the intracellular effects of pEVs and their cargoes on barrier disruption, providing insights into bacterial pathogenesis and disease progression in the host.
Results
S. pneumoniae infections induced lung injuries possibly through the disruption of tight junctions and AEB integrity
To determine whether S. pneumoniae could induce lung injuries, we challenged 3-week-old Balb/c mice intranasally with the S. pneumoniae strain D39 and characterized the lung damages by colony-forming unit (CFU) assay, hematoxylin and eosin (H&E) staining, and cytokine array in the lung tissues of mice. After 24 h post-infection (hpi), we successfully detected S. pneumoniae in the lungs, brains, and blood of mice, which suggested that S. pneumoniae could breach the lung epithelial barrier of mice and disseminate to other tissues (). We further showed that S. pneumoniae infections resulted in the widening of the alveolar septum, fusion of pulmonary alveoli, and infiltration of inflammatory cells (). It was reported that the release of inflammatory cytokines was an important contributor to lung damage [Citation31]. We thus assessed the changes of 111 inflammatory cytokines and chemokines in lung tissues by cytokine array. We showed that a total of 104 proteins were upregulated in the lung tissues of mice after D39 infections compared to control (CTRL) mice (). Specifically, some chemokines such as CXCL10, CXCL9, and inflammatory cytokines IL1A/IL-1α had a most obvious increase. We further examined the expression of TJ proteins in the lung tissues of mice and showed that D39 infections significantly decreased the protein levels of OCLN, a marker of TJs, and that of TJP1/ZO-1 and CDH1/E-cadherin were also decreased ( and Figure S1A). Interestingly, we also found that D39 infections induced a makeable increase of LC3-II, suggesting that S. pneumoniae infections could activate autophagy in the lung tissues of mice ().
Figure 1. S. pneumoniae infections induced lung injuries possibly through the disruption of tight junctions and AEB integrity. (A-F) Balb/c mice were either control-treated or intranasal-infected by D39 at a CFU of 5 × 108 per mouse, and the tissues and whole-cell extracts were collected at 24 h after intranasal infection with bacteria. (A) bacterial burdens in the lungs, brains, and blood were determined respectively (n = 5). (B) Representative histological views of the lungs of mice by H&E staining. Arrows showed examples of disordered alveolar structures and neutrophil infiltration areas. Scale bars: 50 μm. (C and D) cytokine expressions of lung tissues were detected by cytokines array, and the fold changes compared to control were calculated according to gray levels. The indicated numbers from 1 to 10 in both C and D were corresponded to each other. (E and F) WB analysis of OCLN and LC3 levels of lung tissues at 24 hpi. (G) TEER changes of A549 cells in treating multiple dosages of D39 or PBS (mock) monitored by the electrical cell-substrate impedance sensing (ECIS) system. Data were collected and presented as mean ± SD from six replicated wells at each time point. (H) A549 cells were either control-treated or D39-infected at multiple moi (20, 50, and 100 moi), and whole-cell extracts were prepared at the respective time points (2, 4, and 8 hpi) for OCLN determination by WB. (I) Representative images of immunofluorescence staining of A549 cells infected with D39 for OCLN (red) co-stained with DAPI (blue). Scale bars: 20 μm.
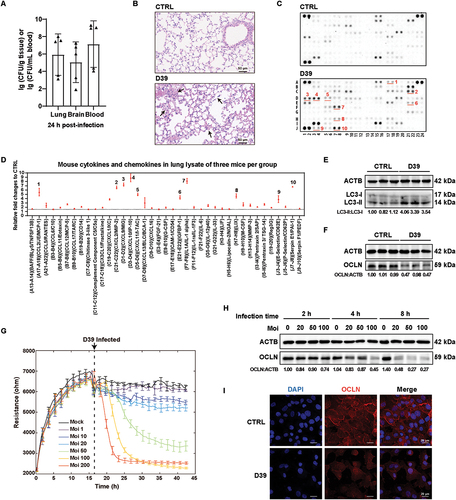
We further validated the impacts of S. pneumoniae strain D39 on A549 cells. We first evaluated the changes in trans-epithelium electrical resistance (TEER) of the A549 monolayer, an indicator of the integrity of the cellular barrier, caused by S. pneumoniae. We found that D39 infections could significantly decrease the TEER of A549 monolayer within a wide range of multiplicity of infection (moi) (), indicating that S. pneumoniae could destroy the integrity of the epithelial cell layer. Furthermore, we examined the changes in protein levels and subcellular localizations of OCLN in A549 cells and found that TJ proteins, especially OCLN, revealed a noticeable decrease after 4 h and 8 h post-infection of D39 at moi of 20, 50, and 100 ( and Figure S1B-C). The result of immunofluorescence staining of OCLN also indicated that D39 infections could decrease the membrane localization of OCLN (). In summary, these results indicated that S. pneumoniae could indeed cause lung injuries in mice, possibly due to the disruption of tight junctions and the dysfunction of AEB integrity.
S. pneumoniae infections induced autophagy in A549 cells
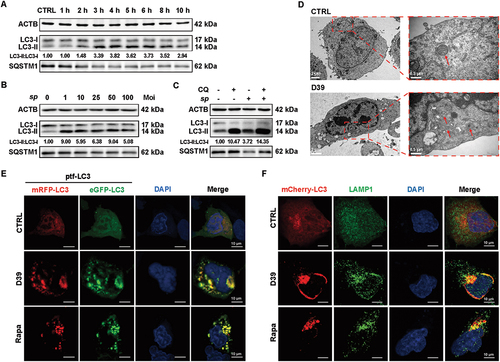
The increase of LC3-II protein in the lung tissues of mice caused by S. pneumoniae strain D39 promoted us to wonder whether autophagy played a role in lung damage by S. pneumoniae infections. We verified the autophagy activation in A549 cells treated with D39 in dose- and time-dependent manners. Similar to the results in mice, we also observed a gradually increased expression of LC3-II in A549 cells from 1 to 6 hpi of D39, while SQSTM1/p62, another marker of autophagy (SQSTM1 could be specifically degraded by autophagy), showed a dose- and time-dependent decrease (, Figure S1D). D39 infections of A549 cells under a moi of 50 at 4 hpi induced the strongest expression of LC3-II (). To further decipher the cellular basis of D39-induced autophagy related to the autophagy-lysosomal pathway, we employed a lysosomal autophagy inhibitor chloroquine (CQ) and monitored the alterations of LC3-II and SQSTM1 in A549 cells. As expected, CQ further increased D39-induced protein expression of LC3‐II with the accumulation of cargo protein SQSTM1, suggesting that the autophagic flux was stimulated by S. pneumoniae, and D39-induced LC3-II accumulation in A549 cells was associated with autophagosomes (). Morphologically, autophagy in mammalian cells is initiated from phagophores which undergo expansion and eventually seal to form an autophagosome. This autophagosome then fuses with a lysosome or vacuole resulting in the degradation of autophagy cargoes [Citation32]. With transmission electron microscope (TEM), we also observed that S. pneumoniae infections in A549 cells resulted in the formation of membrane-like vesicles including autophagosome and autolysosome (). Moreover, we visualized the S. pneumoniae-induced autophagy activation in A549 cells by mRFP-eGFP tandem fluorescence-tagged LC3 (ptf-LC3) followed by confocal microscopy. As expected, D39 infections at 4 h led to a marked increase in the number of autolysosomes, which was comparable to the treatment of rapamycin (Rapa), a classic autophagy activator (). We also showed that the mCherry-positive LC3 could colocalize with the lysosomal protein LAMP1 (lysosomal associated membrane protein 1) ().
Figure 2. S. pneumoniae infections induced autophagy in A549 cells. (A and B) WB analysis of the LC3 and SQSTM1 protein expressions in A549 cells in response to the infection of 50 moi of D39 at the respective time points (1, 2, 3, 4, 5, 6, 8, and 10 hpi). (C) A549 cells were infected with D39 (moi of 50 at 4 hpi) in the presence or absence of CQ (50 μM, 3 h) before LC3 and SQSTM1 WB analysis. (D-F) A549 cells were infected by 50 moi of D39 for 4 h, followed by fixation and imaging preparation. (D) the visualization of the ultrastructure of A549 cells via TEM. In the images, membrane-like vesicles in D39-infected cells were observed. Arrows indicated membrane-like autophagosomes and autolysosomes. Scale bars: 2 μm, 500 nm. (E) immunofluorescence microscopy visualization of LC3 puncta in D39-infected A549 cells. Confocal images of ptfLC3-transfected A549 cells were infected with D39 or treated for 12 h with 100 nM rapa. Scale bars: 10 μm. (F) images of mCherry-LC3 fluorescent dots (red), FITC-LAMP1 fluorescence (green), and the overlay with DAPI (blue), which shows colocalization of mCherry- and FITC-positive puncta. The rapa-treated group was set as the positive control. Scale bars: 10 μm.
Notably, we also found that Streptococcus suis (S. suis), an important zoonotic pathogen [Citation33,Citation34], could also induce LC3-II upregulation in porcine alveolar epithelial cell SJPL (Fig. S1E) and A549 cells (Fig. S1F), suggesting that autophagy was possibly a general phenomenon under genus Streptococcus infections. Previous studies have reported that S. pneumoniae can secrete multiple virulence factors (also called effector proteins) to interfere with the host immune system [Citation35,Citation36]. We therefore investigated the differences among live S. pneumoniae D39, heat-killed S. pneumoniae D39, or concentrated bacterial cultural supernatant fluid (SNF) in inducing autophagy of A549 cells. We found that SNF of D39 could induce an equivalent increase of LC3-II compared to the live bacteria, while heat-killed bacteria induced a weak change of LC3-II (Fig. S1G). Overall, these results indicated that S. pneumoniae could induce autophagy in human alveolar epithelial cells, which might be a result of secreted bacterial components.
S. pneumoniae infections induced autophagosomal degradation of TJ protein OCLN in A549 cells
The above results showed that S. pneumoniae infections in mice and A549 cells significantly decreased the protein level of OCLN (). However, we did not observe the changes in OCLN at the mRNA level by qPCR analysis (Fig. S2A). Previous studies have reported that autophagy can degrade cellular components after infection by pathogenic bacteria [Citation27]. We reasoned that S. pneumoniae could induce the degradation of OCLN protein by autophagy activation. To this end, we detected the level of OCLN in the presence or absence of lysosomal autophagy inhibitor CQ. We found that blocking autophagy by CQ significantly reversed the reduction of OCLN protein expression caused by D39 (), while CQ did not affect the mRNA level of OCLN (Fig. S2A). To further investigate the involvement of S. pneumoniae-induced autophagy in the degradation of OCLN, we generated ATG16L1 (autophagy-related 16 like 1) knockout (KO) A549 cells by CRISPR-Cas9 technology and BECN1 knockdown (KD) A549 cells by RNA interference (RNAi). Both ATG16L1 and BECN1 play crucial roles in phagophore and autophagosome formation [Citation37–40]. Consistent with the inhibitor treatment assay (), we observed that the degradation of OCLN induced by D39 infections was largely prevented in both ATG16L1 KO (Fig. S2B) and BECN1 KD () cells, as compared to wild-type (WT) A549 cells. Besides, we showed a significant colocalization between OCLN and LAMP1 in A549 cells under D39 infections (). These results suggested that S. pneumoniae could induce autophagy-dependent degradation of OCLN. We also examined whether S. pneumoniae-induced autophagy participated in degrading other components of TJs, such as TJP1, CDH1, and CLDN1 (claudin 1), in the presence or absence of CQ. We found that CQ could not affect the protein expression of these proteins in A549 cells after S. pneumoniae infections (Fig. S2C).
Figure 3. S. pneumoniae infections induced autophagosomal degradation of TJ protein OCLN in A549 cells. (A) after the D39 infections, the total OCLN in the A549 cells was quantified by WB analyses. Blockage of autophagy by CQ caused an accumulation of OCLN in A549 cells. (B) after the knockdown of BECN1 in A549 cells, the autophagy activation and OCLN degradation were relieved under S. pneumoniae infections. (C) immunofluorescence microscopy visualization of the colocalization of Cy3-OCLN fluorescence (red) and the FITC-LAMP1 fluorescence (green) in D39-infected A549 cells. Scale bars: 10 μm. (D) schematic overview of autophagosome targeting and labeling using TurboID N-terminally fused to HsLC3B. A: autophagosome; N: nucleus. (E) A549 cells expressing HsLC3B-eGFP-MYC-TurboID were grown in the presence of DOX (12 h), BafA1 (2 h), D39 (4 h), biotin (1 h) and ATP (1 h) followed by fixation and immunolabeling with biotin-546, the colocalization of HsLC3B-eGFP-MYC-TurboID chimeras with biotinylated molecules were observed. Scale bars, 5 μm. (F) homogenates from HsLC3B-eGFP-MYC-TurboID chimera expressing A549 cells grown in the presence of DOX (12 h), BafA1 (2 h), D39 (4 h), biotin (1 h) and ATP (1 h) were left untreated or incubated with protK, Triton X-100, or both followed by immunoblotting. (G and H) lysates from the above protK-treated homogenates were immunoprecipitated with streptavidin magnetic beads, and the whole cell lysates (H: input) and beads-bound proteins from protK-protected proteins (G: IP) were respectively analyzed by immunoblotting. (I and J) A549 cells transiently expressing eGFP-SQSTM1, eGFP-LC3B, or eGFP-empty infected with D39 for 4 h were subjected to IP and assayed using an anti-eGFP antibody and protein A+G agarose. The total proteins (J: input) and bound proteins (I: IP) were analyzed by immunoblotting.
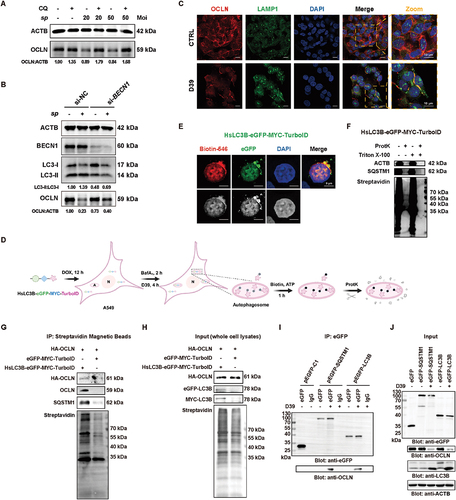
Human LC3 is considered a homolog of yeast Atg8 and is proposed to be a key constituent of the autophagosomal membrane [Citation41]. To further confirm that S. pneumoniae-induced autophagy mediated the degradation of OCLN, we attempted to directly identify OCLN protein inside the autophagosomes by a TurboID-based proximity labeling assay that was based on autophagosomal targeting of TurboID, a biotin ligase able to biotinylated the proximal proteins through fusion with human LC3B (HsLC3B) [Citation42,Citation43]. To this end, we generated a panel of stable A549 cells (doxycycline-inducible) expressing N-terminal eGFP-MYC-TurboID fused with HsLC3B (HsLC3B-eGFP-MYC-TurboID) or not (eGFP-MYC-TurboID as control cells) (). Theoretically, the fusion protein of HsLC3B-eGFP-MYC-TurboID would localize to the lumen of autophagosome under S. pneumoniae infections since HsLC3B undergoes lipidation and localizes to the phagophore (and subsequently the autophagosome) membranes during autophagy, while eGFP-MYC-TurboID would distribute diffusely in A549 cells. By these cells, we first verified that HsLC3B-eGFP-MYC-TurboID chimera could colocalize with biotinylated proteins () and the lysosomal marker LAMP1 (Fig. S2D), respectively. We further performed a proteinase K (protK) protection assay. Briefly, HsLC3B-TurboID-expressing cells were grown in the presence of bafilomycin A1 (BafA1) and doxycycline (DOX) before S. pneumoniae infections. Then, the cells were incubated with 100 μM biotin and 1 mM ATP for 1 h to enable ligase-catalyzed proximity labeling in each cell (). Subsequently, homogenates were left untreated or incubated with protK to effectively degrade cytosol-assessable proteins, Triton X-100 to lyse protective membrane compartments, or a combination of both treatments to digest all proteins within these homogenates. The results of the protK protection assay indicated that in contrast to ACTB/β-actin, a fraction of the lipidated form of SQSTM1 and several biotinylated proteins were protected from protK digestion (). Overall, these results indicated that TurboID fused with HsLC3B could indeed be targeted to the lumen of autolysosomes and readily biotinylated proteins engulfed in these autophagosomes. Next, we combined TurboID-based proximity labeling with streptavidin affinity-isolation and western blotting to identify the biotinylated proteins enclosed in autophagosomes. As shown in , we successfully identified the biotinylated OCLN in HsLC3B-eGFP-MYC-TurboID- but not eGFP-MYC-TurboID-expressing A549 cells after S. pneumoniae infections.
Through immunoprecipitation (IP) coupled with western blot (WB) assays, we found that OCLN could directly interact with SQSTM1 and LC3B in A549 cells (). Obviously, in D39-infected A549 cells, the expression of LC3-II was increased, while that of OCLN was decreased (). In A549 cells over-expressing eGFP-tagged SQSTM1 or LC3B, eGFP-SQSTM1-OCLN, and eGFP-LC3B-OCLN interactions were detected during D39 infections (). Collectively, these results indicated that S. pneumoniae infections could result in the lysosomal degradation of epithelial OCLN protein in an autophagy-dependent manner in A549 cells.
Isolation and characterization of pEVs released by S. pneumoniae
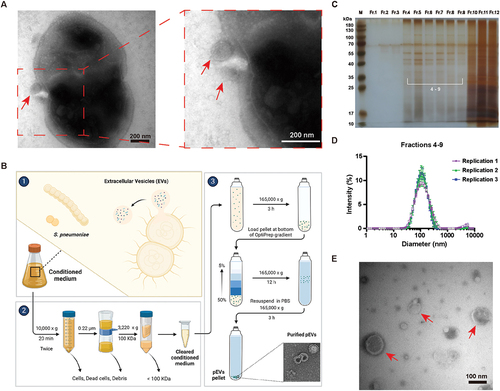
Our previous results showed that concentrated cultural SNF of S. pneumoniae could induce autophagy activation in A549 cells (Fig. S1G). Additionally, it has been suggested that bacteria can actively form OMVs or EVs as transport vehicles to deliver virulence factors to host cells [Citation11,Citation44]. Therefore, we speculated whether S. pneumoniae relied on EVs to deliver some kind of virulence factor involved in the activation of autophagy. To test this hypothesis, we first employed TEM to visualize the morphology of D39 cells and pEVs, and indeed observed the membrane-bound vesicular structures that were releasing from the S. pneumoniae cell surface (). Next, we attempted to isolate and purify these pEVs according to the procedures shown in [Citation15,Citation45]. Briefly, we collected the culture supernatants of D39 bacteria cells when the cellular optical density at 600 nm (OD600) reached 0.9, and then the molecules > 100 kDa were concentrated. After that, the crude extracts of pEVs were pelleted by ultracentrifugation. We used TEM to examine whether the crude extracts contained pEVs. The results suggested that there were numerous vesicular-like structures of varying sizes in these crude extracts (Fig. S3A). To remove non-membranous proteins, protein aggregates, and denatured pEVs, we subjected the crude extracts to Optiprep-based density gradient centrifugation and got different fractions (). Consecutive Optiprep fractions were then analyzed by SDS-PAGE and silver-staining. We found that fractions 1–3, fractions 4–9, and fractions 10–12 revealed different protein banding patterns, suggesting distinct protein contents in these fractions (). Given the similar protein banding pattern, we pooled the fractions 1–3, 4–9, and 10–12, respectively, and subjected the pooled contents to ultracentrifugation and analysis by TEM and Malvern particle size analyzer. We found that pEVs were only observed in fractions 4–9 with diameters ranging from 30 to 300 nm, but not in fractions 1–3 and fractions 10–12, indicating that pEVs were distributed in fractions containing 20–40% Optiprep (). Thus, our data suggested that S. pneumoniae could produce pEVs that possibly represented a potential transport vehicle for delivering bacterial proteins to host cells.
Figure 4. Isolation and characterization of pEvs released by S. pneumoniae cultured in THYE medium. (A) electron micrograph of vesicular structures budding from S. pneumoniae strain D39 (left) and the same vesicles in higher magnification (right). The red arrowheads pointed out the released pEvs from the cell wall. Scale bars: 200 nm. (B) schematic overview of the procedures for isolating, purifying, and identifying pEvs from S. pneumoniae strain D39. (C) pEvs were purified by density gradient ultracentrifugation (optiprep), and then, the separated fractions were visualized by silver-stained SDS-PAGE. (D and E) fractions 4–9 were pooled; OptiPrep was removed by ultracentrifugation, and the samples were determined and imaged by Zetasizer (D) and TEM (E) to observe the morphology and size distribution of pEvs from S. pneumoniae strain D39. pEvs were not visualized in fractions 10–12. Scale bars: 100 nm.
pEVs were internalized into A549 cells to activate autophagy and disrupt the integrity of AEB
Previous studies have reported that bacteria-released pEVs can inherit pathogen-associated molecular patterns of bacteria, and can be effectively recognized and internalized by host cells via endocytosis [Citation11,Citation46]. Alveolar epithelial cells, the first line of defense against pathogens, play a critical role in preventing bacteria from entering the bloodstream [Citation47,Citation48]. We thus wondered whether S. pneumoniae-secreted pEVs could be internalized into alveolar epithelial cells. To prove this, we used two fluorescent lipophilic dyes, PKH67 and 3,3′-dioctadecylox-acarbocyanine perchlorate (DiO) to label the pEVs respectively, and subsequently co-cultured them with A549 cells to trace the internalization of pEVs. Confocal microscopy analysis indicated that after 4, 8, and 12 h after incubation of A549 cells with labeled pEVs, nearly all the A549 cells were positive for PKH67 fluorescence within the cells (), suggesting that the pEVs were internalized by A549 cells. Quantitative analysis by microplate reader also confirmed that S. pneumoniae-secreted pEVs could be efficiently uptake by A549 cells ().
Figure 5. pEvs were internalized into A549 cells to activate autophagy and disrupt the integrity of AEB. (A) detection of intracellular pKH67-labeled (green) pEvs in A549 cells with zeiss LSM800 confocal laser scanning microscope at the indicated time points after exposure to 100 μg/mL pEvs. Scale bars: 20 μm. (B) fluorescence intensity analysis of intracellular DiO-labeled pEvs (excitation: 484 nm, emission: 501 nm); DiO-treated PBS was used in the mock group. (C) TEER changes of A549 monolayer in the treatment of multiple dosages of pEvs or PBS (mock) monitored by the ECIS system. All the data was collected and presented as mean ± SD from three replicated wells at each time point. (D and F) WB analysis of the LC3 and OCLN protein expressions in A549 cells in response to multiple dosages of pEvs. (E and G) A549 cells were incubated with pEvs (100 μg/mL) in the presence or absence of CQ (50 μM, 3 h) before WB analysis of LC3 (E) and OCLN (G).
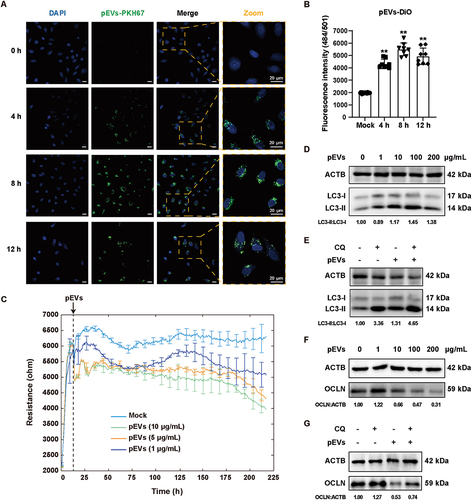
Next, we wondered how would the internalized pEVs affect the A549 cells. We incubated the monolayer A549 cells with pEVs and analyzed the integrity and permeability of the epithelial layer by TEER analysis. As shown in , we found that continuous treatment with 1, 5, and 10 μg/mL pEVs significantly decreased the TEER of the A549 monolayer, suggesting the internalized pEVs could disrupt the epithelial cell-cell junctions and increase its permeability. Notably, we also found that the treatments of pEVs (1, 10, 100, and 200 μg/mL) could markedly increase the ratio of LC3-II:LC3-I and decrease the protein levels of OCLN and several TJ proteins in a dose-dependent manner (, and Figure S3B). Besides, blockage of autophagy by CQ in pEVs-treated A549 cells resulted in an accumulation of LC3-II and reversed expression of OCLN () but not the other TJ proteins (Figure S3C). Overall, these results indicated that S. pneumoniae-secreted pEVs could be efficiently internalized by alveolar epithelial cells, which could disrupt the integrity and increase the permeability of the alveolar epithelial barrier via inducing autophagy activation and OCLN degradation.
pEVs-derived StkP activated autophagy by inducing BECN1 phosphorylation and resulted in autophagic degradation of epithelial OCLN
Next, we wanted to know which components in pEVs were essential in inducing autophagy in A549 cells. We profiled the cargo proteins in purified pEVs by 4D label-free mass spectrometry. We identified a total of 605 proteins shared by three biological replicates (Supplementary material 1), and the top 50 proteins were shown (). We conducted a comprehensive literature survey on the 50 cargo proteins of pEVs and picked eight proteins that were most related to bacterial virulence for further analysis. The selected eight pEVs-associated proteins were expressed individually as eGFP-tagged fusion proteins in A549 cells, and the cytotoxicity was examined. To our surprise, the expression of all of these eight proteins could exert cytotoxic effects in A549 cells to varying degrees (Fig. S3D), suggesting that these proteins were potential virulence factors of S. pneumoniae. Interestingly, we noticed that an S. pneumoniae eSTKs protein StkP could maximally increase the protein level of LC3-II in A549 cells and also markedly decrease the protein level of OCLN, although the other seven pEVs-associated proteins, such as Gap and Eno, could also obviously decrease the OCLN level (). This data suggested that StkP possibly represented the most promising candidate, and we therefore chose this StkP for further experimental verification.
Figure 6. pEvs-derived StkP activated autophagy by inducing BECN1 phosphorylation and resulted in autophagic degradation of epithelial OCLN in A549 cells. (A) purified pEvs from three independent biological replicates were analyzed by LC/MS. A total of 605 proteins were common to all three groups. The top 50 proteins ranked by average intensity (4D label-free) were listed. (B) lysates from A549 cells transiently expressing eGFP-tagged pEvs cargo proteins or eGFP-empty were subjected to SDS-PAGE and analyzed by immunoblotting using antibodies against LC3, eGFP, OCLN, or ACTB. (C) microscopy visualization of mCherry-LC3 fluorescent dots (red), LC3 puncta in eGFP-StkP- or eGFP-expressing A549 cells. Scale bar: 15 μm. (D) immunostaining of Cy3-OCLN (red) in A549 cells instantly expressing eGFP-StkP or eGFP alone. Scale bars: 20 μm. (E and F) lysates from A549 cells transiently expressing eGFP-StkP or eGFP and HA-BECN1 were performed IP with an anti-eGFP antibody and protein A+G agarose (E, upper two panels) or only an anti-HA magnetic beads (E, lower two panels). The total proteins (F: input) and respective bound proteins were analyzed by immunoblotting (E: IP). (G) WB detection of BECN1 phosphorylation (Ser93 and Ser96) level in A549 cells overexpressing eGFP or eGFP-stkP. (H) after overexpressing StkP in A549 cells, LC3 and OCLN were quantified by WB analyses. Blockage of autophagy by CQ caused increased expression of both LC3-II and OCLN. (I) WT BECN1 or BECN1S93,96A were overexpressed in BECN1 knockdown A549 cells (by shRNA), and western blotting analysis of OCLN expression was performed in WT BECN1 or BECN1S93,96A mutated A549 cells following introduction with eGFP or eGFP-StkP.
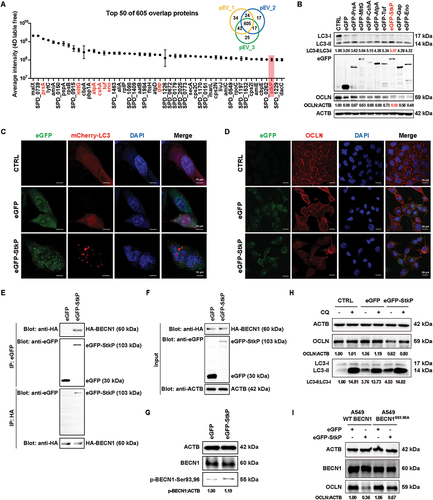
Furthermore, we visualized autophagy by mCherry-LC3 and eGFP-StkP transfection in A549 cells. Compared to CTRL (transfecting mCherry-LC3 alone) and eGFP (transfecting mCherry-LC3 and eGFP) groups, intracellular expression of eGFP-StkP induced an obvious increase in mCherry-LC3 protein puncta aggregation (). We further observed that eGFP-StkP expression resulted in the decrease of OCLN protein (), which was consistent with our previous results that either S. pneumoniae infections or pEVs incubation in A549 cells induced autophagosomal degradation of OCLN (). Then, we wanted to probe how StkP activated autophagy in A549 cells. BECN1 is a fundamental positive regulator of autophagy initiation, which depends on the phosphorylation of BECN1 [Citation49]. Previous studies have reported that multiple serine-threonine kinases, such as DAPK, ULK, and AMPK, could phosphorylate BECN1 to regulate the occurrence of autophagy [Citation50,Citation51]. Given that StkP functioned as a serine-threonine protein kinase [Citation52], we therefore decided to verify whether StkP could phosphorylate BECN1 to initiate autophagy in A549 cells. By co-expressing hemagglutinin-tagged BECN1 (HA-BECN1) and eGFP-StkP in A549 cells, we confirmed the protein-protein interaction between StkP and BECN1 by co-IP assay and western blotting analysis (). Adenosine 5’-triphosphate (ATP) is the main source of cellular energy and disruptions to ATP homeostasis have been linked to autophagy [Citation53]. We investigated the intracellular ATP consumption in A549 cells transfected with eGFP-StkP and HA-BECN1 either alone or in combination. Our findings revealed that transfection of eGFP-StkP alone, but not eGFP or HA-BECN1, was able to significantly decrease the intracellular ATP level, while transfection of eGFP-StkP and HA-BECN1 together resulted in an even greater decrease in ATP levels (Fig. S4A), possibly due to StkP-induced phosphorylation of BECN1. In addition, we analyzed the phosphorylation levels of BECN1 in A549 cells using western blotting and found that transfection of eGFP-StkP in A549 cells increased the level of BECN1 phosphorylation (Ser93 and Ser96) (). Besides, blocking autophagy with CQ in StkP-overexpressed A549 cells led to the accumulation of LC3-II and reversed the expression of OCLN (). Finally, overexpression of StkP increased the level of LC3-II in A549 cells with WT BECN1, but had a much-reduced effect in cells with the BECN1 mutant (Ser93,96Ala), and a similar result was observed with subsequent OCLN degradation (Fig. S4B and ). Overall, these results indicated that S. pneumoniae-secreted protein StkP derived from pEVs could initiate autophagy by phosphorylating the Ser93 and Ser96 sites and consequently result in autophagic degradation of TJ protein OCLN in alveolar epithelial cells.
StkP-laden pEVs contributed to S. pneumoniae virulence and bacterial dissemination by disrupting AEB integrity
To investigate the role of StkP in S. pneumoniae-caused bacterial pneumonia in mice, an stkP-deficient mutant strain (D39-ΔstkP) and a complemented strain (D39-C-ΔstkP) were constructed and verified (Fig. S5A-L). The wild-type D39 (D39-WT) and D39-ΔstkP strains exhibited similar growth in the THYE medium (Fig. S6A-B), suggesting that stkP deficiency would not impair bacterial viability in vitro. Firstly, to assess the in vivo role of the autophagy inhibitor CQ in S. pneumoniae-induced pathogenicity, an intranasal infection of S. pneumoniae was performed in mice. Briefly, mice were pre-treated with CQ (50 mg/kg) via intraperitoneal injection before S. pneumoniae infections of 48 h, 24 h, and 2 h, respectively. The results indicated that CQ treatment improved the survival rate of S. pneumoniae-infected mice (Fig. S7A), reduced the bacterial load in lung tissues and blood (Fig. S7B), and alleviated lung injury (Fig. S7C). In a mouse model of acute pulmonary infection induced by D39-WT, D39-ΔstkP, or D39-C-ΔstkP, we showed that the stkP deficiency completely protected the mice from death compared to the D39-WT and D39-C-ΔstkP groups. Mice infected with the D39-ΔstkP strain showed 100% survival within 96 hpi (). Bacterial loads in the lung tissues were markedly decreased in D39-ΔstkP-infected mice compared to D39-WT-infected mice at 24 hpi, and the bacterial load in the blood of D39-ΔstkP-infected mice was even undetectable (). Besides, the lung injuries of D39-ΔstkP-infected mice were significantly relieved compared to D39-WT-infected mice, and there were no significant differences between D39-WT and D39-C-ΔstkP groups (). These results suggested that stkP played a vital role in the lethality and pathological breakdown of lung tissues in mice during S. pneumoniae infections.
Figure 7. StkP-laden pEvs contributed to S. pneumoniae virulence and bacterial dissemination by disrupting AEB integrity. (A) Balb/c mice were intranasally challenged with D39-WT, D39-ΔstkP, and D39-C-ΔstkP at 5 × 108 CFU in 40 μL PBS; bacterial infected mice were continuously observed to obtain survival data (n = 10). (B) bacterial burdens in the lung tissues and blood were determined at 24 hpi (n = 5). (C) Representative histological views of the lung tissues of mice at 24 hpi by H&E staining. Scale bars: 100 μm. (D) WB analysis of the lung lysates at 24 hpi for indicated antibodies: BECN1, p-BECN1-Ser93,Ser96, LC3, and OCLN. (E) the expression and localization of LC3 and OCLN were observed in the mice’s lung tissues by immunofluorescent staining. Scale bars: 20 μm. Arrows indicated colocalization of increased LC3 and OCLN in lung tissues. (F) TEER changes of A549 monolayer incubated by 100 μg/mL of pEvs derived from D39-WT, D39-ΔstkP, and D39-C-ΔstkP respectively monitored by the ECIS system. All the data was collected and presented as mean ± SD from six replicated wells at each time point. (G) WB analysis of indicated protein in A549 cells treated by pEvs from D39-WT, D39-ΔstkP, and D39-C-ΔstkP, respectively. (H) OCLN integrity observation in different pEvs-treated A549 cells by confocal laser microscopy. Scale bars: 20 μm. (I) confocal imaging of LC3 puncta after incubation with 100 μg/mL of pEvs derived from D39-WT, D39-ΔstkP, or D39-C-ΔstkP, respectively. Scale bars: 10 μm.
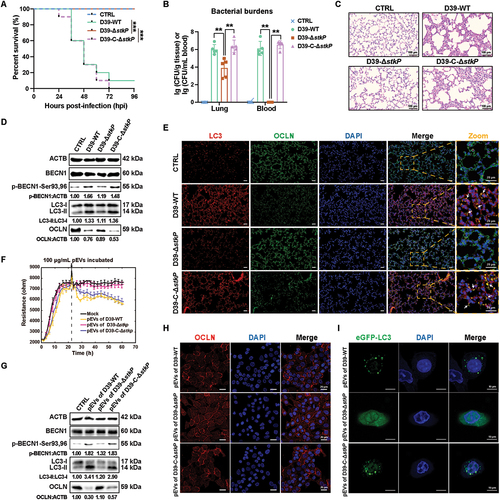
To further confirm the role of stkP in epithelial autophagy in vivo, we examined the changes in BECN1 phosphorylation, LC3-II, and OCLN protein levels in mice infected with D39-WT, D39-ΔstkP, or D39-C-ΔstkP. It was revealed that stkP deficiency significantly reduced both BECN1 phosphorylation level (p-BECN1-Ser93,Ser96) and LC3-II protein level and increased the OCLN protein level in lung tissues of D39-ΔstkP-infected mice in comparison with D39-WT- or D39-C-ΔstkP-challenged mice (). Similarly, through immunofluorescence analysis, we also observed that D39-WT or D39-C-ΔstkP infections could remarkably increase the LC3 level and meantime decrease the OCLN level in lung tissues of mice when compared with the PBS-treated mice, while stkP deficiency could largely reverse these changes (). Interestingly, we observed a higher degree of colocalization between OCLN and LC3 in the lung tissues of mice infected with both D39-WT and D39-C-ΔstkP, as compared to mice infected with D39-ΔstkP (). These results suggested that stkP-induced autophagy contributed to OCLN degradation and alveolar barrier disruption under S. pneumoniae infections.
Then, we employed the A549 monolayer model to simulate the AEB and explored the role of pEVs-derived StkP in affecting the integrity of the epithelial barrier. As was shown in , treatment of A549 monolayer with the pEVs from both D39-WT and D39-C-ΔstkP led to rapid decreases over time in TEER values over time, compared to the PBS-treated Mock group, while pEVs from ΔstkP mutant exhibited a similar trend as the Mock group. Western blotting analysis showed that the incubation of A549 cells with 100 μg/mL pEVs from D39-WT and D39-C-ΔstkP but not D39-ΔstkP for 8 h induced the increases in p-BECN1-Ser93,Ser96 and LC3-II levels and a decrease in OCLN level when compared to PBS control (). Additionally, confocal microscopy analysis also confirmed that treatment of A549 cells with pEVs from D39-WT and D39-C-ΔstkP induced a formation of LC3 puncta and the reduction of OCLN integrity (). In contrast, treatment with pEVs from D39-ΔstkP preserved the OCLN integrity and did not affect the autophagy activity (). Together, these observations suggested that StkP-laden pEVs increased the permeability of AEB through the degradation of TJ protein OCLN, which facilitated bacterial penetration of the alveolar epithelial barrier and dissemination to the bloodstream and promoted the development of pneumonia.
Discussion
S. pneumoniae is an extracellular pathogen, yet its components can be detected intracellularly for the host cells to recognize and respond to infections [Citation54,Citation55]. Our study revealed that S. pneumoniae D39 released StkP-containing pEVs which were able to enter host cells and disrupt the alveolar epithelial barrier integrity by inducing autophagic degradation of OCLN. Through the TurboID-based proximity labeling and IP analysis, we determined that OCLN protein was a critical target of S. pneumoniae-induced autophagy in A549 cells. In detail, intracellular StkP, delivered by pEVs, could interact with BECN1 and enhance its phosphorylation to induce autophagy activation and subsequent dysfunction of the epithelial barrier. Deletion of stkP significantly reduced the AEB disruption caused by S. pneumoniae or S. pneumoniae-derived pEVs. In conclusion, our findings highlighted the significant role of StkP-laden pEVs in the pathogenicity of S. pneumoniae and provided potential insights for the prevention of S. pneumoniae.
EVs formed by several gram-positive bacteria have been found to be essential for the delivery of virulence factors to host cells, which result in various effects during the infection process. For instance, Bacillus subtilis can secrete EVs containing Sublancin immunity protein SunI which is necessary for bacterial survival [Citation56]. Moreover, EVs have been shown to contribute to bacterial pathogenicity by disrupting host barriers and allowing pathogen invasion and dissemination [Citation57]. In the case of S. suis-secreted EVs, 9 out of 46 proteins (19.57%) have been proven or suspected to be virulence factors [Citation58]. These EVs possess intense DNase activity that can degrade neutrophil extracellular traps (NETs) and elicit pro-inflammatory effects on monocytes and macrophages [Citation58]. Furthermore, EVs produced by Group B Streptococcus are able to cause the disruption of amniotic epithelium and the infiltration of leukocytes in the chorion-decidua, leading to fetal demise and preterm birth [Citation59]. A bacterial protein (M protein) delivered by Streptococcus equi EVs has been shown to disrupt the murine BBB [Citation11]. S. pneumoniae-derived EVs (pEVs) can recruit immune cells and induce cytokine production in the lower airways, which contribute to the disease symptoms observed in S. pneumoniae infections [Citation15,Citation60]. The epithelial cells and various immune cells such as neutrophils, monocyte-derived dendritic cells, B cells, T cells, and macrophages have been demonstrated to internalize the pEVs [Citation60,Citation61]. The internalized pEVs can deliver immunomodulatory components into host cells, resulting in pro-inflammatory responses and tissue damage in mammalian hosts [Citation15,Citation61]. Another study has reported that pEVs are rapidly internalized by immune cells, leading to altered cytokine release [Citation62]. These findings indicate that the pro-inflammatory responses triggered by pEVs are important for pathological mechanisms that disrupt biological barriers. Our study focused on the direct effects of pEVs on the disruption of AEB integrity. We found that the majority of pEVs-derived cargo proteins were cytoplasmic proteins and lipoproteins, which was similar to previous reports [Citation15,Citation63]. Some classical virulence factors of bacteria, including PrsA, LytC, PspA, CbpA, eno, and CbpE (ranked in the top 50 of the most abundant proteins), were present in pEVs. We also found that pEVs could be internalized by A549 cells and hijack host autophagy to disrupt the integrity of the alveolar epithelial barrier.
Autophagy, a lysosomal-dependent cellular process, plays a “double-edged sword” role in bacterial infections [Citation20]. Several studies have suggested that bacterial infections can activate autophagy as a protective immune response to promote intracellular bacterial clearance [Citation27,Citation64]. However, other studies have reported that bacteria can hijack the autophagy process for their survival within host cells [Citation65]. In this study, we found that S. pneumoniae infections induced autophagy with increased LC3-II and LC3 puncta in the lungs of mice and A549 cells, which was consistent with the previous report [Citation24]. Notably, it was the first exploration by immunofluorescent staining of mice lungs to show a correlation between increased LC3 and disrupted OCLN, indicating that autophagy might be activated and responsible for S. pneumoniae-caused lung barrier impairment. The early signaling of bacteria-induced autophagy potentiating is still unclear though it overlaps with starvation-induced autophagy. Previous studies have shown that AMPK-mediated phosphorylation of ULK1 and BECN1 is responsible for the induction of autophagy upon infection with Salmonella and its OMVs [Citation66,Citation67]. In addition, OMVs produced by pathogenic strains of Escherichia coli have been found to block autophagic flux and exacerbate inflammasome activation [Citation68]. Furthermore, S. aureus EVs containing immunostimulatory DNA, RNA, and peptidoglycan have been determined to activate innate immune receptors and induce autophagy in epithelial cells [Citation12]. Intracellular S. pneumoniae is also subjected to selective autophagy to promote bacterial clearance [Citation28,Citation29].
Additionally, we discovered that several bacterial proteins presented in pEVs, such as StkP, Gap, AtpA, and CshA, not only decreased cell viability but also induced the downregulation of OCLN ( and Fig. S3D). This promoted us to explore the role of bacterial components in disrupting the alveolar epithelial barrier. The alveolar-capillary barrier is primarily composed of microvascular endothelial cells, alveolar epithelial cells, and cellular junctions [Citation69,Citation70]. Tight junctions (TJs), and adherens junctions (AJs) are essential in maintaining the integrity of the alveolar epithelial barrier and regulating lung permeability. Disruption of this barrier can lead to the accumulation of inflamed cells and leakage of plasma proteins into the alveolar space, resulting in abnormal gas exchange and exacerbation of symptoms related to organ or tissue hypoxia [Citation71]. Therefore, it is essential to explore new pathways to address epithelial barrier dysfunction. OCLN, a key component of tight junctions, plays an essential role in the functioning of the alveolar epithelial barrier [Citation72,Citation73]. In this study, we investigated the impact of S. pneumoniae-induced alterations of OCLN on the dysfunction of the alveolar epithelial barrier. Previous research has shown that S. pneumoniae-induced lung injury is associated with increased epithelial permeability due to rearrangements in the cellular junction proteins (CDH1, CTNNB1/β-catenin, TJP1, and OCLN), as well as alveolar inflammatory responses [Citation5]. These alternations in cellular junctions may contribute to the breach of the epithelial barrier and the development of severe infections. Consistently, our study also observed a decrease in the expression of several TJ proteins in the lung tissues of S. pneumoniae-infected mice and S. pneumoniae-treated A549 cells.
Autophagy has been reported to regulate the expression of TJs and the function of the epithelial barrier [Citation74]. The degradation of CLDN1 through autophagy can do harm to the respiratory tract epithelial barrier during pathogen infections [Citation23,Citation75]. The alveolar epithelium is pivotal in protecting the lungs from inhaled infectious agents, as evidenced by massive tissue damage exclusively in lung alveoli during bacterial pneumonia in mice [Citation76]. Notably, we found that S. pneumoniae secreted pEVs loaded with StkP were responsible for inducing autophagy and the consequent degradation of OCLN, ultimately leading to AEB damages similar to S. pneumoniae infections. Although the levels of other TJs were also reduced in S. pneumoniae-infected cells, this reduction was not due to autophagic degradation. Instead, it occurred through alternative mechanisms that required further investigation.
eSTKs have been identified in prokaryotes and are involved in the regulation of various bacterial cellular processes, such as gene expression, extracellular polysaccharide production, DNA metabolism, cell division, and virulence [Citation77,Citation78]. S. pneumoniae encodes a single copy of the stkP gene, which encodes an eSTKs membrane protein localized at the division septa of S. pneumoniae cells [Citation79]. Similar to eukaryotic cells [Citation52,Citation80], it is thought that bacterial eSTKs interact with other proteins, thus forming interconnected signaling pathways. StkP has been found to be essential for bacterial virulence, lung infections, and the ability to invade and grow in the bloodstream of intranasally infected mice [Citation79], as confirmed in our study. As known, the alveolar epithelium is one of the first barriers that pathogens must cross to establish an infection. and StkP/ComE crosstalk phosphorylation has been shown to affect the intracellular survival of S. pneumoniae in pneumocytes [Citation81]. Additionally, StkP has been found to regulate the pilus, a virulence factor that is essential for bacterial adhesion, and modulate adherence to human brain microvascular endothelial cells and A549 cells [Citation82]. In general, StkP acts as a global regulator in S. pneumoniae, the mutations of stkP have been found to have pleiotropic effects [Citation83]. It was previously unclear whether bacterial StkP could affect a eukaryotic substrate. In this study, we discovered that autophagy was activated in A549 cells by internalized pEVs containing StkP, so we focused on the functions of intracellular StkP derived from pEVs on autophagy. Our results indicated that StkP interacted with host BECN1 in A549 cells and probably increased the phosphorylation of BECN1 (Ser93 and Ser96) to promote autophagy potentiation. The lack of StkP in pEVs or the mutation of BECN1S93,96A failed to induce autophagy activation and OCLN degradation in A549 cells during the incubation of pEVs.
In addition, among the identified proteins in pEVs, ATP synthase AtpA (ATP synthase subunit alpha) has been reported to promote biofilm persistence and influence innate immunity in S. aureus [Citation84]. In both C. elegans and mammalian cells, inhibition of ATP synthase leads to reduced ATP contents, decreased oxygen consumption, and increased autophagy [Citation85]. However, the influence of S. pneumoniae ATP synthase on host autophagy and subsequent effects on host response remains unexplored. Additionally, another pEVs-associated protein Gap (Glyceraldehyde-3-phosphate dehydrogenase, GAPDH) is a common virulence factor among pathogenic bacteria of the genera Listeria, Mycobacterium, and Streptococcus [Citation86]. It serves as a key enzyme in metabolism and plays an important role in cell autophagy [Citation87]. Further studies are necessary to elucidate the underlying pathway involved in the regulation of autophagy and other related pathways.
In conclusion, our research highlighted pEVs as important cargo vehicles of virulence factors for S. pneumoniae infections. One of the cargo proteins, StkP conferred hyper-virulent property to pEVs of S. pneumoniae, as it could hijack the host cell autophagic responses for the own benefit of S. pneumoniae. This finding suggested the possibility that taking advantage of epithelial autophagy could be beneficial in the clinical therapy of infectious diseases associated with the breakdown of biological barriers.
Materials and methods
Bacterial strains and cell culture
S. pneumoniae D39 was cultured in THYE (THB [Hopebio, HB0311–3] supplemented with 0.5% yeast extract [OXOID, LP0021]) medium or on tryptic soy agar (TSA; BD Bioscience 236,950) plates containing 3% (v:v) sheep blood (Hopebio 1,001,339–1) with 5% carbon dioxide at 37°C. Growth was measured by monitoring the OD600. The concentrations of antibiotics were added as needed: erythromycin (Ery; Solarbio, E8100; 2.5 μg/mL). S. suis SC19 was cultured in TSA plates or TSB medium supplemented with 5% (v:v) bovine serum (Zhejiang Tianhang Biotechnology Co., Ltd., 23022–8615) at 37°C.
The A549 cells (ATCC, CCL-185), A549-ATG16L1 KO cells, A549-shCtrl cells, A549-shBECN1 cells, A549-WT BECN1 cells, A549-BECN1S93,96A cells, A549-HsLC3B-eGFP-MYC-TurboID cells, and A549-eGFP-MYC-TurboID cells (all of these cells were constructed in this study. The primers, shRNA, and sgRNA used in this research were listed in Table S1) were cultured in RPMI-1640 medium (Gibco 31,800,022) supplemented with 10% heat-inactivated fetal bovine serum (SORFA, SX1101), 2 mM L-glutamine, and penicillin and streptomycin (100 units/mL) at 37°C, 95% humidity, and 5% CO2. After confluent monolayer achievement, A549 cells were washed with PBS (Gibco 70,011,044) and starved in serum-free RPMI-1640 medium (Gibco 31,800,022) for 12 h before the experiments. The drugs were used as needed: CQ (MedChemExpress, HY-17589A; 50 μM, 3 h), Rapa (MedChemExpress, HY-10219; 100 nM, 12 h), DOX (MedChemExpress, HY-N0565; 0.5 μg/mL, 12 h), BafA1 (MedChemExpress, HY-100558; 10 nM, 2 h), Biotin (Sangon Biotech, A100340; 100 μM, 1 h), ATP (Sangon Biotech, A600020–0005; 1 mM, 1 h).
Bacterial strain construction
S. pneumoniae D39-ΔstkP was constructed by replacing the entire coding sequence of stkP with an erythromycin-resistance expression cassette (ermR) through homologous recombination. A DNA fragment including stkP upstream flanking, ermR, and stkP downstream flanking was introduced into competent D39 by natural transformation as previously described [Citation88]. Briefly, S. pneumoniae was grown in a THY1 medium (THYE medium supplemented with 4 mM HCl and 0.5% glycine) at 37°C until an OD600 of 0.03 was reached. S. pneumoniae was additionally incubated in THYII medium (THYE +1 mM CaCl2 +10 mM NaOH + 0.2% bovine serum albumin [Biosharp, BS114-500 g] + 1 ng/μL competence-stimulating peptide [Anaspec, AS-63779]) at 37°C for 14 min. Then donor DNA was added, and the transformation mix was incubated at 37°C for 1 h, then diluted 4-fold with THYE medium and incubated at 37°C for another 2 h before plating. The homologous recombination events (D39-ΔstkP) were selected by plating on a medium containing Ery and confirmed by PCR and DNA sequencing. The complemented strain (D39-C-ΔstkP) was constructed by integrating the stkP gene into the chromosome by homologous recombination as described above. The stkP flanked with the upstream and downstream fragment was PCR-amplified and then introduced into the D39-ΔstkP strain. The complemented strain was only grown in TSA blood agar plates without Ery due to the exchange of stkP with ermR. D39-C-ΔstkP strain was further identified by PCR and DNA sequencing. Primers used in this study were provided in Table S1.
Electrical cell-substrate impedance sensing
The ECIS Z-Theta system (Applied BioPhysics, Troy, MI, USA) was used to measure the TEER values in A549 cells seeded on collagen-coated, gold-plate 96W1E+ type electrodes (Applied BioPhysics, B002046). This method was used to monitor the real-time changes in the monolayer cell barrier function as previously described [Citation89]. Bacterial infections or pEVs treatments were conducted when the confluent epithelial monolayers had achieved a stable TEER (around 7000 ohms).
RNA extraction and qRT-PCR
Total RNA was extracted from the samples using TRIzol reagent (Thermo Fisher Scientific 15,596,026) according to the manufacturer’s protocol. 500 ng of the total RNA in each sample was then used for cDNA synthesis (Vazyme, R223–01). qRT-PCR was performed with the real-time PCR thermal cycler QuantStudio 3 (Applied BioSystems, Foster City, CA, USA) using ChamQ Blue Universal SYBR qPCR Master Mix (Vazyme, Q312–02) according to the manufacturers’ recommendations.
Western blot analysis
Cells and homogenized lung tissues were lysed in ice‐cold Cell lysis buffer (Beyotime, P0013) or RIPA buffer (MedChemExpress, HY-K1001) containing protease inhibitor (MedChemExpress, HY-K0010), then the samples with loading buffer were heated to 100°C for 10 min, and then ran in 12% SDS-PAGE gels. The separated proteins were transferred to PVDF Western Blotting Membranes (Merck Millipore, IPVH00010), and incubated with indicated primary antibodies overnight. Afterward, relevant secondary antibodies conjugated to HRP (Biodragon, BF03008, and BF03001) were applied. Finally, the chemiluminescent signals were acquired using a ChemiDoc Imaging system (Bio-Rad, Hercules, CA, USA). The quantitative analysis of bands in western blot was performed by Quantity One (Bio-Rad).
RNA interference and gene transfection
The transfection of shRNA, siRNA, or DNA was performed using jetPRIME® transfection reagent according to the manufacturer’s protocol. Human BECN1 siRNA, GAPDH siRNA, and negative control (NC) siRNA were designed and synthesized by JTS Scientific, the sequence targeting the BECN1 gene was provided in Table S1. The knockdown efficiency was determined at 48 h post-transfection using western blotting analysis. ATG16L1 KO A549 cells were constructed using the Cas9-single guide RNA (sgRNA) method. The sgRNA sequences targeting the human ATG16L1 gene (Table S1) were predicted using the online CRISPR-Cas9 design tool (http://crispr.cos.uni-heidelberg.de) and individually cloned into an all-in-one pYSY-SpCas9-sgRNA-eGFP plasmid (YSY Biotech Co., Ltd., Nanjing, China). Puromycin (5 μg/mL) was used for the selection of transduced cells. To construct stable A549 cells simultaneously expressing shRNA targeting human BECN1 and the rescue gene of wild-type human BECN1 or BECN1 with both Ser93 and Ser96 mutated to alanine (Ser93,96Ala), the shRNA (driven by the U6 promoter, the sequence was provided in Table S1) was firstly cloned into the lentiviral plasmid pSico-U6-EF1α-IRES-eGFP using restriction enzymes HpaI and PsPxI and T4 DNA ligase. Then shRNA-resistant WT human BECN1 or mutated BECN1 of Ser93,96Ala (driven by the EF1A promoter) were PCR-amplified from cDNA of A549 cells and were cloned into a modified lentiviral plasmid pSico (a multiple cloning site [MCS]-IRES2 sequence was inserted on the N terminus of eGFP in pSico [Addgene 11,578; deposited by Tyler Jacks]) using Gibson assembly [Citation90]. Lentivirus was produced by co-transfecting 293T cells with the lentiviral plasmid, pMD2.G (Addgene 12,259; deposited by Didier Trono) and pSpAX2 (Addgene 12,260; deposited by Didier Trono). The supernatant was harvested and concentrated after 48 h and 72 h of transfection. For lentivirus infection of A549 cells, cells were infected with lentivirus at a moi of 5 in the presence of 4 μg/mL polybrene. After 48 h, the eGFP+ cells were sorted using MoFlo Astrios EQ (Beckman Coulter).
Isolation and identification of pEVs
S. pneumoniae were grown in a THYE medium until an OD600 of 0.9 was reached and were pelleted using centrifugation (10,000 × g for 20 min at 4°C). Culture supernatants were filtered (0.22-μm pore size) and concentrated 25-fold with 100 kDa Ultra 15 mL Centrifugal Filters (Merck Millipore, UFC9100). The sedimentation of the vesicles was obtained by ultracentrifuging (165,000 × g for 3 h at 4°C) and resuspended in PBS. Crude vesicle preparations were further purified by centrifugation on a density gradient using OptiPrep medium (Sigma-Aldrich, D1556). Pelleted EVs were adjusted to 50% OptiPrep in a total volume of 2 mL and overlaid with gradient layers of Optiprep ranging from 45 to 5%. Gradients were centrifuged at 165,000 × g for 12 h at 4°C, and then 1 mL of each fraction was subjected to SDS-PAGE and stained with a Fast Silver Stain Kit (Beyotime, P0017s). The fractions 4–9 (containing the pEVs) were pooled, and the Optiprep medium was replaced with PBS via ultracentrifugation (165,000 × g for 3 h at 4°C). Protein concentrations of pEVs samples were determined using a BCA Protein Assay Kit (Biosharp, BL521A). The size distribution and particle diameter data of purified pEVs particles were generated with a Zetasizer Nano ZS device and analyzed by Zetasizer software (Malvern Panalytical, Malvern, Worcestershire, UK).
Proteomic analysis of pEVs by 4D label-free mass spectrometry
To identify the protein components of purified pEVs, the protein samples of pEVs were prepared for liquid chromatography-mass spectrometry (LC/MS) analysis performed mindlessly by PTM Biolabs (Hangzhou, China). Protein samples were washed with ice-cold acetone and diluted with TEAB for digestion. Then trypsin was added in a 1:50 ratio to the protein mass and left to digest overnight. The protein solutions were reduced with DTT for 30 min at 56°C and alkylated with iodoacetamide for 15 min in darkness at room temperature. The tryptic peptides were finally dissolved in a solution of 0.1% formic acid and 2% acetonitrile in water (solvent A) and separated with a gradient from 5% to 25% solvent B (0.1% formic acid in 90% acetonitrile) on an EASY-nLC 1200 UPLC system (ThermoFisher Scientific). The peptides were then subjected to nano-electrospray ionization and analyzed by an Orbitrap Exploris™ 480 (ThermoFisher Scientific) mass spectrometer. Data from the MS/MS analysis were processed using the MaxQuant search engine (v.1.6.15.0) against an S. pneumoniae database (Streptococcus_pneumoniae_serotype_2_strain_D39_373153_PR_20221020.fasta) concatenated with reverse decoy database. The mass tolerance for precursor and fragment ions were set at 20 ppm and 0.02 Da respectively, and variable modifications such as acetylation on protein N-terminal and oxidation on Met were specified.
TEM imaging
For imaging vesicle budding from S. pneumoniae strain D39, the bacteria were cultured to an OD600 of 0.9 ± 0.1, collected by centrifugation at 3,220 × g for 15 min, and resuspended in sterile deionized water. For TEM, samples of bacteria or pEVs were placed on 200-mesh carbon-coated copper grids (Beijing Zhongjingkeyi, BZ11022a) and incubated for 30 min at room temperature. The excess samples were wiped with filter paper, left to dry for 1 h, and then negatively stained with filtered aqueous 2% uranyl acetate for 30 s. The negatively stained excess dye was then blotted dry with filter paper and placed in a vacuum drying oven. Samples were then sent to the Electron Microscope Core of Huazhong Agricultural University for further treatment and observation with TEM at an accelerating voltage of 100 kV (HT7650, HITACHI, Tokyo, Japan).
For A549 cells ultrastructure visualization, cells were cultured on 6-well plates at a density of 1.5 × 106 cells/well one day before the experiment. After the D39 infections, cells were resuspended in an electron microscope fixative (Wuhan Pinuofei, PN0018). After this primary fixation, the cells were secondarily fixed with 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 2 h at room temperature. Then, cells were rinsed thrice in a phosphate buffer for 15 min each time. The samples were then dehydrated in a graded ethanol series and embedded in an acetone/812 embedding agent (Structure Probe, Inc., 90529-77-4). After 48 h of polymerization at 60°C, ultrathin sections (80 nm thick) were mounted on 200-mesh carbon-coated copper grids. Finally, sections were stained with 2% uranyl acetate and lead citrate by sequence. Grids were examined with a TEM at 100 kV.
Uptake experiments of pEVs
For uptake experiments of pEVs, D39-derived pEVs were labeled with the PKH67 Green Fluorescent Cell Linker Kit (Sigma Aldrich, PKH67GL): pEVs diluted in PBS were added to 1 ml Diluent C. Then the 30-kDa Ultra Centrifugal Filters (Merck Millipore, UFC9030) was used to remove the unbound dye. The labeled pEVs were filtered with a 0.45 μm filter and then used for uptake experiments. To observe the localization of pEVs in A549 cells, PKH67-labeled pEVs (100 μg/mL) were used to treat A549 cells for 4, 8, and 12 h. Samples were washed and stained for 5 min with DAPI before imaging with a Zeiss LSM800 confocal laser scanning microscope (Carl Zeiss AG, Oberkochen, BW, Germany). Additionally, we performed the labeling of pEVs with DiO (20 μg/mL, Beyotime, C1038) for 30 min at 37°C. DiO-labeled pEVs were treated as described above and then used to treat A549 cells. At indicated time points, the fluorescence intensity of the intracellular DiO signal was measured (excitation: 484 nm, emission: 501 nm) by a fluorescence microplate reader (Perkin-Elmer, Waltham, MA, USA).
Immunofluorescence microscopy
A549 cells were cultured as a monolayer on a confocal dish (NEST 801,002), pretreated with drugs, or transfected with plasmids before S. pneumoniae infections. After the infection, the cells were washed three times with pre-cooled PBS, fixed with 4% paraformaldehyde (Biosharp, BL539A) for 15 min, then incubated with blocking buffer (1× PBS, 5% normal goat serum [Solarbio, SL038], 0.3% Triton X-100 [Solarbio, IT9100]) at 37°C for 1 h. The blocked samples were incubated with the primary antibody as needed (OCLN/occludin, Cell Signaling Technology, 91131S, rAb, 1:200; LAMP1, abcam, ab289548, mAb. 1:100; Biotin-546, Santa Cruz Biotechnology, sc -101,339 AF546, 1:50) overnight at 4°C. The next day, the unbound primary antibody was washed with PBS. The cells were incubated with CoraLite488-conjugated Goat Anti-Mouse IgG(H+L) (Proteintech, SA00013–1; 1:200), CoraLite488-conjugated Goat Anti-Rabbit IgG(H+L) (Proteintech, SA00013–2; 1:200), Cy3-conjugated Affinipure Goat Anti-Mouse IgG(H+L) (Proteintech, SA00009–1; 1:100), or Cy3-conjugated Affinipure Goat Anti-Rabbit IgG(H+L) (Proteintech, SA00009–2; 1:100) as secondary antibodies, at room temperature for 1 h. Unbound secondary antibodies were washed with PBS, and the nuclei were stained with DAPI (Beyotime, P0131-25 ml). The A549 samples were observed and photographed under the confocal microscope (Zeiss LSM800).
IP assays
For , A549 cells cultured in a cell culture dish (100 mm) were transfected with pEGFP-C1 (Addgene 36,412; deposited by Benjamin Glick), pEGFP-SQSTM1 (SQSTM1 were PCR-amplified from cDNA of A549 cells and were cloned into pEGFP-C1 plasmid), or pEGFP-LC3B (LC3B was PCR-amplified from cDNA of A549 cells and was cloned into the pEGFP-C1 plasmid) for 24 h before D39 infections. For , the A549 cells were co-transfected with HA-fusion expressed BECN1 plasmid (HA-BECN1) and eGFP-fusion expressed StkP plasmid (eGFP-StkP) for 24 h, co-transfection of eGFP-empty vector and HA-BECN1 used as a control. For IP analysis, cells were washed three times with PBS and lysed with Cell lysis buffer for Western and IP (Biosharp, BL509A) and incubated with anti-IgG rAb (Proteintech 30,000–0-AP), anti-HA rAb (Proteintech 51,064–2-AP) or anti-eGFP rAb (Proteintech 50,430–2-AP) overnight, then the mixture was incubated with protein A+G agarose (Beyotime, P2012) for 2 h, all the procedures were performed at 4°C. The beads were collected through centrifugation, washed 5 times with chilled Cell lysis buffer for Western and IP, boiled in protein loading buffer for 10 min, and then loaded onto an SDS-PAGE gel for immunoblot analysis with indicated antibodies as above indicated, except HA detected using Anti-HA VHH HRP (AlpaLifeBio, KTSM1315; 1:5000). Non-immunoprecipitated samples (input samples) were processed in the same way without performing the IP steps.
Proximity labeling, protK digest, and streptavidin affinity isolation
Open-reading frames (ORF) of rtTA, the reverse Tet trans-activator for the Tet-On system, was cloned into pLVX-puro lentiviral plasmid (Clontech 632,164) through Gibson assembly (rtTA was driven constitutively under CMV promoter). ORFs of eGFP-MYC-TurboID and human LC3B were PCR-amplified and were cloned into modified pLVX-puro lentiviral plasmid (Clontech 632,164; the CMV promoter was replaced by TRE3GV promoter) through Gibson assembly (eGFP-MYC-TurboID was fused with human LC3B or not on the C-terminus of LC3B, which was driven by TRE3GV promoter under rtTA and DOX). To produce lentivirus, 293T cells were co-transfected with lentiviral plasmid, pMD2.G (Addgene 12,259; deposited by Didier Trono) and pSpAX2 (Addgene 12,260; deposited by Didier Trono). The supernatant was harvested and concentrated after 48 h and 72 h of transfection. For the lentivirus infection of A549 cells, cells were infected with lentivirus at a moi of 5 in the presence of 4 μg/mL polybrene. To produce stable A549 cells for the TurboID-based proximity labeling assay [Citation42,Citation43], the A549 cells were infected by lentivirus expressing rtTA (A549-rtTA) and then were screened with 1 μg/mL puromycin for 3 days. Then, the A549-rtTA cell was infected by lentivirus expressing HsLC3B-eGFP-MYC-TurboID (as an experimental group) or eGFP-MYC-TurboID (as background control), and the eGFP+ cells were sorted by MoFlo Astrios EQ (Beckman Coulter) after 48 h of infection.
A549 cells stably expressing HsLC3B-eGFP-MYC-TurboID or eGFP-MYC-TurboID were induced with doxycycline for 12 h and treated with BafA1 for 2 h before D39 infection. Biotinylation by TurboID was triggered as described previously [Citation43,Citation91]. Cells were incubated with 100 μM biotin and 1 mM ATP for 1 h at 37°C. All steps described below were performed at 4°C unless stated otherwise. As described in previous research [Citation43], cells were washed in homogenization buffer I (10 mM KCl, 1.5 mM MgCl2, 10 mM HEPES-KOH, and 1 mM DTT pH 7.5) and suspended in an overhead shaker for 20 min. Cells were homogenized using a pellet pestle before resuspension in homogenization buffer II (375 mM KCl, 22.5 mM MgCl2, 220 mM HEPES-KOH and 0.5 mM DTT pH 7.5) and centrifugation for 5 min at 600 g. 30 mg protK (Solarbio, P9460), 0.5% Triton X-100 (Solarbio, IT9100), or both were added to cleared lysate for 30 min followed by its inactivation through the addition of 100 mM PMSF (Beyotime, ST2573). The four samples were collected for immunoblotting analysis. Importantly, we incubated cell homogenates with protK to degrade all proteins not protected by intact membranes, thereby specifically enriching for biotinylated proteins enclosed in autophagosomes. Subsequently, the protK-treated sample was subjected to streptavidin affinity-isolation according to the protocol (Streptavidin Magnetic Beads; MedChemExpress, HY-K0208). The beads continued to be treated for WB analysis as described in the IP assay. Non-immunoprecipitated samples (input samples) were processed in the same way without performing the IP steps.
Animal experiments
Four-week-old female Balb/c mice obtained from Huazhong Agricultural University Experimental Animal Center were used for all animal experiments. Mice were intranasally infected with 5 × 108 CFUs of S. pneumoniae in 40 μL PBS. The bacteria D39-WT, D39-ΔstkP, and D39-C-ΔstkP were re-suspended in PBS, respectively, and the control group was instilled with the same volume of PBS alone. At 24 hpi, animal blood was obtained by cardiac blood sampling on the sacrificed mouse, and the lungs and brain tissue were homogenized with PBS for bacteria number counts or RIPA lysis for WB analysis. For the H&E assay, mice lung tissues were immersed in 4% paraformaldehyde for 4 h and transferred to 70% ethanol. Lung tissues were embedded in paraffin and then processed for H&E staining according to the routine histologic procedure. For tissue immunostaining, the lung sections were incubated with primary antibodies of OCLN/occludin (Proteintech 66,378–1-Ig; mAb, 1:800), LC3A/B (Cell Signaling Technology, 12741S, rAb; 1:50), and secondary antibodies of CoraLite488-conjugated Goat Anti-Mouse IgG(H+L) or Cy3-conjugated Affinipure Goat Anti-Rabbit IgG(H+L), and DAPI. Images were obtained using a fluorescence microscope. For the cytokine protein assay, the Proteome Profiler Mouse XL Cytokine Array (R&D Systems, ARY028) was used for measuring cytokines of lung lysates according to the manufacturer’s instructions. For animal survival analysis, 10 mice were included in each group and the mice’s survival was monitored over 96 h. For need, mice were administered intraperitoneal injections of PBS or CQ (50 mg/kg) three times at 48 h, 24 h, and 2 h prior to intranasal infections. All animal experiments were performed with protocols approved by the Laboratory Animal Welfare and Ethics Committee of Huazhong Agricultural University following the Laboratory Animal Guideline for ethical review of animal welfare (ID number: HZAUMO-2023–0290).
Statistical analysis
The results were presented as mean ± SD using Graphpad Prism 9.0.0. All the experiments were conducted at least three times. Two-tailed Student’s t-test (for two groups) or one-way analysis of variance (ANOVA followed by Tukey’s test for more than two groups) was applied to determine statistical significance. p < 0.05, p < 0.01 and p < 0.001 were indicated by *, ** and ***, respectively.
Revised Supplementary figures R2.docx
Download MS Word (2.5 MB)Revised Table S1 R1.docx
Download MS Word (22 KB)Acknowledgements
This project was supported by National Key Research and Development Program of China (2021YFD1800405), the National Natural Science Foundation of China (NSFC) (32122086), the Natural Science Foundation of Hubei Province (2021CFA016), the Fundamental Research Funds for the Central Universities (2662023PY005), and the earmarked fund for CARS-35.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data of mass spectrometry proteomics obtained in this study have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD046576.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2024.2330043
Additional information
Funding
References
- Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med. 2013 Jul 1;3(7):a010215. doi: 10.1101/cshperspect.a010215
- van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. 2009 Oct 31;374(9700):1543–1556. doi: 10.1016/S0140-6736(09)61114-4
- Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nature Rev Microbiol. 2018 Mar 29;16(6):355–367. doi: 10.1038/s41579-018-0001-8
- Liu X, Kimmey JM, Matarazzo L, et al. Exploration of bacterial bottlenecks and streptococcus pneumoniae Pathogenesis by CRISPRi-seq. Cell Host Microbe. 2021 Jan 13;29(1):107–120.e106. doi: 10.1016/j.chom.2020.10.001
- Prevotat A, Rouyer C, Gosset P, et al. Biphasic lung injury during Streptococcus pneumoniae infection in a murine model. Med Maladies Infect. 2018 Mar 1;48(2):103–113. doi: 10.1016/j.medmal.2017.11.001
- Umstead TM, Hewage EK, Mathewson M, et al. Lower respiratory tract delivery, airway clearance, and preclinical efficacy of inhaled GM-CSF in a postinfluenza pneumococcal pneumonia model. Am J Physiol Lung Cell Mol Physiol. 2020 Apr 1;318(4):L571–l579. doi: 10.1152/ajplung.00296.2019
- Erfinanda L, Zou L, Gutbier B, et al. Loss of endothelial CFTR drives barrier failure and edema formation in lung infection and can be targeted by CFTR potentiation. Sci, Trans Med. 2022 Dec 7;14(674):eabg8577. doi: 10.1126/scitranslmed.abg8577
- Guerrero-Mandujano A, Hernández-Cortez C, Ibarra JA, et al. The outer membrane vesicles: secretion system type zero. Traffic. 2017 Jul;18(7):425–432.
- Mug-Opstelten D, Witholt B. Preferential release of new outer membrane fragments by exponentially growing Escherichia coli. Biochimi Biophys Acta (BBA) Biomembr. 1978 Apr 4;508(2):287–295. doi: 10.1016/0005-2736(78)90331-0
- Toyofuku M, Nomura N, Eberl L. Types and origins of bacterial membrane vesicles. Nat Rev Microbiol. 2018 Jan;17(1):13–24. doi: 10.1038/s41579-018-0112-2
- Pan F, Zhu ML, Liang Y, et al. Membrane vesicle delivery of a streptococcal M protein disrupts the blood–brain barrier by inducing autophagic endothelial cell death. P Natl Acad Sci USA. 2023 Jun 13;120(24):e2219435120. doi: 10.1073/pnas.2219435120
- Bitto NJ, Cheng L, Johnston EL, et al. Staphylococcus aureus membrane vesicles contain immunostimulatory DNA, RNA and peptidoglycan that activate innate immune receptors and induce autophagy. J Extracell Vesicles. 2021 Apr 10;10(6):e12080. doi: 10.1002/jev2.12080
- Marsollier L, Brodin P, Jackson M, et al. Impact of Mycobacterium ulcerans Biofilm on Transmissibility to Ecological Niches and Buruli Ulcer Pathogenesis. PLoS Pathog. 2007 May 4;3(5):582–594. doi: 10.1371/journal.ppat.0030062
- Rivera J, Cordero RJB, Nakouzi AS, et al. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. P Natl Acad Sci USA. 2010 Nov 2;107(44):19002–19007. doi: 10.1073/pnas.1008843107
- Codemo M, Muschiol S, Iovino F, et al. Immunomodulatory effects of pneumococcal extracellular vesicles on cellular and humoral host defenses. MBio. 2018 Apr 10;9(2):e00559–00518. doi: 10.1128/mBio.00559-18
- Toyofuku M, Schild S, Kaparakis-Liaskos M, et al. Composition and functions of bacterial membrane vesicles. Nature Rev Microbiol. 2023 Mar 17;21(7):415–430. doi: 10.1038/s41579-023-00875-5
- Wang X, Eagen WJ, Lee JC. Orchestration of human macrophage NLRP3 inflammasome activation by staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci USA. 2020 Feb 11;117(6):3174–3184. doi: 10.1073/pnas.1915829117
- Racanelli AC, Kikkers SA, Choi AMK, et al. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018 Feb 8;14(2):221–232. doi: 10.1080/15548627.2017.1389823
- Feng YC, Yao ZY, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015 Jun 1;25(6):354–363. doi: 10.1016/j.tcb.2015.02.002
- Yang Z, Lin P, Chen B, et al. Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5). Autophagy. 2021 Oct 3;17(10):3048–3067. doi: 10.1080/15548627.2020.1851897
- Nighot PK, CAA H, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem. 2015 Mar 13;290(11):7234–7246. doi: 10.1074/jbc.M114.597492
- Foerster EG, Mukherjee T, Cabral-Fernandes L, et al. How autophagy controls the intestinal epithelial barrier. Autophagy. 2022 Jan 2;18(1):86–103. doi: 10.1080/15548627.2021.1909406
- Blanke SR, Liu M, Wang Q, et al. Glaesserella parasuis serotype 5 breaches the porcine respiratory epithelial barrier by inducing autophagy and blocking the cell membrane claudin-1 replenishment. PLoS Pathog. 2022 Oct 13;18(10):e1010912. doi: 10.1371/journal.ppat.1010912
- Luo J, Li P, Shi J, et al. Streptococcus pneumoniae induces autophagy through the inhibition of the PI3K-I/Akt/mTOR pathway and ROS hypergeneration in A549 cells. PLoS One. 2015 Mar 24;10(3):e0122753. doi: 10.1371/journal.pone.0122753
- Sharma P, Roy S. Streptococcus pneumoniae exerts oxidative stress, subverts antioxidant signaling and autophagy in human corneal epithelial cells that is alleviated by tert-butylhydroquinone. Med Microbiol Immunol. 2022 Mar 24;211(2–3):119–132. doi: 10.1007/s00430-022-00731-y
- Dong Y, Jin C, Ding Z, et al. TLR4 regulates ROS and autophagy to control neutrophil extracellular traps formation against Streptococcus pneumoniae in acute otitis media. Pediat Res. 2020 May 21;89(4):785–794. doi: 10.1038/s41390-020-0964-9
- Shizukuishi S, Ogawa M, Matsunaga S, et al. Streptococcus pneumoniae hijacks host autophagy by deploying CbpC as a decoy for Atg14 depletion. EMBO Rep. 2020 May 6;21(5):e49232. doi: 10.15252/embr.201949232
- Ogawa M, Matsuda R, Takada N, et al. Molecular mechanisms of streptococcus pneumoniae-targeted autophagy via pneumolysin, golgi-resident Rab41, and Nedd4-1-mediated K63-linked ubiquitination. Cell Microbiol. 2018 Apr 22;20(8):e12846. doi: 10.1111/cmi.12846
- Ogawa M, Takada N, Shizukuishi S, et al. Streptococcus pneumoniae triggers hierarchical autophagy through reprogramming of LAPosome-like vesicles via NDP52-delocalization. Commun Biol. 2020 Jan 13;3(1):25. doi: 10.1038/s42003-020-0753-3
- Shu ZC, Yuan J, Wang H, et al. Streptococcus pneumoniae PepO promotes host anti-infection defense via autophagy in a Toll-like receptor 2/4 dependent manner. Virulence. 2020 Jan 1;11(1):270–282. doi: 10.1080/21505594.2020.1739411
- Li S, Zhang Y, Guan Z, et al. SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res. 2023 Jan 17;33(3):201–214. doi: 10.1038/s41422-022-00775-y
- Ding X, Jiang X, Tian R, et al. RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy. 2019 Oct;15(10):1774–1786.
- Liu P, Zhang Y, Tang H, et al. Prevalence of streptococcus suis in pigs in China during 2000-2021: a systematic review and meta-analysis. One Health. 2023 Feb 18;16:100513. doi: 10.1016/j.onehlt.2023.100513
- Lun ZR, Wang QP, Chen XG, et al. Streptococcus suis: an emerging zoonotic pathogen. Lancet Infect Dis. 2007 Mar;7(3):201–209.
- Kadioglu A, Weiser JN, Paton JC, et al. The role of streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008 Apr;6(4):288–301.
- Subramanian K, Henriques-Normark B, Normark S. Emerging concepts in the pathogenesis of the streptococcus pneumoniae: from nasopharyngeal colonizer to intracellular pathogen. Cell Microbiol. 2019 Nov 7;21(11):e13077. doi: 10.1111/cmi.13077
- Magné J, Green DR. LC3-associated endocytosis and the functions of rubicon and ATG16L1. Sci Adv. 2022 Oct 28;8(43):eabo5600. doi: 10.1126/sciadv.abo5600
- Gammoh N. The multifaceted functions of ATG16L1 in autophagy and related processes. J Cell Sci. 2020 Oct 30;133(20):jcs249227. doi: 10.1242/jcs.249227
- Hill SM, Wrobel L, Ashkenazi A, et al. VCP/p97 regulates Beclin-1-dependent autophagy initiation. Nat Chem Biol. 2021 04 01;17(4):448–455. doi: 10.1038/s41589-020-00726-x
- Lei Y, Klionsky DJ. New functions of a known autophagy regulator: VCP and autophagy initiation. Autophagy. 2021 May;17(5):1063–1064. doi: 10.1080/15548627.2021.1905974
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000 Nov 1;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720
- Branon TC, Bosch JA, Sanchez AD, et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 2018 Aug 20;36(9):880–887. doi: 10.1038/nbt.4201
- Guerroué F L, Eck F, Jung J, et al. Autophagosomal content profiling reveals an LC3C-dependent piecemeal mitophagy pathway. Molecular Cell. 2017 Nov 16;68(4):786–796. doi: 10.1016/j.molcel.2017.10.029
- Zingl FG, Thapa HB, Scharf M, et al. Outer membrane vesicles of Vibrio cholerae protect and deliver active cholera toxin to host cells via porin-dependent uptake. MBio. 2021 Jun 29;12(3):e0053421. doi: 10.1128/mBio.00534-21
- Wang X, Thompson CD, Weidenmaier C, et al. Release of staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat Commun. 2018 Apr 11;9(1):1379. doi: 10.1038/s41467-018-03847-z
- Martin-Gallausiaux C, Malabirade A, Habier J, et al. Fusobacterium nucleatum Extracellular Vesicles Modulate Gut Epithelial Cell Innate Immunity via FomA and TLR2. Front Immunol. 2020 Dec 21;11:583644. doi: 10.3389/fimmu.2020.583644
- Xue Y, Wang M, Han H. Interaction between alveolar macrophages and epithelial cells during mycoplasma pneumoniae infection. Front Cell Infect Microbiol. 2023 Apr 11;13:1052020. doi: 10.3389/fcimb.2023.1052020
- Ryndak MB, Laal S. Mycobacterium tuberculosis Primary Infection and Dissemination: A Critical Role for Alveolar Epithelial Cells. Front Cell Infect Microbiol. 2019 Aug 21;9:299. doi: 10.3389/fcimb.2019.00299
- Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013 Jan 17;152(1–2):290–303. doi: 10.1016/j.cell.2012.12.016
- Li J, Zhang T, Ren T, et al. Oxygen-sensitive methylation of ULK1 is required for hypoxia-induced autophagy. Nat Commun. 2022 Mar 4;13(1):1172. doi: 10.1038/s41467-022-28831-6
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2014 Nov 17;11(1):28–45. doi: 10.4161/15548627.2014.984267
- Ulrych A, Fabrik I, Kupčík R, et al. Cell wall stress stimulates the activity of the protein kinase StkP of streptococcus pneumoniae, leading to multiple phosphorylation. J Mol Biol. 2021 Dec 3;433(24):167319. doi: 10.1016/j.jmb.2021.167319
- Bressan C, Pecora A, Gagnon D, et al. The dynamic interplay between ATP/ADP levels and autophagy sustain neuronal migration in vivo. Elife. 2020 Sep 28;9:e56006. doi: 10.7554/eLife.56006
- Lemon JK, Weiser JN, McDaniel LS. Degradation products of the extracellular pathogen streptococcus pneumoniae access the cytosol via its pore-forming toxin. MBio. 2015 Jan 20;6(1):e02110–02114. doi: 10.1128/mBio.02110-14
- Mitchell AM, Mitchell TJ. Streptococcus pneumoniae: virulence factors and variation. Clin Microbiol Infect. 2010 May;16(5):411–418. doi: 10.1111/j.1469-0691.2010.03183.x
- Brown L, Kessler A, Cabezas-Sanchez P, et al. Extracellular vesicles produced by the gram-positive bacterium are disrupted by the lipopeptide surfactin. Mol Microbiol. 2014 May 15;93(1):183–198. doi: 10.1111/mmi.12650
- Brown L, Wolf JM, Prados-Rosales R, et al. Through the wall: extracellular vesicles in gram-positive bacteria, mycobacteria and fungi. Nature Rev Microbiol. 2015 Sep 1;13(10):620–630. doi: 10.1038/nrmicro3480
- Haas B, Grenier D, Chaussee MS. Isolation, characterization and biological properties of membrane vesicles produced by the swine pathogen streptococcus suis. PLoS One. 2015 Jun 25;10(6):e0130528. doi: 10.1371/journal.pone.0130528
- Surve MV, Anil A, Kamath KG, et al. Membrane vesicles of group B streptococcus disrupt feto-maternal barrier leading to preterm birth. PLoS Pathog. 2016 Sep 1;12(9):e1005816. doi: 10.1371/journal.ppat.1005816
- Parveen S, Subramanian K. Emerging roles of extracellular vesicles in pneumococcal infections: immunomodulators to potential novel vaccine candidates. Front Cell Infect Mi. 2022 Feb 14;12:836070. doi: 10.3389/fcimb.2022.836070
- Yerneni SS, Werner S, Azambuja JH, et al. Pneumococcal extracellular vesicles modulate host immunity. MBio. 2021 Aug 31;12(4):e0165721. doi: 10.1128/mBio.01657-21
- Mehanny M, Koch M, Lehr CM, et al. Streptococcal extracellular membrane vesicles are rapidly internalized by immune cells and alter their cytokine release. Front Immunol. 2020 Feb 14;11:80. doi: 10.3389/fimmu.2020.00080
- Olaya-Abril A, Prados-Rosales R, McConnell MJ, et al. Characterization of protective extracellular membrane-derived vesicles produced by Streptococcus pneumoniae. J Proteomics. 2014 Jun 25;106:46–60. doi: 10.1016/j.jprot.2014.04.023
- Wei S, Xu T, Chen Y, et al. Autophagy, cell death, and cytokines in K. pneumoniae infection: therapeutic perspectives. Emerg Microbes Infect. 2022 Dec 18;12(1):2140607. doi: 10.1080/22221751.2022.2140607
- Song YJ, Ge X, Chen YL, et al. Mycobacterium bovis induces mitophagy to suppress host xenophagy for its intracellular survival. Autophagy. 2022 Jun 3;18(6):1401–1415. doi: 10.1080/15548627.2021.1987671
- Losier TT, Russell RC. Bacterial outer membrane vesicles trigger pre-activation of a xenophagic response via AMPK. Autophagy. 2019 Aug 3;15(8):1489–1491. doi: 10.1080/15548627.2019.1618640
- Losier TT, Akuma M, McKee-Muir OC, et al. AMPK promotes xenophagy through priming of autophagic kinases upon detection of bacterial outer membrane vesicles. Cell Rep. 2019 Feb 19;26(8):2150–2165. doi: 10.1016/j.celrep.2019.01.062
- David L, Taieb F, Pénary M, et al. Outer membrane vesicles produced by pathogenic strains of Escherichia coli block autophagic flux and exacerbate inflammasome activation. Autophagy. 2022 Apr 9;18(12):2913–2925. doi: 10.1080/15548627.2022.2054040
- Li X, Jamal M, Guo P, et al. Irisin alleviates pulmonary epithelial barrier dysfunction in sepsis-induced acute lung injury via activation of AMPK/SIRT1 pathways. Biomed Pharmacother. 2019 Oct 1;118:109363. doi: 10.1016/j.biopha.2019.109363
- Dudek SM, Garcia JGN. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001 Oct 1;91(4):1487–1500. doi: 10.1152/jappl.2001.91.4.1487
- Sim TY, Harith HH, Tham CL, et al. The protective effects of a synthetic geranyl acetophenone in a cellular model of TNF-α-induced pulmonary epithelial barrier dysfunction. Molecules. 2018 Jun 5;23(6):1355. doi: 10.3390/molecules23061355
- Cummins PM. Occludin: one protein, many forms. Mol Cell Biol. 2012 Jan;32(2):242–250. doi: 10.1128/MCB.06029-11
- Becker G, Da Silva S, Sabo AN, et al. Blood–brain barrier permeability: is 5-hydroxytryptamine receptor type 4 a game changer? Pharmaceutics. 2021 Nov 3;13(11):1856. doi: 10.3390/pharmaceutics13111856
- Nighot P, Ma T. Role of autophagy in the regulation of epithelial cell junctions. Tissue Barr. 2016 Jun 9;4(3):e1171284. doi: 10.1080/21688370.2016.1171284
- Wu J, Gao FX, Xu T, et al. CLDN1 induces autophagy to promote proliferation and metastasis of esophageal squamous carcinoma through AMPK/STAT1/ULK1 signaling. J Cell Physiol. 2019 Sep 9;235(3):2245–2259. doi: 10.1002/jcp.29133
- LaCanna R, Liccardo D, Zhang P, et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J Clin Invest. 2019 May 1;129(5):2107–2122. doi: 10.1172/JCI125014
- Grangeasse C. Rewiring the pneumococcal cell cycle with serine/threonine- and tyrosine-kinases. Trends Microbiol. 2016 Apr 26;24(9):713–724. doi: 10.1016/j.tim.2016.04.004
- Dworkin J. Ser/Thr phosphorylation as a regulatory mechanism in bacteria. Curr Opin Microbiol. 2015 Apr 1;24:47–52. doi: 10.1016/j.mib.2015.01.005
- Echenique J, Kadioglu A, Romao S, et al. Protein serine/threonine kinase StkP positively controls virulence and competence in streptococcus pneumoniae. Infect Immun. 2004 Apr 1;72(4):2434–2437. doi: 10.1128/IAI.72.4.2434-2437.2004
- Shi L, Pigeonneau N, Ravikumar V, et al. Cross-phosphorylation of bacterial serine/threonine and tyrosine protein kinases on key regulatory residues. Front Microbiol. 2014 Sep 17;5:495. doi: 10.3389/fmicb.2014.00495
- Mitchell TJ, PiñPiñAs GE, Reinoso-Vizcaino NM, et al. Crosstalk between the serine/threonine kinase StkP and the response regulator ComE controls the stress response and intracellular survival of streptococcus pneumoniae. PLoS Pathog. 2018 Jun 8;14(6):e1007118. doi: 10.1371/journal.ppat.1007118
- Herbert JA, Mitchell AM, Mitchell TJ, et al. A serine-threonine kinase (StkP) regulates expression of the pneumococcal Pilus and modulates bacterial adherence to human epithelial and endothelial cells in vitro. PLoS One. 2015 Jun 19;10(6):e0127212. doi: 10.1371/journal.pone.0127212
- Saskova L, Novakova L, Basler M, et al. Eukaryotic-type serine/threonine protein kinase StkP is a global regulator of gene expression in streptococcus pneumoniae. J Bacteriol. 2007 Apr 6;189(11):4168–4179. doi: 10.1128/JB.01616-06
- Bosch ME, Bertrand BP, Heim CE, et al. Staphylococcus aureus ATP synthase promotes biofilm persistence by influencing innate immunity. MBio. 2020 Sep 8;11(5):e01581–01520. doi: 10.1128/mBio.01581-20
- Chin RM, Fu X, Pai MY, et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014 Jun 19;510(7505):397–401. doi: 10.1038/nature13264
- Salcines-Cuevas D, Terán-Navarro H, Calderón-Gonzalez R, et al. Glyceraldehyde-3-phosphate dehydrogenase common peptides of listeria monocytogenes, mycobacterium marinum and streptococcus pneumoniae as universal vaccines. Vaccines (Basel). 2021 Mar 17;9(3):269. doi: 10.3390/vaccines9030269
- Huo J, Dong W, Xu J, et al. Role of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in autophagy activation following subarachnoid hemorrhage. Exp Neurol. 2024 Jan;371:114577. doi: 10.1016/j.expneurol.2023.114577
- Bricker AL, Camilli A. Transformation of a type 4 encapsulated strain of streptococcus pneumoniae. FEMS Microbiol Lett. 1999 Mar 15;172(2):131–135. doi: 10.1111/j.1574-6968.1999.tb13460.x
- Xu BJ, Chen JQ, Fu JY, et al. Meningitic Escherichia coli-Induced Interleukin-17A Facilitates Blood–Brain Barrier Disruption via Inhibiting Proteinase 3/Protease-Activated Receptor 2 Axis. Front Cell Neurosci. 2022 Feb 11;16:814867. doi: 10.3389/fncel.2022.814867
- Avilan L. Assembling multiple fragments: the Gibson Assembly. In: Scarlett G, editor. DNA manipulation and analysis. New York, NY: Springer US; 2023. p. 45–53.
- Zhang YL, Song GY, Lai NK, et al. TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat Commun. 2019 Jul 19;10(1):3252. doi: 10.1038/s41467-019-11202-z