
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The noncanonical ubiquitin-like conjugation cascade involving the E1 (Atg7), E2 (Atg3, Atg10), and E3 (Atg12–Atg5-Atg16 complex) enzymes is essential for incorporation of Atg8 into the growing phagophore via covalent linkage to PE. This process is an indispensable step in autophagy. Atg8 and E1-E3 enzymes are the first subset from the core autophagy protein machinery structures that were investigated in earlier studies by crystallographic analyses of globular domains. However, research over the past decade shows that many important functions in the conjugation machinery are mediated by intrinsically disordered protein regions (IDPRs) – parts of the protein that do not adopt a stable secondary or tertiary structure, which are inherently dynamic and well suited for protein-membrane interactions but are invisible in protein crystals. Here, we summarize earlier and recent findings on the autophagy conjugation machinery by focusing on the IDPRs. This summary reveals that IDPRs, originally considered dispensable, are in fact major players and a driving force in the function of the autophagy conjugation system. Abbreviation: AD, activation domain of Atg7; AH, amphipathic helix; AIM, Atg8-family interacting motif; CL, catalytic loop (of Atg7); CTD, C-terminal domain; FR, flexible region (of Atg3 or Atg10); GUV, giant unilammelar vesicles; HR, handle region (of Atg3); IDPR, intrinsically disordered protein region; IDPs: intrinsically disordered proteins; LIR, LC3-interacting region; NHD: N-terminal helical domain; NMR, nuclear magnetic resonance; PE, phosphatidylethanolamine; UBL, ubiquitin like.
Conjugation of the Atg8 family of proteins (Atg8/LC3/GABARAP) to phosphatidylethanolamine (PE) embedded in the phagophore membrane is an essential step in the autophagy process. This covalent linkage is mediated by the noncanonical E1 (yeast Atg7/mammalian ATG7), E2 (Atg3/ATG3), and E3 (Atg12–Atg5-Atg16/ATG12–ATG5-ATG16L1 complex) enzymatic cascade, which is highly conserved from yeast to mammals. The indispensability of Atg8–PE formation for autophagosome biogenesis prompted significant efforts to visualize the components of the Ag8 conjugation system. Protein crystallography played an important role in this endeavor as it revealed the architecture of ATG4B, Atg5/ATG5, Atg7, Atg8-family proteins, and Atg10 [Citation1–10] with only several missing residues. However, Atg3/ATG3 and Atg12/ATG12 were visualized from protein crystals with significant gaps or absent large domains [Citation3,Citation6,Citation8,Citation11] and Atg16/ATG16L1 in the monomeric form was impossible to capture. Only a partial homodimeric Atg16/ATG16L1 or WD40 in ATG16L1 [Citation12–14] were solved by x-ray crystallography.
The reason for these incomplete protein images is the presence of domains lacking a well-defined 3D structure under physiological conditions. These domains are called intrinsically disordered protein regions (IDPRs). IDPRs have a distinct amino acid composition [Citation15] endowing them with specific physicochemical properties and behavior [Citation16–18], which makes them often a challenge in purification and characterization. Earlier studies disregarded IDPRs as nonfunctional because their lack of a well-defined 3D structure excludes them from the structure-function paradigm, an assumption that the function of a protein is closely related to its 3D architecture. In comparison to IDPRs, intrinsically disordered proteins (IDPs) are entirely disordered polypeptides in a monomeric form but can adopt an inducible 3D structure upon interaction with a physiological partner or by oligomerization.
Within the last two decades, IDPs/IDPRs have been extensively studied and characterized because they are associated with many human diseases [Citation19–23]. In fact, diseases linked to disrupted IDPs/IDPRs and to dysfunctional autophagy overlap [Citation24,Citation25]. Therefore, it is not surprising that the dynamic function of the autophagy machinery relies on IDPRs [Citation26,Citation27]. One of the many possible reasons for defective autophagy is a failure in the Atg8 conjugation machinery [Citation28]. Here, we highlight IDPRs acting in the mechanisms involved in Atg8–PE conjugation. We describe these mechanisms in detail to display an array of functions executed by flexible regions residing in the autophagy conjugation system. This summary emphasizes the importance of IDPRs in protein function and, hopefully, encourages the autophagy community in exploring disordered protein domains further and beyond the Atg8–PE conjugation machinery.
Atg8: The Atg8 family of proteins is evolutionarily conserved from yeast to humans. Yeast has only one Atg8, whereas human cells have at least six distinct Atg8-family proteins separated into two subfamilies, LC3s (LC3A, LC3B, and LC3C) and GABARAPs (GABARAP, GABARAPL1, and GABARAPL2). Before entering the enzymatic conjugation cascade, each Atg8 molecule needs to be processed by Atg4, an enzyme that removes a C-terminal amino acid sequence downstream of the glycine residue of Atg8 that is ultimately linked to PE. In yeast, unprocessed Atg8 has R117 downstream of the penultimate C-terminal glycine, G116. The nuclear magnetic resonance (NMR) structure of unprocessed Atg8 in solution [Citation29] reveals that the N-terminal helical domain (NHD) has a significantly higher mobility than the ubiquitin-like (UBL) domain and that the NHD exists in a dynamic conformational ensemble with only a partial α2-helix (). Removal of R117 increases rigidity of the ubiquitin-like domain [Citation29] and appears to be associated with a conformational change in the NHD that bestows its lower mobility, fully folded α2-helix, and an additional α1-helix in processed Atg8 (). The human processed LC3C with a polyproline II motif in the N-terminal sequence extension relative to LC3A and LC3B has its NHD in a dynamic partially unfolded conformation, whereas LC3B is a compact molecule with the fully folded NHD similar to that in processed Atg8 [Citation30–32] (). A question of why there is a need for a partially disordered NHD conformation associated with the N- or C-terminal extensions in LC3C or Atg8, respectively, remains to be elucidated. From a study on the autophagy machinery in yeast, we know that the fully folded NHD is important for the E1 enzyme, Atg7, which utilizes an induced hydrophobic pocket between α1 and α2 helices for initial recognition of the Atg8 substrate [Citation7]. However, even the processed Atg8 must maintain some conformational mobility of its NHD in order to unmask a UBL-domain surface that is engaged in membrane tethering by Atg8–PE [Citation33]. The tethering surface has two highly conserved aromatic amino acid residues, phenylalanine and tyrosine. None of the NMR structures of non-lipidated Atg8s (, RCSB PDB [https://www.rcsb.org]) has this surface fully accessible. This suggests that an “active” conformation with a fully accessible membrane-tethering surface in Atg8–PE in trans-membrane association may be somewhat different from the conformations revealed so far in the NMR structures of non-lipidated Atg8s. Human Atg8-family proteins may adopt the “active” conformation for membrane tethering in trans-membrane association as well. This possibility has not been excluded; however, LC3B–PE and GABARAP–PE can insert the tip of the NHD into the lipid bilayer and these lipidated Atg8s can be in cis-membrane association with the membrane [Citation34]. Atg8s contain a second disordered region at the C terminus (). Studies with S. cerevisiae show that structural plasticity and an extended conformation of this short region are utilized in the activation of Atg8, when the C terminal tail enters the hydrophobic cavity of the Atg7 activation domain (AD) [Citation2,Citation7]. It is conceivable that the protruding flexible C terminus also has an advantage in maneuvering the C-terminal glycine to PE, as a rigid structure would require precise navigation of the Atg8 UBL domain very close to the membrane.
Figure 1. Atg8 and LC3 proteins and the human cysteine protease ATG4B. (A) Multistate NMR structures of yeast Atg8 and human LC3B and LC3C. The NMR structure of unprocessed Atg8 with a C-terminal R117 (PDB ID: 2KQ7) reveals the disordered regions (black), a partially folded N-terminal α2-helix (gray), and the UBL domain (green). The N terminus is a dynamic conformational ensemble with a high mobility. The NMR structure of processed Atg8 in solution (PDB ID: 2KWC) shows a compact conformation with a folded NHD. The NMR structure of LC3C (PDB ID: 2NCN) reveals that the polyproline II motif in the N-terminal extension replaces the α1 helix with a disordered conformation, whereas LC3B (PDB ID: 2N9X) without this extension is a compact molecule. Conserved phenylalanine and tyrosine (yellow) form a membrane tethering surface in the UBL domain. (B) Crystal structure of human ATG4B (PDB ID: 2CY7). Trp142, the regulatory loop near the catalytic triad (C74, D278, and H280) and the N-terminal domain are involved in enzymatic autoinhibition. The APEAR motif may target the protein to the membrane.
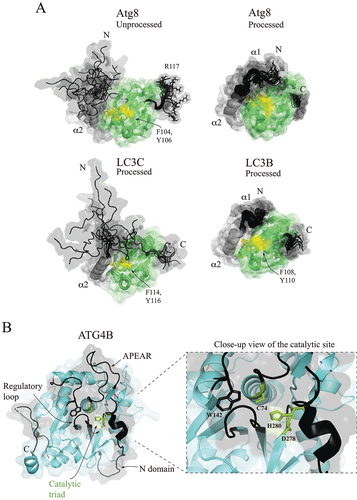
Atg4: The sole cysteine protease in the autophagy protein machinery, Atg4 proteolytically processes the Atg8 family of proteins to bring it into the conjugation cascade. Human ATG4B, is the only homolog that has been structurally analyzed, and will be used here for the description of IDPRs in this protease. ATG4B () cleaves its substrates via the catalytic triad C74, D278, and H280. The enzyme is assumed to utilize two autoinhibitory mechanisms mediated by the flexible regulatory loop and disordered N terminus. In particular, the interaction between W142 and the regulatory loop shields the catalytic C74 from the solvent, whereas the N-terminal tail in a closed conformation blocks the exit of the catalytic site. These two autoinhibitory regions must undergo a significant conformational change upon LC3 binding. F119 of LC3 lifts the regulatory loop and, thereby, forms a groove for the LC3 tail to enter. It has been reported that autoinhibition by the N-terminal tail of ATG4B is released through binding to a second, nonsubstrate, LC3 molecule via the LC3-interacting region (LIR) motif and that the repositioned N terminus in an open conformation unblocks the exit of the catalytic site. A limitation of this interpretation of crystallographic data is that the interaction between the N terminus of ATG4B and the nonsubstrate LC3 molecule may be triggered by crystallization and may not be biologically relevant. ATG4B has an additional loop called APEAR, which was proposed to target the protein to membranes [Citation4,Citation9,Citation35].
E1-like enzyme: Atg7/ATG7 functions as a noncanonical activating enzyme [Citation2,Citation3,Citation7] for the ubiquitin-like substrates, Atg8-family proteins and Atg12/ATG12. Structure and function of this E1-like enzyme, operating as a homodimer (), has been extensively studied in yeast. NMR shows that Atg7 utilizes its C-terminal disordered tail (amino acids 601–630) to initially recognize the substrate. Approximately 13 C-terminal residues bind the ubiquitin-like molecule by attaching to three hydrophobic pockets on the surface of Atg8; namely, the W and L site, also recognized by the Atg8-family interacting motif (AIM) in autophagy receptors and adaptors, and an induced pocket between the N-terminal α1 and α2 helices [Citation7]. Comparison of the Atg7 C-terminal domain (CTD) in the crystal structures of the Atg7 apo-enzyme (4GSL) and Atg7CTD-Atg8 complex (3VH4) shows that, in the second binding phase, Atg7 uses its C-terminal IDPR as a retractable leash to pull the substrate close to the activation domain. A mechanism through which this action is achieved is a partial folding of the C-terminal tail into the α17 helix. This helix then becomes a part of the binding interface between Atg7 and Atg8 () [Citation3,Citation7,Citation36]. Another important IDPR in Atg7 is a long disordered catalytic loop (CL; residues 471–511) that carries the catalytic cysteine C507 and undergoes a large conformational change regulating the entrance of the C-terminal tail of Atg8 into the activation domain of Atg7 [Citation36] (). When Atg7 forms α17 to pull Atg8 close to the AD, the catalytic loop folds residues 487–492 into the α12 helix, leaving the rest of the loop disordered and exposing the central β-sheet. The Atg8 UBL domain then binds to the central β-sheet, which triggers a flip of the disordered CL to a new position where C507 becomes proximal to Atg8 G116. With the help of ATP, Atg7 C507 in the CL and Atg8 G116 in the tail form a thioester bond in the Atg7–Atg8 conjugate that is a hallmark of the interaction between Atg7 and Atg8 ().
Figure 2. E1 and E2 enzymes in the Atg8 conjugation machinery in yeast. (A) Crystal structure of the yeast Atg7 homodimer (PDB ID: 4GSL) (light blue) dimerizing via the C-terminal domain. The catalytic C507 of the Atg7 (1) protomer is close to the N-terminal domain of the Atg7 (2) protomer, and vice versa. Disordered regions (black) visible in the crystal are highlighted in a solid line and invisible regions are in a dashed line. (B) Structural alignement in Pymol of the monomeric C-terminal domains in the Atg7 apo-enzyme (light blue; PDB ID: 4GSL) and Atg7CTD (brown)-Atg8 (green) complex (PDB ID: 3VH4). The IDPRs are highlighted in black for the apo-enzyme and in red for the complex. (C) Crystal structure of yeast Atg3 free of its binding partners (PDB ID: 2DYT) shows that the protein is enriched in IDPRs. C234 is the catalytic cysteine that engages in thioester bonds. Disordered regions (black) visible in the crystal are highlighted in a solid line and invisible regions are in a dashed line. (D) Crystal structure of free Atg10 from yeast (PDB ID: 4EBR) shows the flexible region (FR), analogous to the Atg3 FR and invisible in the crystal structure, that is involved in binding to Atg7. The catalytic cysteine C133 forms the thioester bond with the C-terminal glycine of Atg12. This bond protects Atg10 from proteolytic cleavage near F112 in the absence of Atg7. Disordered regions (black) visible in the crystal are highlighted in a solid line and invisible regions are in a dashed line.
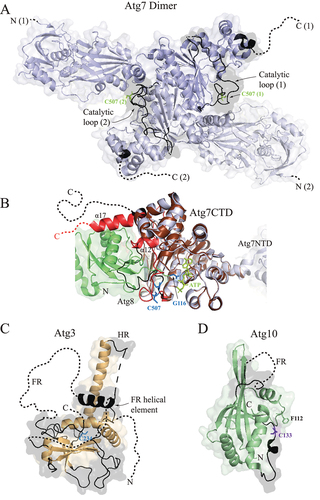
E2-like enzymes: One of the most dynamic proteins in the autophagy conjugation cascade is Atg3, a protein with a unique evolutionarily conserved architecture that takes Atg8 from Atg7 and brings it to the membrane. Highly flexible Atg3 [Citation11,Citation37] () encompasses multiple IDPRs that are essential for protein function; namely, the large flexible region (FR), handle region (HR), and N terminus (). It has been shown for the yeast protein that the Atg3 FR functions as an allosteric regulator of enzymatic activity [Citation38]. Autoinhibited Atg3 attaches the FR on its own surface. Atg7 releases this autoinhibition by triggering a flip and binding of the FR to the N-terminal domain of Atg7. This conformational change of the FR induces a helical extension in the HR and, thereby, repositions the catalytic cysteine to a location favorable for formation of a thioester bond with the C-terminal glycine of Atg8, which at that moment resides in the AD of Atg7. Studies on human ATG3 show that the protein in the thioester conjugate with LC3 attaches to the phagophore via an N-terminal amphipathic helix formed upon a contact with the membrane [Citation39–41]. Molecular dynamics simulations with the human ATG3–LC3 conjugate suggest that a fine-tuned amino acid composition of the ATG3 amphipathic helix (AH) ensures only a transient association of the conjugate with the membrane. Mutagenesis enriching the ATG3 AH with bulky hydrophobic residues that insert deep into the lipid bilayer yields a prolonged retention of ATG3 on the membrane, which interferes with efficient LC3 lipidation [Citation42]. A flexible evolutionarily conserved segment downstream of the AH in the ATG3 N terminus communicates the membrane attachment to the catalytic loop of ATG3 and structurally rearranges the catalytic cysteine in order to facilitate the LC3–PE linkage [Citation39]. Previous studies showed that the human ATG3 FR binds to the ATG12 UBL domain [Citation43] and the AIM motif in the HR of Atg3 binds to Atg8 in yeast [Citation44]. The ATG3 FR-ATG12 UBL interaction is assumed to facilitate downward (toward the membrane) orientation of LC3B in the membrane-associated ATG3–LC3B conjugate [Citation42]. The AIM-mediated interaction between Atg3 and Atg8 is needed for the cytoplasm-to-vacuole targeting pathway, but not for starvation-induced autophagy in yeast [Citation44]. A recent study probing membrane scaffolding and shaping by the Atg8 conjugation machinery strengthens indispensability of the FR and HR in Atg3. The FR and HR target Atg7 and Atg12–Atg5-Atg16 to the membrane and mediate weak interactions with Atg8, E1 and E3 to create a flexible scaffolding web on the phagophore membrane [Citation45].
Atg10 in yeast () and ATG10 in humans function as an E2-like enzyme that takes the ubiquitin-like substrate Atg12/ATG12 from Atg7/ATG7 and transfers it to Atg5/ATG5 in an E3-independent manner. This transfer requires establishing a thioester bond between the catalytic cysteine of Atg10/ATG10 and the C-terminal glycine of Atg12/ATG12. The catalytic cysteine in Atg10 from yeast (C133) is located in a disordered loop that is not visible in the Atg7-Atg10 crystal structure [Citation3] but is important for the transthiolation reaction. We recently showed with yeast proteins [Citation46] that, without Atg7, the thioester between Atg10 and Atg12 protects the intactness of Atg10 because, in the absence of this bond, the protein is susceptible to proteolysis near Phe112 (S. cerevisiae residue numbering). This finding suggests that the disordered catalytic loop of free Atg10 is highly accessible and relies on protection by binding partners. Analogously to Atg3, the crystal structure of free Atg10 from yeast [Citation1] reveals that the protein carries a flexible loop, although much shorter than that seen in Atg3. This loop is invisible in the crystal structure but is indispensable in the interaction with Atg7. Comparison of two crystal structures (PDB ID: 4GSK and 4EBR) shows that the Atg10 FR folds into a helix in the Atg7-Atg10 complex but is disordered in the separate protein, suggesting that the Atg10 FR helix is a secondary structure inducible by Atg7.
E3-like enzyme: The Atg12–Atg5-Atg16 complex in yeast () and ATG12–ATG5-ATG16L1 in humans () facilitates lipidation of Atg8-family proteins through an Atg3/ATG3-Atg12/ATG12 interaction [Citation41,Citation47]. The complex is a dimeric heterotrimer with an architecture very distinct from canonical E3s. The reason(s) for this unique structure remains elusive. Atg12/ATG12 carries a large, disordered region at the N terminus upstream of the UBL domain. We have shown recently that in yeast the N-terminal IDPR packs near the globular UBL domain of Atg12 unconjugated to Atg5 and, thereby, acts as a protecting shield for the disordered C-terminal tail of Atg12. Furthermore, a short α-helix within the IDPR is important for the interaction of Atg12 with Atg7, presumably during the initial recognition, in analogy to α1 and α2 helices in Atg8. The same N-terminal helix of Atg12 is also essential for noncovalent binding to Atg10 [Citation46]. When Atg12 is in the Atg12–Atg5 conjugate, the first half of the N terminus, consisting of approximately 55 amino acids, binds to Atg17 to recruit Atg12–Atg5-Atg16 to the PAS [Citation48]. The overall conformation of the Atg12/ATG12 IDPR () in the Atg12–Atg5-Atg16 and ATG12–ATG5-ATG16L1 complexes has not been elucidated.
Figure 3. The architecture of the E3 enzyme in yeast and humans. (A) The Atg12–Atg5-Atg16 complex in yeast. The crystal structure of the Atg12[UBL]–Atg5-Atg16N (PDB ID: 3W1S) visualizes the Atg12 UBL domain (pale cyan) in the conjugate with Atg5 (light pink) and the Atg16 N-terminal helix bound to Atg5. In the yeast S. cerevisiae, the covalent bond is between G186 of Atg12 and K149 of Atg5. Atg12 contains a long IDPR (black dashed line) of unknown conformation in the conjugate. Monomeric Atg16 is an intrinsically disordered protein (black), where the interaction with Atg5, dimerization, and membrane binding induce α-helical structures. The dimerizing coiled-coil domain (PDB ID: 3A7P) in the middle of Atg16 brings two heterotrimers together. The C-terminal region of Atg16 (residues 113–131) forms an amphipathic helix (dashed line) that inserts into the lipid bilayer and anchors the Atg12–Atg5-Atg16 complex in the membrane. (B) Monomeric ATG12–ATG5-ATG16L1 in a complex with the ATG3 FR. The crystal structure of human ATG3 FR-ATG12[UBL]–ATG5-ATG16L1N (PDB ID: 4NAW) visualizes the FR of ATG3 (black helix) bound to the UBL domain of ATG12 (pale cyan). The disordered N terminus of ATG12 has an unknown conformation in the conjugate with ATG5 (light pink). The covalent bond is between G140 of ATG12 and K130 of ATG5. An amphipathic helix downstream of the ATG5-binding helix (black) is formed upon membrane binding. The coiled-coil domain (black) (PDB ID: 7XFR), which dimerizes, carries at the C terminus a binding region (BR) for one molecule of WIPI2B. The second binding site for WIPI2B is downstream of the coiled-coil domain. RAB33B binds to the coiled-coil domain in between the two WIPI2B-binding sites. The coiled-coil domain is connected via a disordered region to the WD40 domain (gray) that exhibits a β-propeller architecture (PDB ID: 5NUV). The relatively long connecting IDPR carries two membrane-binding regions that are preceded by the RB1CC1 binding site.
![Figure 3. The architecture of the E3 enzyme in yeast and humans. (A) The Atg12–Atg5-Atg16 complex in yeast. The crystal structure of the Atg12[UBL]–Atg5-Atg16N (PDB ID: 3W1S) visualizes the Atg12 UBL domain (pale cyan) in the conjugate with Atg5 (light pink) and the Atg16 N-terminal helix bound to Atg5. In the yeast S. cerevisiae, the covalent bond is between G186 of Atg12 and K149 of Atg5. Atg12 contains a long IDPR (black dashed line) of unknown conformation in the conjugate. Monomeric Atg16 is an intrinsically disordered protein (black), where the interaction with Atg5, dimerization, and membrane binding induce α-helical structures. The dimerizing coiled-coil domain (PDB ID: 3A7P) in the middle of Atg16 brings two heterotrimers together. The C-terminal region of Atg16 (residues 113–131) forms an amphipathic helix (dashed line) that inserts into the lipid bilayer and anchors the Atg12–Atg5-Atg16 complex in the membrane. (B) Monomeric ATG12–ATG5-ATG16L1 in a complex with the ATG3 FR. The crystal structure of human ATG3 FR-ATG12[UBL]–ATG5-ATG16L1N (PDB ID: 4NAW) visualizes the FR of ATG3 (black helix) bound to the UBL domain of ATG12 (pale cyan). The disordered N terminus of ATG12 has an unknown conformation in the conjugate with ATG5 (light pink). The covalent bond is between G140 of ATG12 and K130 of ATG5. An amphipathic helix downstream of the ATG5-binding helix (black) is formed upon membrane binding. The coiled-coil domain (black) (PDB ID: 7XFR), which dimerizes, carries at the C terminus a binding region (BR) for one molecule of WIPI2B. The second binding site for WIPI2B is downstream of the coiled-coil domain. RAB33B binds to the coiled-coil domain in between the two WIPI2B-binding sites. The coiled-coil domain is connected via a disordered region to the WD40 domain (gray) that exhibits a β-propeller architecture (PDB ID: 5NUV). The relatively long connecting IDPR carries two membrane-binding regions that are preceded by the RB1CC1 binding site.](/cms/asset/ee3ba72e-8def-4ac0-99ec-b89066a68039/kaup_a_2357496_f0003_oc.jpg)
Although Atg16/ATG16L1 functions as a stable dimer, monomeric Atg16 from yeast is the most dynamic and unstable protein of all components in the Atg8 conjugation machinery. This behavior originates in the amino acid sequence that endows Atg16 with the characteristics of an intrinsically disordered protein [Citation49]. A portion of the disordered N terminus in Atg16 folds into an α-helix upon binding to Atg5 that is coupled with Atg12 in the Atg12–Atg5 covalent conjugate [Citation5,Citation6,Citation8] (). Before the Atg5-Atg16 interaction takes place, a disordered segment between residues D101 and V112 of Atg16 binds the Atg21 -propellers that bind phosphoinositides/PROPPIN, which recruits Atg16 to phosphatidylinositol-3-phosphate-enriched membranes [Citation50]. This segment is located in the middle part of the protein containing a heptad repeat that is, typically for coiled-coil domains [Citation18], disordered in a monomeric form and folds into a parallel coiled-coil domain in the homodimer [Citation12]. We showed [Citation49] that the C-terminal sequence between residues 113–131 (S. cervisiae numbering) forms an amphipathic helix that has a coiled-coil-like propensity and a hydrophobic face that inserts into the membrane. In comparison, the coiled-coil domain that homodimerizes is not amphipathic and is dispensable for membrane binding. Disruption by mutagenesis in the amphipathic helix or homodimerization domain makes Atg16 dysfunctional in Atg8 lipidation and autophagy. A recent study [Citation45] confirmed the membrane-binding capability of the C-terminal region in Atg16 when it showed that deletion of 32 residues at the Atg16 C terminus abolishes localization of the Atg12–Atg5-Atg16 complex on giant unilammelar vesicles (GUVs) but retains a dimeric structure of the complex. A membrane-bound model of the monomeric AlphaFold2 structure of Atg16 visualizes the residues in the protein-membrane interface. Multisite mutations of these Atg16 residues (C-terminal segments L117-V128 and W131-I146) to alanine cause defects in autophagy activity, Atg8–PE conjugation, and recruitment of the Atg12–Atg5-Atg16 complex on GUVs.
Association of the E3-like enzyme with the membrane has significant implications. Atg8, E1, E2, and E3 together form a membrane scaffold and transform the morphology of the phagophore. The membrane scaffold, associating with membranes via the membrane-binding region of Atg16, is proposed to be an essential force in driving morphological changes of prolate GUVs toward the in-bud shaped GUVs, where the in-bud form is reminiscent of the phagophore [Citation45]. A mammalian homolog of Atg16, ATG16L1, has a more complex architecture. ATG16L1 carries a long IDPR upstream of a well-folded globular domain, WD40 (). The IDPR serves as a binding site for multiple physiological partners of ATG16L1 and for protein dimerization via a coiled-coil domain. In particular, residues 11–28 fold into an α-helix upon binding to ATG5, whereas residues 29–46 fold into an amphipathic α-helix upon insertion into the phagophore [Citation51]. The coiled-coil domain binds RAB33B [Citation52] and WIPI2B [Citation53] and disordered sequences downstream of the coiled-coil domain bind another WIPI2B (residues 207–230), RB1CC1/FIP200 (residues 229–249) [Citation54,Citation55], and the endosomal membrane (residues 266–319) [Citation51]. Thus, a large IDPR functions as a flexible hub organizing and engaging ATG16L1 in the autophagy machinery. If membrane shaping by an interacting web among human homologs of Atg8, E1, E2, and E3 exists as it does in yeast, then scaffolding on the membrane could be one of the reasons for the unique architecture of the E3-like enzyme in autophagy.
In this commentary, we focus on the Atg8 conjugation machinery through an IDPR lens to reveal a functional impact of protein structural flexibility in the Atg8 lipidation reactions. This summary of mechanisms mediated by high-plasticity regions illustrates how IDPRs bring an action and movement to a globular protein body, analogously to the manner in which hands and legs allow a task to be accomplished and provide mobility to the torso of a human body. Because a rigid body is a carrier of flexible parts, one cannot exist without the other. In the case of proteins, the rigid structural parts cannot be studied without also examining the intrinsically disordered regions to be able to understand how proteinaceous machines operate.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hong SB, Kim BW, Kim JH, et al. Structure of the autophagic E2 enzyme Atg10. Acta Crystallogr D Biol Crystallogr. 2012 Oct;68(Pt 10):1409–1417. doi: 10.1107/S0907444912034166 PubMed PMID: 22993095
- Hong SB, Kim BW, Lee KE, et al. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol. 2011 Nov 6;18(12):1323–1330. doi: 10.1038/nsmb.2165 PubMed PMID: 22056771
- Kaiser SE, Mao K, Taherbhoy AM, et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nat Struct Mol Biol. 2012 Dec;19(12):1242–1249. doi: 10.1038/nsmb.2415 PubMed PMID: 23142976
- Kumanomidou T, Mizushima T, Komatsu M, et al. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J Mol Biol. 2006 Jan 27;355(4):612–618. doi: 10.1016/j.jmb.2005.11.018 PubMed PMID: 16325851
- Matsushita M, Suzuki NN, Obara K, et al. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem. 2007 Mar 2;282(9):6763–6772. doi: 10.1074/jbc.M609876200 PubMed PMID: 17192262
- Noda NN, Fujioka Y, Hanada T, et al. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013 Feb;14(2):206–211. doi: 10.1038/embor.2012.208 PubMed PMID: 23238393
- Noda NN, Satoo K, Fujioka Y, et al. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell. 2011 Nov 4;44(3):462–475. doi: 10.1016/j.molcel.2011.08.035 PubMed PMID: 22055191
- Otomo C, Metlagel Z, Takaesu G, et al. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013 Jan;20(1):59–66. doi: 10.1038/nsmb.2431 PubMed PMID: 23202584
- Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J. 2009 May 6;28(9):1341–1350. doi: 10.1038/emboj.2009.80 PubMed PMID: 19322194
- Sugawara K, Suzuki NN, Fujioka Y, et al. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J Biol Chem. 2005 Dec 2;280(48):40058–40065. doi: 10.1074/jbc.M509158200 PubMed PMID: 16183633
- Yamada Y, Suzuki NN, Hanada T, et al. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem. 2007 Mar 16;282(11):8036–8043. doi: 10.1074/jbc.M611473200 PubMed PMID: 17227760
- Fujioka Y, Noda NN, Nakatogawa H, et al. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. 2010 Jan 8;285(2):1508–1515. doi: 10.1074/jbc.M109.053520 PubMed PMID: 19889643
- Bajagic M, Archna A, Busing P, et al. Structure of the WD40-domain of human ATG16L1. Protein Sci. 2017 Sep;26(9):1828–1837. doi: 10.1002/pro.3222 PubMed PMID: 28685931
- Pantoom S, Konstantinidis G, Voss S, et al. RAB33B recruits the ATG16L1 complex to the phagophore via a noncanonical RAB binding protein. Autophagy. 2021 Sep;17(9):2290–2304. doi: 10.1080/15548627.2020.1822629 PubMed PMID: 32960676
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000 Nov 15;41(3):415–427. doi: 10.1002/1097-0134(20001115)41:3<415:AID-PROT130>3.0.CO;2-7 PubMed PMID: 11025552
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005 Mar;6(3):197–208. doi: 10.1038/nrm1589 PubMed PMID: 15738986
- Habchi J, Tompa P, Longhi S, et al. Introducing protein intrinsic disorder. Chem Rev. 2014 Jul 9;114(13):6561–6588. doi: 10.1021/cr400514h PubMed PMID: 24739139
- van der Lee R, Buljan M, Lang B, et al. classification of intrinsically disordered regions and proteins. Chem Rev. 2014 Jul 9;114(13):6589–6631. doi: 10.1021/cr400525m PubMed PMID: WOS:000338980600003
- Ambadipudi S, Zweckstetter M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin Drug Discov. 2016;11(1):65–77. doi: 10.1517/17460441.2016.1107041 PubMed PMID: 26549326
- Babu MM, van der Lee R, de Groot NS, et al. Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol. 2011 Jun;21(3):432–440. doi: 10.1016/j.sbi.2011.03.011 PubMed PMID: 21514144
- Biesaga M, Frigole-Vivas M, Salvatella X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr Opin Chem Biol. 2021 Jun;62:90–100. doi: 10.1016/j.cbpa.2021.02.009 PubMed PMID: 33812316
- Santofimia-Castano P, Rizzuti B, Xia Y, et al. Targeting intrinsically disordered proteins involved in cancer. Cell Mol Life Sci. 2020 May;77(9):1695–1707. doi: 10.1007/s00018-019-03347-3 PubMed PMID: 31667555
- Uversky VN, Dave V, Iakoucheva LM, et al. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem Rev. 2014 Jul 9;114(13):6844–6879. doi: 10.1021/cr400713r PubMed PMID: 24830552
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019 Jan 10;176(1–2):11–42. doi: 10.1016/j.cell.2018.09.048 PubMed PMID: WOS:000455410800006
- Saha S, Panigrahi DP, Patil S, et al. Autophagy in health and disease: A comprehensive review. Biomed Pharmacother. 2018 Aug;104:485–495. doi: 10.1016/j.biopha.2018.05.007 PubMed PMID: 29800913
- Popelka H. Dancing while self-eating: Protein intrinsic disorder in autophagy. Prog Mol Biol Transl Sci. 2020;174:263–305. PubMed PMID: 32828468.
- Popelka H, Uversky VN. Theater in the self-cleaning cell: intrinsically disordered proteins or protein regions acting with membranes in autophagy. Membranes. 2022 Apr 24;12(5). 457. doi: 10.3390/membranes12050457 PubMed PMID: 35629783
- Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000 Nov 23;408(6811):488–492. doi: 10.1038/35044114 PubMed PMID: 11100732
- Schwarten M, Stoldt M, Mohrluder J, et al. Solution structure of Atg8 reveals conformational polymorphism of the N-terminal domain. Biochem Biophys Res Commun. 2010 May 7;395(3):426–431. doi: 10.1016/j.bbrc.2010.04.043 PubMed PMID: WOS:000277801200026
- Krichel C, Mockel C, Schillinger O, et al. Solution structure of the autophagy-related protein LC3C reveals a polyproline II motif on a mobile tether with phosphorylation site. Sci Rep. 2019 Oct 2;9(1):14167. doi: 10.1038/s41598-019-48155-8 PubMed PMID: 31578424
- Kuang Y, Ma K, Zhou C, et al. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy. 2016 Dec;12(12):2363–2373. doi: 10.1080/15548627.2016.1238552 PubMed PMID: 27653272
- Kumeta H, Watanabe M, Nakatogawa H, et al. The NMR structure of the autophagy-related protein Atg8. J Biomol NMR. 2010 Jul;47(3):237–241. doi: 10.1007/s10858-010-9420-1 PubMed PMID: 20428927
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007 Jul 13;130(1):165–178. doi: 10.1016/j.cell.2007.05.021 PubMed PMID: 1763206
- Zhang W, Nishimura T, Gahlot D, et al. Autophagosome membrane expansion is mediated by the N-terminus and cis-membrane association of human ATG8s. Elife. 2023 Jun 8;12. doi: 10.7554/eLife.89185 PubMed PMID: 37288820
- Maruyama T, Noda NN. Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. J Antibiot (Tokyo). 2017 Sep 13;71(1):72–78. doi: 10.1038/ja.2017.104 PubMed PMID: 28901328
- Yamaguchi M, Satoo K, Suzuki H, et al. Atg7 activates an autophagy-essential ubiquitin-like protein Atg8 through multi-step recognition. J Mol Biol. 2018 Feb 2;430(3):249–257. doi: 10.1016/j.jmb.2017.12.002 PubMed PMID: 29237558
- Popelka H, Uversky VN, Klionsky DJ. Identification of Atg3 as an intrinsically disordered polypeptide yields insights into the molecular dynamics of autophagy-related proteins in yeast. Autophagy. 2014 Jun;10(6):1093–1104. doi: 10.4161/auto.28616 PubMed PMID: 24879155
- Zheng YM, Qiu Y, Grace CRR, et al. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat Commun. 2019 Aug 9;10. PubMed PMID: WOS:000480234500014. doi: 10.1038/s41467-019-11435-y
- Ye Y, Tyndall ER, Bui V, et al. An N-terminal conserved region in human Atg3 couples membrane curvature sensitivity to conjugase activity during autophagy. Nat Commun. 2021 Jan 14;12(1):374. doi: 10.1038/s41467-020-20607-0 PubMed PMID: 33446636
- Nath S, Dancourt J, Shteyn V, et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol. 2014 May;16(5):415–424. doi: 10.1038/ncb2940 PubMed PMID: 24747438
- Rao S, Skulsuppaisarn M, Strong LM, et al. Three-step docking by WIPI2, ATG16L1, and ATG3 delivers LC3 to the phagophore. Sci Adv. 2024 Feb 9;10(6):eadj8027. doi: 10.1126/sciadv.adj8027 PubMed PMID: 38324698
- Nishimura T, Lazzeri G, Mizushima N, et al. Unique amphipathic alpha helix drives membrane insertion and enzymatic activity of ATG3. Sci Adv. 2023 Jun 23;9(25):eadh1281. doi: 10.1126/sciadv.adh1281 PubMed PMID: 37352354
- Metlagel Z, Otomo C, Takaesu G, et al. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Natl Acad Sci USA. 2013 Nov 19;110(47):18844–18849. doi: 10.1073/pnas.1314755110 PubMed PMID: 24191030
- Yamaguchi M, Noda NN, Nakatogawa H, et al. Autophagy-related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3-Atg8 interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. J Biol Chem. 2010 Sep 17;285(38):29599–29607. doi: 10.1074/jbc.M110.113670 PubMed PMID: 20615880
- Alam JM, Maruyama T, Noshiro D, et al. Complete set of the Atg8–E1–E2–E3 conjugation machinery forms an interaction web that mediates membrane shaping. Nat Struct Mol Biol. 2023 Dec 6. 31(1):170–178. doi: 10.1038/s41594-023-01132-2 PubMed PMID: 38057553
- Popelka H, Lahiri V, Hawkins WD, et al. The intrinsically disordered N Terminus in Atg12 from Yeast is necessary for 565 the functional structure of the protein. Int J Mol Sci. 2023 Oct 10;24(20). 15036. doi: 10.3390/ijms242015036 PubMed PMID: 37894717
- Sakoh-Nakatogawa M, Matoba K, Asai E, et al. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol. 2013 Apr;20(4):433–439. doi: 10.1038/nsmb.2527 PubMed PMID: 23503366
- Harada K, Kotani T, Kirisako H, et al. Two distinct mechanisms target the autophagy-related E3 complex to the pre-autophagosomal structure. Elife. 2019 Feb 27;8. doi: 10.7554/eLife.43088 PubMed PMID: 30810528
- Popelka H, Reinhart EF, Metur SP, et al. Membrane binding and homodimerization of Atg16 via two distinct protein regions is essential for autophagy in Yeast. J Mol Biol. 2021 Mar 5;433(5). 166809. doi: 10.1016/j.jmb.2021.166809 PubMed PMID: WOS:000617675100005
- Munzel L, Neumann P, Otto FB, et al. Atg21 organizes Atg8 lipidation at the contact of the vacuole with the phagophore. Autophagy. 2021 Jun;17(6):1458–1478. doi: 10.1080/15548627.2020.1766332 PubMed PMID: 32515645
- Lystad AH, Carlsson SR, de la Ballina LR, et al. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. 2019 Mar;21(3):372–383. doi: 10.1038/s41556-019-0274-9 PubMed PMID: 30778222
- Metje-Sprink J, Groffmann J, Neumann P, et al. Crystal structure of the Rab33B/Atg16L1 effector complex. Sci Rep. 2020 Jul 31;10(1):12956. doi: 10.1038/s41598-020-69637-0 PubMed PMID: 32737358
- Gong X, Wang Y, Tang Y, et al. ATG16L1 adopts a dual-binding site mode to interact with WIPI2b in autophagy. Sci Adv. 2023 Mar;9(9):eadf0824. doi: 10.1126/sciadv.adf0824 PubMed PMID: 36857448
- Dooley HC, Razi M, Polson HE, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014 Jul 17;55(2):238–252. doi: 10.1016/j.molcel.2014.05.021 PubMed PMID: 24954904
- Gammoh N, Florey O, Overholtzer M, et al. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol. 2013 Feb;20(2):144–149. doi: 10.1038/nsmb.2475 PubMed PMID: 23262492