
Abstract
Several signaling pathways important for the proliferation and growth of brain cells are pathologically dysregulated in gliomas, including the epidermal growth factor receptor (EGFR). Expression of EGFR is high in neural progenitors during development and in gliomas but decreases significantly in most adult brain regions. Here we show that EGFR expression is maintained in the astrocyte ribbon of the adult human subventricular zone. The transcriptional regulation of EGFR expression is poorly understood. To investigate the role of epigenetics on EGFR regulation in the contexts of neural development and gliomagenesis, we measured levels of DNA methylation and histone H3 modifications at the EGFR promoter in human brain tissues, glioma specimens, and EGFR-expressing neural cells, acutely isolated from their native niche. While DNA was constitutively hypomethylated in non-neoplastic and glioma samples, regardless of their EGFR-expression status, the activating histone modifications H3K27ac and H3K4me3 were enriched only when EGFR is highly expressed (developing germinal matrix and gliomas). Conversely, repressive H3K27me3 marks predominated in adult white matter where EGFR is repressed. Furthermore, the histone methyltransferase core enzyme ASH2L was bound at EGFR in the germinal matrix and in gliomas where levels of H3K4me3 are high, and the histone acetyltransferase P300 was bound in samples with H3K27ac enrichment. Our studies use human cells and tissues undisturbed by cell-culture artifact, and point to an important, locus-specific role for chromatin remodeling in EGFR expression in human neural development that may be dysregulated during gliomagenesis, unraveling potential novel targets for future drug therapy.
Introduction
Diffusely infiltrating gliomas are the most frequent primary brain tumors in adults, and are universally fatal once they undergo anaplastic progression. In recent years, the molecular complexity of gliomas has emerged, with the identification of new driver mutations and epigenetic phenotypes that stratify these tumors into distinct subgroups with defined therapeutic and prognostic implications.Citation1-3 However, the prime events in gliomagenesis are still poorly understood. To appreciate how gliomas arise, we must learn more about their pre-neoplastic precursors, believed to resemble neural stem cells (NSC) and/or more committed glial progenitors, which are widely abundant and proliferative in the human brain during development, but become quiescent in adulthood.Citation4-8 Glioma cells commonly share abnormalities in pathways that normally control proliferation, migration and differentiation of neural progenitorsCitation4; one such example is signaling through the epidermal growth factor receptorCitation9,10 (EGFR) and its downstream mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) pathways.Citation11 EGFR, a member of the ErbB receptor tyrosine kinase family, is highly expressed in neural progenitors during brain developmentCitation12,13 and in activated stem cell astrocytes and transit amplifying cells in the adult rodent subventricular zone (SVZ).Citation14-16 Its expression diminishes significantly in the adult human cortex (Cx) and white matter (WM) under non-reactive conditionsCitation17-20 but is retained within the adult human SVZ astrocyte ribbonCitation17 (our data). The mechanisms maintaining high EGFR expression during human neural development and its silencing upon differentiation are not well understood and have not been previously studied at the epigenetic level. Interestingly, most diffuse gliomas, both low- and high-grade tumors, show pathological expression of EGFR.Citation21-23 Historically, EGFR overexpression in gliomas has been largely attributed to gene amplification, the activating mutation v3, and gene fusion events,Citation24,25 which overall comprise approximately half of glioblastomas (GBM) and are rarely seen in lower grade gliomas.Citation19 A few studies have begun to investigate mechanisms of EGFR overexpression in gliomas outside of genetic alterations,Citation26,27 including the role of epigenetics,Citation28,29 but none so far have scrutinized the EGFR promoter in human glioma samples. Epigenetic regulation of EGFR expression at its promoter has been recognized in other solid tumors.Citation30,31 In colorectal cancer cells, for example, dissociation of SP1 and the CREB-binding protein (CBP) from the EGFR promoter leads to its hypoacetylation and transcriptional downregulation.Citation32
We hypothesized that EGFR silencing during neural differentiation and its aberrant expression in gliomas are at least partly mediated via local epigenetic mechanisms at its proximal promoter, modulating accessibility of transcription factors for regulation of gene expression. The basic promoter region regulating expression of the EGFR gene has been previously characterized.Citation33,34 It spans the region from −450 to −170 from codon 1, which is contained within a larger ∼2 kb CpG island, lacks a classical TATA box with multiple transcriptional initiation sites, and contains transcription factor binding sites for SP1, AP1 and AP2, p53 and WT1.Citation35-40
To elucidate a possible role for epigenetics on EGFR expression in the context of human neural development and gliomagenesis, we performed a unique comparative analysis using non-cultured neural cells and germinal matrix/SVZ or WM tissues isolated from their native niche, as well as human gliomas deliberately chosen not to contain EGFR amplification/v3 mutation. We found that the EGFR promoter is a direct target for the histone modifiers ASH2L and P300, remodeling the landscape via enrichment of H3K4 trimethylation and H3K27 acetylation, respectively, in neural tissues with high expression of EGFR. Our results point to novel epigenetic mechanisms allowing for EGFR regulation that are not only important during neural development but may also mark vulnerable cell populations with tumorigenic potential.
Results
Characterization of EGFR expression in developing and adult human SVZ and in gliomas
In order to study the role of epigenetic regulation on EGFR in the contexts of neural development and gliomagenesis, we first confirmed previous findings of EGFR downregulation in adult brainCitation17 and high-expression in gliomasCitation19,21,24 in the experimental human autopsy brain tissues and glioma specimens, by immunohistochemistry (IHC). In agreement with previous literature,Citation17,18,41 we saw abundant and strongly EGFR-positive cells within the developing germinal matrix (GM) (22–40 weeks gestation, n = 5, score 3 = 80%) and postnatal infant SVZ (3 weeks-12 months, n = 6, score 3 = 100%), with a prominent decrease in both intensity of staining and number of EGFR-positive cells in the adult caudate nucleus (Cd) and WM internal capsule fibers where only rare, weakly- EGFR-positive cells were seen (23-77 years, n = 5, score 1 = 60% and score 0 = 40%) (). While EGFR-positive cells from pediatric SVZ displayed strong membranous and cytoplasmic process reactivity, cells in the adjacent gray and white matter showed weak cytoplasmic staining only (). Unlike adult Cd and WM, the astrocyte ribbon within the adult SVZ region, highlighted with immunostain for glial fibrillary acidic protein (GFAP) (Fig. S1), retained moderate-to-strong staining in most of the traversing processes in a band-like fashion, occasionally showing prominent membranous staining around nuclei of cells with astrocytic morphology (23-77 years, n = 5, score 2 = 80% and 3 = 20%) (). We also found traversing GFAP-positive and EGFR-positive processes between the astrocytic ribbon and the ependymal surface in pediatric and less frequently in adult SVZ, reminiscent of radial glia-like cell morphology (arrows in and brown inset, and in Fig. S1). Notably, the EGFR-positive cells in adult human were negative for the proliferation marker Ki67 (Fig. S1), in contrast to EGFR-positive cells described in the adult rodent SVZ.Citation14,15 Similar to protein expression, EGFR mRNA levels were high in the fetal GM, lower in the adult SVZ, and decreased drastically in the adult WM compared to fetal GM, and relative to adult Cx where expression was lowest ().
Figure 1. EGFR expression analysis in the developing and adult human SVZ and in gliomas. (A, B) Immunohistochemical (IHC) analysis of EGFR expression in non-neoplastic tissues reveals strong immunoreactivity throughout the fetal germinal matrix (GM) (score (Sc) 3 in 80%) and infant subventricular zone (SVZ) where it localizes to the astrocytic ribbon (AR) (Sc of 3 in 100%). In the adult SVZ, moderate band-like immunoreactivity is seen throughout the AR, with strongly positive membranous staining around some cells within it (inset). In the adult white matter (WM), only rare, weakly stainined cytoplasmic processes are visible, typically in association with blood vessels. Ependymal cell layer (E) indicates the beginning of the GM/ SVZ. Traversing EGFR-positive processes between the astrocytic ribbon and the ependymal surface are noted as arrows. In gliomas, both LGG and HGG show strong immunoreactivity for EGFR (Sc of 3 in ∼50% and ∼80%, respectively). Scale bar = 100 μM (20X objective). Distribution of EGFR protein expression is evaluated by semiquantitative IHC scoring as previously defined.Citation19 The presence of membraneous and cytoplasmic staining is scored on a scale from 0 (no staining) to 3 (strong staining) in adult Cd (n = 5), fetal GM (n = 5), infant SVZ at AR (n = 6), adult SVZ at AR (n = 5), adult WM (n = 5), LGG (n = 44), and HGG (n = 43). (C) The changes of EGFR mRNA expression parallel those for its protein, with significantly higher levels of EGFR in fetal GM, LGG gliomas, and HGG gliomas, compared to adult Cx and adult WM. EGFR mRNA levels are expressed as average Log2 fold change over Cx ± SE, with statistical significance relative to adult Cx (##P < 0.01; ### P < 0.001), adult WM (*P < 0.05; *** P < 0.001; one-tailed t test: *’ P < 0.05), and adult SVZ (^^P < 0.01).
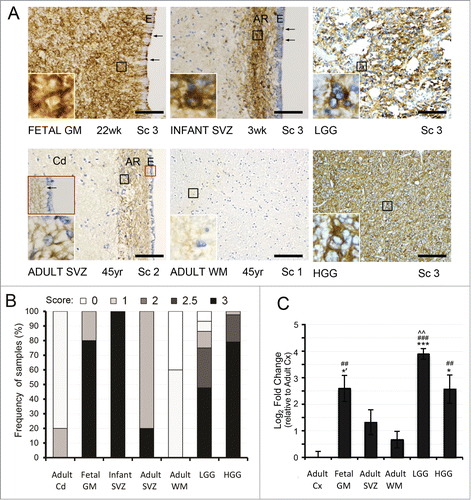
Immunohistochemical analysis of low-grade glioma (LGG) and high-grade glioma (HGG) microarrays revealed strong EGFR expression in both LGG (score 3 = 48%) and HGG (score 3 = 79%). Some LGGs also showed weak or moderate EGFR immunostaining (), in line with previously published data showing EGFR expression in many low-grade gliomas with increasing intensity of staining during high-grade progression and with the presence of EGFR gene amplification.Citation19,21-23,27 In general, EGFR mRNA levels in adult glioma specimens paralleled the expression pattern of the protein, with several fold higher transcript levels compared to adult non-neoplastic Cx and WM (). High-grade gliomas in particular showed lower than expected EGFR gene expression, likely due to the deliberate exclusion of EGFR-amplified HGG from the qRT-PCR analysis. Overall, the findings indicated concomitant changes in EGFR transcript and protein levels in the tissues examined, indicating that expression of EGFR is at least partially regulated at the level of gene transcription.
Conserved patterns of DNA methylation at EGFR during neural development and in gliomas
Given the well-established role of epigenetic control on gene expression,Citation42,43 including in the brain,Citation44 the responsiveness of EGFR to several cis and trans transcriptional regulators, and the presence of a large CpG island in its promoter region,Citation33-40 we asked if local epigenetic remodeling at the EGFR promoter plays a role in its transcriptional regulation in the developing and adult human brain and in gliomas.
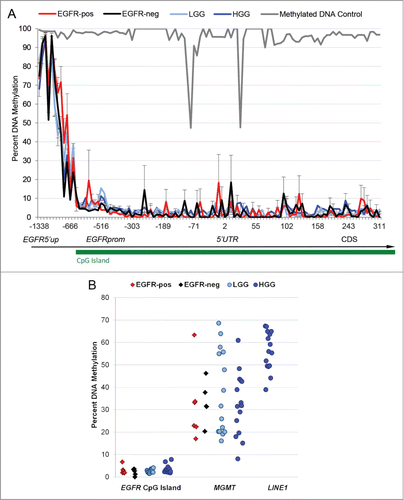
We have developed a novel method for isolation of EGFR-positive and EGFR-negative neural populations directly from human SVZ postmortem brains by fluorescence-activated cell sorting (FACS), using fluorochrome-conjugated EGF ligand to differentiate EGFR-expression status and CD44 for regional SVZ specificity, while excluding endothelium (CD34+), ependyma (CD24+), and blood cells (CD45+, CD34+) (Fig. S2). Taking advantage of this technique, we compared levels of DNA methylation at EGFR in freshly isolated EGFR+ CD44+ CD24–CD34–CD45– (EGFR-pos) and EGFR–CD44+ CD24–CD34–CD45– (EGFR-neg) cells from infant to adult human SVZ to a large group of LGGs and HGGs with heterogeneous patterns of EGFR expression. We targeted the 5′ upstream shore as well as the body of the ∼2 kb CpG island of EGFR, spanning its proximal promoter region and 5′UTR (). Next generation targeted bisulfite sequencing with depth of several thousand reads at a single base pair resolution revealed a unique and previously unreported pattern of DNA methylation at EGFR: a small hypermethylated region located ∼1.3 kb upstream of the transcriptional start site (TSS) in the 5′ CpG island shore, and a much larger hypomethylated region within the CpG island containing the EGFR basic promoter and 5′UTR regions.Citation33 Intriguingly, methylation patterns at EGFR appeared conserved among non-neoplastic neural populations, low-grade gliomas, and high-grade gliomas regardless of their EGFR expression status (). Comparing this data to published databases of global DNA methylation analyses by reduced representation bisulfite sequencing, albeit at lower depth of resolution, a very similar pattern of DNA hyper- and hypomethylation was seen within the same EGFR region in cultured undifferentiated human embryonic stem cells,45 whereas terminally differentiated NeuN + neurons showed somewhat higher CpG methylation levels at the island's boundaries.Citation46 Our analysis of several biological control genes revealed consistent hypermethylation at the promoter of the transposon element LINE-1 and an artificially methylated DNA control, and expected variable methylation at the MGMT promoterCitation24,47 ().
Figure 2. Conserved patterns of DNA methylation at the EGFR promoter in acutely isolated human neural cells and in gliomas. (A) DNA hypermethylation at the EGFR CpG island shore (EGFR5′up) and DNA hypomethylation at the CpG island spanning the EGFR promoter (EGFRprom) are noted across EGFR-positive and EGFR-negative neural populations (EGFR-pos and EGFR-neg) and in low- and high-grade glioma samples (LGG and HGG). Artificially bisulfite-methylated DNA serves as positive control at EGFR. (B) Variable diversity with higher levels of DNA methylation is seen across samples at the MGMT (-528 to -382 from TSS) and LINE1 (-327 to -27 from TSS) promoters, compared to the EGFR CpG island (-604 to 311 from TSS).
Dynamic chromatin changes at histone H3 parallel EGFR expression patterns during neural development and in gliomas
The finding of conserved CpG hypomethylation at the EGFR promoter even in EGFR-negative populations led us to speculate that chromatin remodeling, rather than simply DNA methylation, may play a dynamic role in allowing transcriptional regulation of EGFR. Posttranslational modifications at the tail of histone H3, including the activating marks of open chromatin H3K27 acetylation (H3K27ac) and H3K4 trimethylation (H3K4me3) and the repressive mark H3K27 trimethylation (H3K27me3), are important for the regulation of stem cell maintenance and differentiation by modulating accessibility to the transcriptional machinery.Citation42,43 Although these modifications have been implicated in rodent SVZ neurogenesis and differentiation,Citation48 their relevance to human EGFR promoter regulation has not been specifically addressed, despite the availability of data showing their variable presence in different cell lines.Citation49,50 We therefore asked if the chromatin landscape differs between EGFR-positive (fetal GM, adult SVZ, and gliomas) and EGFR-negative (adult WM) tissues. We focused on the region within the EGFR promoter (EGFRprom), shown to have variable enrichment for H3K27ac, H3K4me3, and, to a lesser extent, H3K27me3 in other cell lines deposited in ENCODE or published otherwise, including in cultured human neural and glioma cells with exogenous EGF stimulation.Citation49-51 Two other regions within the EGFR gene were also assessed: the 5′ CpG island shore upstream of the EGFR promoter (EGFR5′up), where DNA methylation was higher (), and the downstream EGFR intron 2 (EGFRIn2), found to be devoid of H3K27ac or H3K4me3 enrichment in a wide range of different cell lines deposited in the ENCODE database. We first measured enrichment of the activating marks H3K27ac and H3K4me3 at EGFR via chromatin immunoprecipitation (ChIP) in non-neoplastic human brain regions. We observed that in fetal GM with a high number of EGFR-expressing and actively proliferating neural progenitors, EGFRprom was highly and significantly enriched in both the activating histone modifications H3K27ac () and H3K4me3 (), compared to adult WM. In contrast, these marks were not significantly higher at EGFRIn2, and H3K4me3 was also insignificant at EGFR5′up. In adult SVZ, where many cells within the astrocyte ribbon retain moderate EGFR expression (), these activating histone modifications were also significantly enriched over WM at the EGFR promoter (). To ascertain if repression of EGFR in adult WM is mediated via repressive histone deposition, we measured levels of H3K27me3, which is often coupled with decreases in H3K27ac or H3K4me3 in differentiated cells.Citation43 Indeed, levels of H3K27me3 were significantly higher compared to H3K27ac and H3K4me3 in adult WM at the dynamic domain of the EGFR promoter (Fig. S3, top panel). In fetal GM, H3K27me3 was significantly lower than its activating counterparts H3K27ac and H3K4me3, whereas in adult SVZ, the enrichment of all marks at H3 was not statistically different (Fig. S3, middle and bottom panels). We also included positive controls for H3K27ac/H3K4me3 (beta2-microglobulin, B2M) and for H3K27me3 (HOXB7), which also served as reciprocal negative controls.Citation49-51 Scaling our data to these suggested that indeed fetal GM has much stronger enrichment for H3K27ac and H3K4me3 than for H3K27me3, and that the enrichment of repressive H3K27me3 is modest in all non-neoplastic tissues.
Figure 3. Dynamic histone modifications at the EGFR promoter in developing and adult SVZ and in gliomas. (A) The activating mark H3K27ac is significantly enriched at the EGFR promoter region (EGFRprom) and to a lesser extent at the CpG island shore (EGFR5′up) in fetal GM, adult SVZ, and LGG gliomas, compared to adult WM. The second intron of EGFR (EGFRIn2) represents internal negative control as a region lacking H3K27ac and H3K4me3 in a wide range of cell lines deposited in the ENCODE database. The 5′UTR of the housekeeping gene B2M represents positive control as a region showing high enrichment of H3K27ac and H3K4me3 in a wide range of cell lines in the ENCODE database and published.Citation43,49-51,83 IgG pull-down represents non-specific binding. (B) Significant enrichment of H3K4me3 is seen in fetal GM, adult SVZ, HGG, and LGG compared to adult WM. In A-B, the data is expressed as average of percent enrichment over input ± SE, with statistical significance relative to adult WM (*P < 0.05; ** P < 0.01; *** P < 0.001; one-tailed t test: *’ P < 0.05).
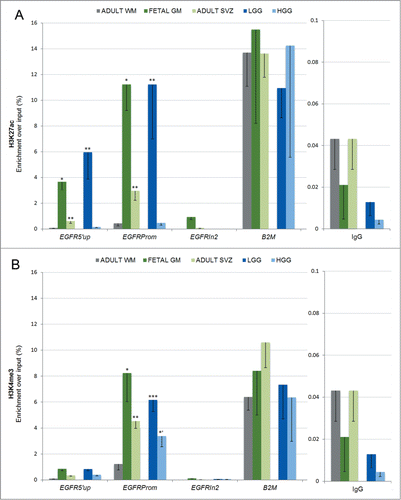
To study the possible epigenetic mechanisms mediating EGFR upregulation in the context of gliomagenesis, we selected to analyze two types of gliomas with distinct molecular phenotypeCitation3 and grade: LGG with mutation in isocitrate dehydrogenase 1 (IDH1) and HGG with wild-type IDH1, both of which showed increased EGFR expression () in the absence of EGFR gene amplification/v3 mutation. We first measured enrichment of the same activating H3 modifications: H3K27ac and H3K4me3. Interestingly, similar to fetal GM, there was robust enrichment of H3K27ac and H3K4me3 in adult LGG, whereas adult HGG specimens showed a unique pattern of H3K4me3 enrichment in the absence of acetylation at K27 (). The level of H3K27me3 at EGFRprom in glioma tissues was comparable to that of adult WM, appearing more pronounced if scaled to its positive control HOXB7 (Fig. S4). Some enrichment was also noted at EGFR5′up.
In vivo binding of ASH2L and P300 at the EGFR promoter correlates with H3K4me3 and H3K27ac enrichments
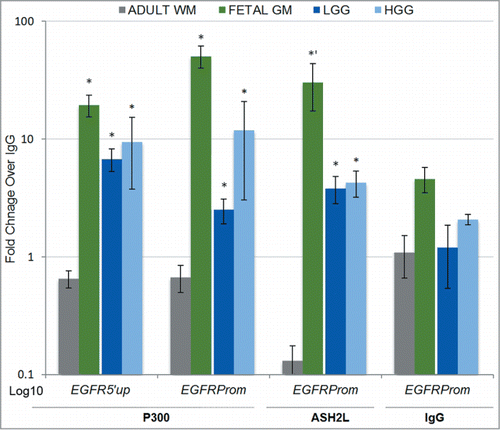
The finding of robust accumulation of the histone modifications H3K27ac and H3K4me3 at EGFRprom during neural development and in gliomas prompted us to delve deeper into the epigenetic mechanisms regulating these chromatin changes at EGFR. The P300/CBP subunit is a co-activator with intrinsic histone acetyltransferase activity for H3K27, well known for its dynamic presence at the promoters of transcriptionally active genes.Citation42 The Trithorax group (TrxG) protein ASH2L is a conserved core enzymatic subunit of the mixed lineage leukemia (MLL) histone methyltransferase complex,Citation52 and is necessary for its H3K4 trimethylation activity.Citation53 We therefore measured the in vivo binding of the P300 and ASH2L histone modifiers in tissues that showed robust enrichment of their respective catalytic targets, H3K27 acetylation and H3K4 methylation, in comparison to adult WM. P300 and ASH2L fold enrichment over IgG was significantly higher in fetal GM and in gliomas, where H3K4me3 and H3K27ac were abundant, compared to adult white matter, where these activating marks were lower and repressive H3K27 methylation predominated (). Overall, these findings implicate a role for ASH2L and P300 in maintaining inducible open epigenetic landscape at EGFR in the human fetal germinal matrix and in gliomas, which may be at least partially responsible for the high expression of this molecule in these tissues.
Figure 4. The EGFR promoter is a direct in vivo target of P300 and ASH2L during neural development and in gliomas. The histone modifiers P300 and ASH2L show significant binding above IgG background at the EGFR promoter in fetal germinal matrix and in LGG and HGG gliomas compared to adult WM, in correlation with the respectively enriched H3K27ac and H3K4me3 histone marks in these tissues. Data is expressed as average fold change enrichment over IgG ± SE with statistical signficance relative to adult WM (*P < 0.05; one-tailed t-test: *’ P < 0.05).
Discussion
To gain deeper understanding into the mechanisms of EGFR regulation in the contexts of human neural development and gliomagenesis, we performed a novel in vivo characterization of the local epigenetic landscape at the EGFR promoter using human cells/tissues derived from the fetal germinal matrix, adult SVZ, and adult white matter, as well as from glioma specimens stratified by grade and IDH1-mutation status. Epigenetic analysis using normal and diseased human tissues has revealed distinct DNA methylation profiles, serving as potentially useful translational tools for identifying tumor cell type of origin.Citation54 To our knowledge, our study is the first to undertake similar translational comparative epigenetic survey specifically at EGFR using freshly isolated brain cells or tissue from their endogenous niche, undisturbed by cell culture artifact. Although our analysis was challenging given inherent variability of age, tissue heterogeneity, and postsurgical/postmortem time, we obtained statistically significant results for robust H3K27ac and H3K4me3 enrichment during brain development and in gliomas. An important caveat in our ChIP assay results was the use of tissue, rather than isolated neural populations as done for our DNA methylation analysis. Although we attempted to perform ChIP with cell isolates, which ranged between 1,000 and 50,000 events per sort, we were unable to recover DNA above non-specific IgG pull-down. To minimize cellular heterogeneity in our tissue starting material, we used 2 opposing niches: the developing germinal matrix where the majority of cells are EGFR-expressing progenitors, and the adult white matter, where the majority of cells are EGFR-negative glia with predominance of oligodendrocytes over astrocytes.
Within these limitations, we discovered stable DNA methylation and more dynamic histone H3 modifications at the EGFR promoter, the combination of which likely mediates transcriptional regulation of EGFR expression during neural development in the germinal matrix. In essence, we found that while DNA remains hypomethylated at the EGFR promoter in both EGFR-positive and EGFR-negative FACS-isolated cells, activating H3K27ac and H3K4me3 marks are enriched in EGFR-expressing populations only. Promoter CpG islands in actively transcribed genes, such as during development, tend to be hypomethylated; in such regions tissue-specific methylation patterns strongly correlate with transcriptional activity.Citation55,56 It is therefore possible that, in adult, differentiated tissues, DNA methylation may reside somewhere else within or adjacent to the EGFR gene, and still exert a repressive effect.
Based on our data, we believe maintenance of high EGFR expression in the developing germinal matrix is achieved through an “open” chromatin state with high levels of H3K27ac and H3K4me3 upon a hypomethylated DNA promoter. In these tissues, enrichment of H3K27ac showed some permissiveness to the 5′upstream shore, while H3K4me3 was more tightly associated within one nucleosome of the TSS. This pattern of distribution is consistent with genome wide studies showing H3K4me3 enrichment primarily at the TSSCitation57 and a broader presence of H3K27ac in active promoter and enhancer regions without predilection for the TSS.Citation58 In the adult human SVZ, where only a subset of cells continues to express EGFR, the chromatin landscape showed equivalent levels of H3K27ac, H3K4me3, and H3K27me3. Given the inherent diversity of cells within the adult SVZ, it remains to be determined whether this pattern represents true bivalent H3K27me3/H3K4me3 domain of a “poised” EGFR-positive progenitor or merely reflects a mixed population of cells in a region with more heterogeneous EGFR expression. Analysis of published databases for these bivalent marks reveals predominance of H3K4me3 over H3K27me3 in cultured human astrocytes and fetal-derived neural progenitors at the EGFR promoter,Citation50,51 although exogenous administration of EGF ligand may have contributed to these enrichments. Therefore, the role of bivalency in regulating transcription at the native EGFR promoter during neural differentiation in humans remains to be fully established. Our findings in fetal GM and gliomas are also in general accord with a recently published study, which demonstrates elevated levels of H3K4me3 in undifferentiated progenitor cells within the adult baboon SVZ and in GBM tissue from patients whose tumors were located near the SVZ,Citation59 implicating a cell of origin link between germinal matrix progenitors and glioma subtypes that show similar epigenetic landscapes. Our study demonstrates the use of locus-specific epigenetic analysis for potentially linking glioma subtypes to distinct human brain regions of origin, providing evidence for comparable epigenetic landscape at EGFR between the fetal germinal matrix (but not the adult subcortical white matter) and low-grade IDH1mutant gliomas (and to a lesser extent HGG). More research is clearly needed to firmly establish this relationship.
Overall, our results suggest that DNA methylation is a stable epigenetic mark at the EGFR promoter, established early during neural development, and support the idea that DNA hypomethylation could serve as an epigenetic “primer” for subsequent dynamic modulation of EGFR expression by chromatin remodeling. “Gene priming” has been previously described in embryonic stem cells as an epigenetic phenomenon that bestows properties to a gene prior to its activation to facilitate its “on” switch when exposed to the appropriate transcription factor environment.Citation60,61 Some of these properties include DNA methylation-free windowsCitation62 and bivalent histone methylation marks that modulate Polycomb repressor binding.Citation43 We extend this concept to human neural development and speculate that maintenance of DNA hypomethylation, coupled to dynamic H3K27acetylation and H3K4 trimethylation at a specific site within the EGFR promoter, may “prime” a subset of cells within their neurogenic niche with a permissive chromatin state for this proto-oncogene throughout life, rendering survival and growth advantage as well as potential malignant transformation given further oncogenic hits.Citation63 This phenomenon is likely true for many different proto-oncogenes and/or tumor suppressors. Understanding the upstream modifying enzymes inducing such a permissive epigenetic state in tumor precursor cells, then, could have important future therapeutic implications for tumorigenesis.
Here we provide novel evidence for the role of 2 histone modifiers, P300 and ASH2L, in modulating activating chromatin modifications at EGFR during neural development and in gliomas. The P300/CBP co-activator has intrinsic histone acetyltransferase activity and has been shown to play a role in neural fate determination.Citation64 A requirement for the ASH2L complex in ESC pluripotency and maintenance of open chromatin was also recently demonstrated.Citation65 The binding of ASH2L at EGFR implies the presence of the MLL histone methyltransferase complex but future studies await determination of which of its family members, Set1a-2a and MLL1-5, is involved. MLL1 is a likely candidate, given its presence in NSC of the postnatal rodent SVZCitation66 and its implication in glioma stem cell tumorigenicity.Citation67 Whether ASH2L and P300/CBP have independent or additive effects on EGFR expression is also not yet clear but the latter seems more likely. The MLL complex, of which ASH2L is a core component, contains a transactivation domain, which recruits the co-activator P300/CBPCitation68,69 and a motif for preferentially binding at non-methylated CpG DNA sites.Citation70 In addition to its role in making genes permissive for transcriptional activation by H3K4 trimethylation and P300/CBP sequestration, the MLL complex also recruits H3K27 demethylases.Citation71,72 This could explain the lower levels of repressive H3K27me3 seen in the ASH2L-bound fetal samples at EGFRprom and its enrichment in adult white matter where the modifier was off-target. The main catalyzer of H3K27 trimethylation is the polycomb repressive complex PRC2 through its active core subunit enhancer of zeste homolog 2 (EZH2), which was shown to induce a permanent repressive switch from neurogenic to gliogenic fate at the Neurog1 promoter in mouse stem cells.Citation73 In future experiments, it would be important to assess if EZH2 is similarly recruited to the human EGFR promoter in adult white matter glia to catalyze H3K27 trimethylation and initiate terminal differentiation toward chromatin compaction and stable EGFR gene silencing. In general, identifying the transcriptional regulators bound at EGFR during its open, closed, and potentially poised state in freshly isolated human brain cells will elucidate greatly the mechanisms of its regulation during human neural development.
Within the context of glioma heterogeneity, our study provides a unique analysis of epigenetics at EGFR using both IDH1-mutant low-grade gliomas, likely of the “proneural type,” and IDH1-wild type glioblastomas, which lack EGFR-amplification.Citation3,74 Given the role of mutant IDH1 in hypermethylation of DNA and histones,Citation75 we expected to see higher levels of methylation for both DNA and repressive histone marks in our IDH1-mutant LGG tumor group. Instead, we found similar levels of DNA methylation and robust H3K27me3 enrichment in IDH1-wild type HGGs and IDH1-mutant LGGs. Enrichment of H3K27me3 was not confined to EGFRprom only but was present at EGFR5′up as well, consistent with a more global rather than promoter-specific repressive chromatin accumulation, as is frequently seen in cancer.Citation3,76,77 Our results are in contrast to 2 recent studies, which find DNA hypermethylation at EGFR ex18 in primary and recurrent high-grade gliomasCitation28 and induction of H3K9me3/H3K27me3 at EGFR upon exogenous expression of IDH1-R132H in a GBM cell line.Citation29 Regional differences of analysis (exon 18Citation28 vs. promoter region in our study), method of analysis (MSP-PCR restriction enzyme assayCitation28 vs. deep bisulfite sequencing in our study), and model system (U87MG cell linesCitation29 vs. tumor specimens in our study) most likely explain the differences seen between the above 2 studies and ours. The lack of differential hypermethylation at the EGFR proto-oncogene in IDH1-mutant vs. IDH1-wild type gliomas underscores the importance of locus-specific epigenetic control of tumor suppressor genes and proto-oncogenes in oncogenesis.Citation78
Lastly, our immunohistochemical analysis elaborated on the presence of a unique subpopulation of EGFR-positive cells within the adult SVZ, which had been previously quantitated by in-situ hybridizationCitation17,18 but not by immunohistochemistry or qRT-PCR. The cells showed astrocytic morphology and displayed moderate-to-strong membranous and cytoplasmic EGFR reactivity throughout the ventral-dorsal axis of the adult astrocyte ribbon. Interestingly, in human, these EGFR-positive cells were not actively dividing, unlike EGFR-positive cells in the adult rodent SVZ.Citation14,15 Whether EGFR-positive human SVZ cells exhibit quiescent stem cell potential in vivo remains to be determined. If so, chromatin modulation at EGFR may play an important role in keeping them “poised” for future activation.
Conclusions
The current study provides novel information regarding a dynamic epigenetic landscape at EGFR both during neural development and in gliomas, using native human tissues/cells, and opens the road for further studies to elucidate how epigenetic modifications in vulnerable cells and vulnerable genes, such as EGFR in neural cells, may play a role in gliomagenesis. EGFR tyrosine kinase inhibitors have been disappointing for glioma treatment and EGFR overexpression in general carries worse prognosis.Citation24 Therefore, inhibition of EGFR expression remains an underexplored and potentially better strategy for therapy. Discovering novel epigenetic regulatory modalities of EGFR expression in our proposed model of gliomagenesis could unravel new targets with therapeutic potential in gliomas, and offer a paradigm for further research to help us understand how gliomas may arise in susceptible individuals.
Materials and Methods
Sample collection
For non-neoplastic analyses, cells and tissues were obtained from de-identified pediatric to adult postmortem autopsy material [16 weeks gestation – 77 years, of either sex (43% male and 57% female], post mortem time < 24 h, without gross brain pathology), with available formal consent signed by appropriate next of kin, granting the use of autopsy tissue for diagnostic and research purposes, under the policy and regulation at Columbia University Medical Center and at Mount Sinai School of Medicine and tissue Biorepository with the available IRB exemption (HS#14-01007). Tissue from specific zones: fetal germinal matrix (GM), adult subventricular zone (SVZ), adult periventricular white matter (WM), and sometimes adult frontal lobe neocortex (Cx) were collected at the time of autopsy, and were either flash-frozen or fixed in 10% formalin. The same flash-frozen samples were used for chromatin and mRNA analyses. Non-neoplastic glial populations used for DNA methylation analysis were isolated from the SVZ of postnatal pediatric and adult autopsies with postmortem time of 12 hours or less. Tissue digestion was performed as described with some modifications.Citation14,15 Fluorochrome-conjugated EGF ligand and several antibodies against surface antigens were used in fluorescence-activated cell sorting (FACS) to isolate distinct populations of cells from SVZ tissue that had or lacked EGFR expression (EGFR-pos, n = 6, and EGFR-neg, n = 5), while excluding endothelium (CD34+), ependyma (CD24+), and blood cells (CD45+, CD34+). Upon FACS, cells were snap-frozen in PBS. For immunohistochemical analysis, similar regions were fixed in 10% formalin for ∼1 week and then paraffin embedded.
For neoplastic analyses, low-grade (LGG) and high-grade (HGG) human glioma samples were obtained snap-frozen from the Columbia Neurooncology tumor bank (de-identified, IRB AAAJ9652-Y1M00). Tumor specimens for DNA methylation analysis included 16 LGG (WHO grade I & II: pilocytic astrocytomas, diffuse astrocytomas, oligodendrogliomas, oligoastrocytomas, and pleomorphic xanthoastrocytomas) and 15 HGG (WHO grade III & IV: anaplastic astrocytomas, anaplastic oligodendrogliomas, anaplastic oligoastrocytomas, and glioblastomas). For immunohistochemical analysis of gliomas, a tissue microarray was used, which contained 1 mm cores from 44 LGG (WHO grade I & II: pilocytic astrocytomas, diffuse astrocytomas, oligodendrogliomas, oligoastrocytomas, and pleomorphic xanthoastrocytomas) and 43 HGG (WHO grade III & IV: anaplastic astrocytomas, anaplastic oligodendrogliomas, anaplastic oligoastrocytomas, and glioblastomas with and without EGFR gene amplifications/v3 mutations), in triplicates.
Immunohistochemical staining and semi-quantitative scoring analysis
Paraffin embedded sections (4 μM) fixed in 10% formalin were deparaffinized in xylene, with subsequent rehydration in decreasing gradient of ethanol. Antigen retrieval was performed using cell-conditioning solution 1 (Ventana Medical Systems, AZ) for 60 min at 95°C. After blocking, sections were incubated with primary anti-EGFR antibody (280005, Life Technologies, 1:100) for 32 min, and after washes, with a secondary anti-mouse antibody for 20 min on automated strainers at room temperature (Ventana XT). Anti-GFAP and anti-Ki67 antibodies were also used (760-4345/ 790-4286, Ventana Medical Systems, AZ, ready-to-use) with anti-rabbit secondary antibody. EGFR/GFAP/Ki67 immunoreactivity was detected by means of the Ultraview Universal DAB Detection Kit (760-500, Ventana Medical Systems, AZ). Nuclei were counterstained with hematoxylin. For analysis, the presence of membranous or cytoplasmic EGFR staining was scored on a scale of 0 to 3, as follows: 0, absence of immunoreactivity; 1, at least 10% of cells with weak immunoreactivity in a 200× microscopic field; 2, at least 10% of cells with moderate immunoreactivity in a 200× microscopic field; 2.5, < 50% of strongly immunoreactive cells and 3, ≥ 50% of strongly immunoreactive cells within a 200× microscopic field.Citation19,79
DNA methylation analysis
DNA methylation levels were analyzed via targeted bisulfite deep sequencing.Citation80 Briefly, 1 μg of genomic DNA from ∼15,000 or more sorted human SVZ glial cells or glioma tissues was bisulfite-converted, captured by amplification via the Fluidigm Access Array system using bisulfite-specific primers that spanned CpG islands of interest at EGFR and control genes (Table S1), was subjected to deep sequencing using Illumina MiSeq, and was computationally decoded into BedGraph files. In rare populations with lower DNA yield, an additional amplification step was performed using the Fluidigm Access Array chip prior to sequencing.
Chromatin immunoprecipitation and qPCR analysis
Chromatin modifications were assessed via chromatin immunoprecipitation (ChIP) assays. Chromatin was extracted from freshly snap-frozen gliomas (six 1-mm3 punches: LGG, n = 6; HGG, n = 11; Ash2L, n = 5; P300, n = 2) or from frozen post-mortem brain tissues (twelve 1-mm3 punches: adult WM, n = 6; adult SVZ, n = 6; fetal GM, n = 4), sheared to approximately 150-400 bp, and subjected to immunoprecipitation with ChIP-grade antibodies against H3K27ac (ab4729, Abcam, 5 μg), H3K4me3 (07-473, Millipore, 5 μg), H3K27me3 (ab6002, Abcam, 5 μg), P300 (sc-585, Santa Cruz, 5 μg), ASH2L (MABE72, Millipore, 5 μg), or with IgG mouse/rabbit as negative control (12-371/12-370, Millipore, 5 μg) after pre-incubated with magnetic Dynabeads from the appropriate species (11201D or 11203D, Invitrogen). We followed a modified microChIP protocol optimized for small tissue biopsies.Citation81,82 Ten percent of the lysate was saved as “input” for normalization analysis. The chromatin-associated DNA was purified using the QIAquick PCR Purification Kit (28104, Qiagen, Valencia, CA) and quantified by qPCR (Stratagene Mx3000P) using primers designed within (EGFRprom) and outside (EGFR5′up, EGFRIn2) of the EGFR promoter and within the promoter/5′UTR of B2M, HOXB7, and Olig2 (Table S1). The data was analyzed as percent input recovery or as fold change relative to IgG (2−ΔΔCt method) whenever recovery was less than 1%. Melting curves were analyzed to ensure specificity of the primers. The efficiency for all primers was calculated, deemed appropriate, and accounted for in the analysis. Mean and standard error (SE) values were determined for each percent input/fold difference.
RNA isolation and mRNA expression analysis
Frozen brain tissue (∼60 mg) of the same specimens used for chromatin analysis was also used for EGFR mRNA quantification by qRT-PCR. Total RNA was isolated using the Trizol extraction method following manufacturer's instructions (15596, Life Technologies, Carlsbad, CA). Briefly, tissues were homogenized in Trizol using dounce glass grinders (Wheaton, Millville, NJ) until no visible traces of tissue were seen. RNA was separated from DNA and proteins after addition of 0.2 V chloroform and centrifugation at 12,000 g for 15 min at 4°C. Total RNA contained in the aqueous part was precipitated with 0.5 V of isopropanol and centrifuged at 12,000 g for 10 min at 4°C. After washing the pellet with 75% ethanol, RNA was dissolved in water and DNase treated for 15 min at room temperature using the RNase-Free DNase Kit (79254, Qiagen, Valencia, CA), cleaned with the RNeasy MinElute Cleanup Kit (74204, Qiagen, Valencia, CA), and quantified with the Nanodrop 1000 spectrophotometer (Thermo Scientific, Asheville, NC). Total RNA of 160 ng was used to generate cDNA using the High-Capacity RNA-to-cDNA Kit (4387406, Life Technologies, Carlsbad, CA). EGFR mRNA was quantitated by qRT-PCR in adult Cx (n = 6), fetal GM (n = 4), adult SVZ (n = 6), adult WM (n = 6), LGG (n = 6), and HGG (n = 7). PCR reactions were carried out in duplicates using a SYBR-Green based system (101414, Quanta Biosciences, Gaithersburg, MD) in a 7900HT Fast Real-Time PCR System (Life Technologies, Carlsbad, CA). Expression of the housekeeping gene GAPDH was used as an internal control. All primers used for the reactions were designed to span exon-exon junctions in order to minimize genomic contamination (Table S1). Melting curves were analyzed to ensure specificity of the primers.
Statistical analysis
For qRT-PCR analyses, statistical significance was determined using ANOVA and Bonferroni correction. For ChIP analyses, statistical significance was determined by 2-tailed Student's t-test with a significance threshold of P < 0.05. For analyses including non-parametric samples (P < 0.05 by Shapiro-Wilk test for n < 30) we used Kruskal-Wallis one-way analysis of variance instead of ANOVA and posterior post-hoc Mann-Whitney U test instead of Student's t-test. For those samples showing a normal distribution but non-homogeneous variances (P < 0.05 by Levene's test), the Student's t-tests were corrected.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank the following groups and people: the epigenetics core facility at Columbia University Medical Center (CUMC), director Dr. Benjamin Tycko and Dr. Claire LeFave, for performing the sequencing and computational analysis of the DNA methylation data; Jordan Goldberg for helping with the DNA methylation data analysis; the Autopsy service at CUMC and the Pathology department at Mount Sinai, including the autopsy director Dr. Mary Fowkes, the pediatric pathologist Dr. Margret Magid, and the tissue biorepository director Dr. Michael Donovan, for facilitating tissue collection; the FACS core facilities at CUMC Immunology (CCTI) and Herbert Irving Centers for help with sorting human neural populations; Georgi Petrov for assistance with graphics; the Biostatistics Shared Core Facility at Mount Sinai for statistical consultation and assistance; and Drs. Martin Walsh and Eric Nestler for critical review of the manuscript.
Supplemental_Material.docx
Download MS Word (22.4 MB)Funding
The research was supported by the Irving Institute Trials Office Pilot Award grant, NIH NCATS UL1 TR000040 (NMT) and by NIH NINDS R01 NS074039 (FD). Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10OD020056.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Yong RL, Tsankova NM. Emerging interplay of genetics and epigenetics in gliomas: a new hope for targeted therapy. Semin Pediatr Neurol 2015; 22:14-22; http://dx.doi.org/10.1016/j.spen.2014.12.004
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 2014; 14:92-107; PMID:24457416; http://dx.doi.org/10.1038/nrc3655
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155:462-77; PMID:24120142; http://dx.doi.org/10.1016/j.cell.2013.09.034
- Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med 2005; 353:811-22; PMID:16120861; http://dx.doi.org/10.1056/NEJMra043666
- Canoll P, Goldman JE. The interface between glial progenitors and gliomas. Acta Neuropathol 2008; 116:465-77; PMID:18784926; http://dx.doi.org/10.1007/s00401-008-0432-9
- Quinones-Hinojosa A, Chaichana K. The human subventricular zone: a source of new cells and a potential source of brain tumors. Exp Neurol 2007; 205:313-24; PMID:17459377; http://dx.doi.org/10.1016/j.expneurol.2007.03.016
- Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, Lawton MT, McDermott MW, Parsa AT, Manuel-Garcia Verdugo J, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 2004; 427:740-4; PMID:14973487; http://dx.doi.org/10.1038/nature02301
- Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 2012; 149:36-47; PMID:22464322; http://dx.doi.org/10.1016/j.cell.2012.03.009
- Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J Neurosci 2008; 28:914-22; PMID:18216199; http://dx.doi.org/10.1523/JNEUROSCI.4327-07.2008
- Galvez-Contreras AY, Quinones-Hinojosa A, Gonzalez-Perez O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front Cell Neurosci 2013; 7:258; PMID:24381541; http://dx.doi.org/10.3389/fncel.2013.00258
- Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer 2010; 10:319-31; PMID:20414201; http://dx.doi.org/10.1038/nrc2818
- Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 1996; 175:1-13; PMID:8608856; http://dx.doi.org/10.1006/dbio.1996.0090
- Burrows RC, Wancio D, Levitt P, Lillien L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron 1997; 19:251-67; PMID:9292717; http://dx.doi.org/10.1016/S0896-6273(00)80937-X
- Pastrana E, Cheng LC, Doetsch F. Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc Natl Acad Sci U S A 2009; 106:6387-92; PMID:19332781; http://dx.doi.org/10.1073/pnas.0810407106
- Codega P, Silva-Vargas V, Paul A, Maldonado-Soto AR, Deleo AM, Pastrana E, Doetsch F. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron 2014; 82:545-59; PMID:24811379; http://dx.doi.org/10.1016/j.neuron.2014.02.039
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 2002; 36:1021-34; PMID:12495619; http://dx.doi.org/10.1016/S0896-6273(02)01133-9
- Weickert CS, Webster MJ, Colvin SM, Herman MM, Hyde TM, Weinberger DR, Kleinman JE. Localization of epidermal growth factor receptors and putative neuroblasts in human subependymal zone. J Comp Neurol 2000; 423:359-72; PMID:10870078; http://dx.doi.org/10.1002/1096-9861(20000731)423:3%3c359::AID-CNE1%3e3.0.CO;2-0
- Chong VZ, Webster MJ, Rothmond DA, Weickert CS. Specific developmental reductions in subventricular zone ErbB1 and ErbB4 mRNA in the human brain. Int J Dev Neurosci 2008; 26:791-803; PMID:18662768; http://dx.doi.org/10.1016/j.ijdevneu.2008.06.004
- Burel-Vandenbos F, Benchetrit M, Miquel C, Fontaine D, Auvergne R, Lebrun-Frenay C, Cardot-Leccia N, Michiels JF, Paquis-Flucklinger V, Virolle T. EGFR immunolabeling pattern may discriminate low-grade gliomas from gliosis. J Neurooncol 2011; 102:171-8; PMID:20652725; http://dx.doi.org/10.1007/s11060-010-0308-4
- Ferrer I, Alcantara S, Ballabriga J, Olive M, Blanco R, Rivera R, Carmona M, Berruezo M, Pitarch S, Planas AM. Transforming growth factor-alpha (TGF-alpha) and epidermal growth factor-receptor (EGF-R) immunoreactivity in normal and pathologic brain. Prog Neurobiol 1996; 49:99-123; PMID:8844822; http://dx.doi.org/10.1016/0301-0082(96)00009-3
- Mottolese M, Natali PG, Coli A, Bigotti G, Benevolo M, Cione A, Raus L, Carapella CM. Comparative analysis of proliferating cell nuclear antigen and epidermal growth factor receptor expression in glial tumours: correlation with histological grading. Anticancer Res 1998; 18:1951-6; PMID:9677449
- Orian JM, Vasilopoulos K, Yoshida S, Kaye AH, Chow CW, Gonzales MF. Overexpression of multiple oncogenes related to histological grade of astrocytic glioma. Br J Cancer 1992; 66:106-12; PMID:1322154; http://dx.doi.org/10.1038/bjc.1992.225
- Reifenberger J, Reifenberger G, Ichimura K, Schmidt EE, Wechsler W, Collins VP. Epidermal growth factor receptor expression in oligodendroglial tumors. Am J Pathol 1996; 149:29-35; PMID:8686753
- Jansen M, Yip S, Louis DN. Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol 2010; 9:717-26; http://dx.doi.org/10.1016/S1474-4422(10)70105-8
- Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 2013; 45:1141-9; PMID:23917401; http://dx.doi.org/10.1038/ng.2734
- Zhang J, Antonyak MA, Singh G, Cerione RA. A mechanism for the upregulation of EGF receptor levels in glioblastomas. Cell Rep 2013; 3:2008-20; PMID:23770238; http://dx.doi.org/10.1016/j.celrep.2013.05.021
- Burel-Vandenbos F, Turchi L, Benchetrit M, Fontas E, Pedeutour Z, Rigau V, Almairac F, Ambrosetti D, Michiels JF, Virolle T. Cells with intense EGFR staining and a high nuclear to cytoplasmic ratio are specific for infiltrative glioma: a useful marker in neuropathological practice. Neuro Oncol 2013; 15:1278-88; PMID:23935154; http://dx.doi.org/10.1093/neuonc/not094
- Gomori E, Pal J, Kovacs B, Doczi T. Concurrent hypermethylation of DNMT1, MGMT and EGFR genes in progression of gliomas. Diagn Pathol 2012; 7:8; PMID:22264301; http://dx.doi.org/10.1186/1746-1596-7-8
- Li J, Taich ZJ, Goyal A, Gonda D, Akers J, Adhikari B, Patel K, Vandenberg S, Yan W, Bao Z, et al. Epigenetic suppression of EGFR signaling in G-CIMP+ glioblastomas. Oncotarget 2014; 5:7342-56; PMID:25277177
- Montero AJ, Diaz-Montero CM, Mao L, Youssef EM, Estecio M, Shen L, Issa JP. Epigenetic inactivation of EGFR by CpG island hypermethylation in cancer. Cancer Biol Ther 2006; 5:1494-501; PMID:17369752; http://dx.doi.org/10.4161/cbt.5.11.3299
- Wang J, Huang SK, Marzese DM, Hsu SC, Kawas NP, Chong KK, Long GV, Menzies AM, Scolyer RA, Izraely S, et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J Invest Dermatol 2015; 135:532-41; PMID:25243790; http://dx.doi.org/10.1038/jid.2014.418
- Chou CW, Wu MS, Huang WC, Chen CC. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS One 2011; 6:e18087; PMID:21464950; http://dx.doi.org/10.1371/journal.pone.0018087
- Ishii S, Xu YH, Stratton RH, Roe BA, Merlino GT, Pastan I. Characterization and sequence of the promoter region of the human epidermal growth factor receptor gene. Proc Natl Acad Sci U S A 1985; 82:4920-4; PMID:2991899; http://dx.doi.org/10.1073/pnas.82.15.4920
- Haley J, Whittle N, Bennet P, Kinchington D, Ullrich A, Waterfield M. The human EGF receptor gene: structure of the 110 kb locus and identification of sequences regulating its transcription. Oncogene Res 1987; 1:375-96; PMID:3329716
- Johnson AC, Murphy BA, Matelis CM, Rubinstein Y, Piebenga EC, Akers LM, Neta G, Vinson C, Birrer M. Activator protein-1 mediates induced but not basal epidermal growth factor receptor gene expression. Mol Med 2000; 6:17-27; PMID:10803405
- Wang X, Bolotin D, Chu DH, Polak L, Williams T, Fuchs E. AP-2alpha: a regulator of EGF receptor signaling and proliferation in skin epidermis. J Cell Biol 2006; 172:409-21; PMID:16449191; http://dx.doi.org/10.1083/jcb.200510002
- Johnson AC, Ishii S, Jinno Y, Pastan I, Merlino GT. Epidermal growth factor receptor gene promoter. Deletion analysis and identification of nuclear protein binding sites. J Biol Chem 1988; 263:5693-9; PMID:2833511
- Kageyama R, Merlino GT, Pastan I. Epidermal growth factor (EGF) receptor gene transcription. Requirement for Sp1 and an EGF receptor-specific factor. J Biol Chem 1988; 263:6329-36; PMID:3283124
- Deb SP, Munoz RM, Brown DR, Subler MA, Deb S. Wild-type human p53 activates the human epidermal growth factor receptor promoter. Oncogene 1994; 9:1341-9; PMID:8152794
- Englert C, Hou X, Maheswaran S, Bennett P, Ngwu C, Re GG, Garvin AJ, Rosner MR, Haber DA. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. EMBO J 1995; 14:4662-75; PMID:7588596
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, Gupta N, Berger MS, Huang E, Garcia-Verdugo JM, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011; 478:382-6; PMID:21964341; http://dx.doi.org/10.1038/nature10487
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074-80; PMID:11498575; http://dx.doi.org/10.1126/science.1063127
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315-26; PMID:16630819; http://dx.doi.org/10.1016/j.cell.2006.02.041
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007; 8:355-67; PMID:17453016; http://dx.doi.org/10.1038/nrn2132
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013; 153:1149-63; PMID:23664763; http://dx.doi.org/10.1016/j.cell.2013.04.037
- Kozlenkov A, Roussos P, Timashpolsky A, Barbu M, Rudchenko S, Bibikova M, Klotzle B, Byne W, Lyddon R, Di Narzo AF, et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res 2013; 42:109-27; PMID:24057217
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350-4; PMID:11070098; http://dx.doi.org/10.1056/NEJM200011093431901
- Lim DA, Alvarez-Buylla A. Adult neural stem cells stake their ground. Trends Neurosci 2014; 37:563-71; PMID:25223700; http://dx.doi.org/10.1016/j.tins.2014.08.006
- Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014; 157:580-94; PMID:24726434; http://dx.doi.org/10.1016/j.cell.2014.02.030
- Rheinbay E, Suva ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, Oksuz O, Rabkin SD, Martuza RL, Rivera MN, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep 2013; 3:1567-79; PMID:23707066; http://dx.doi.org/10.1016/j.celrep.2013.04.021
- Yoo S, Bieda MC. Differences among brain tumor stem cell types and fetal neural stem cells in focal regions of histone modifications and DNA methylation, broad regions of modifications, and bivalent promoters. BMC Genomics 2014; 15:724; PMID:25163646; http://dx.doi.org/10.1186/1471-2164-15-724
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 2006; 13:713-9; PMID:16878130; http://dx.doi.org/10.1038/nsmb1128
- Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 2006; 13:852-4; PMID:16892064; http://dx.doi.org/10.1038/nsmb1131
- Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan AC, Galm O, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res 2012; 22:407-19; PMID:21613409; http://dx.doi.org/10.1101/gr.119867.110
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128:669-81; PMID:17320505; http://dx.doi.org/10.1016/j.cell.2007.01.033
- Das R, Dimitrova N, Xuan Z, Rollins RA, Haghighi F, Edwards JR, Ju J, Bestor TH, Zhang MQ. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci U S A 2006; 103:10713-6; PMID:16818882; http://dx.doi.org/10.1073/pnas.0602949103
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science 2008; 322:1849-51; PMID:19056940; http://dx.doi.org/10.1126/science.1162253
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931-6; PMID:21106759; http://dx.doi.org/10.1073/pnas.1016071107
- Sandstrom RS, Foret MR, Grow DA, Haugen E, Rhodes CT, Cardona AE, Phelix CF, Wang Y, Berger MS, Lin CH. Epigenetic regulation by chromatin activation mark H3K4me3 in primate progenitor cells within adult neurogenic niche. Sci Rep 2014; 4:5371; PMID:24947819; http://dx.doi.org/10.1038/srep05371
- Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, Schier AF. Chromatin signature of embryonic pluripotency is established during genome activation. Nature 2010; 464:922-6; PMID:20336069; http://dx.doi.org/10.1038/nature08866
- Dillon N. Factor mediated gene priming in pluripotent stem cells sets the stage for lineage specification. Bioessays 2012; 34:194-204; PMID:22247014; http://dx.doi.org/10.1002/bies.201100137
- Xu J, Pope SD, Jazirehi AR, Attema JL, Papathanasiou P, Watts JA, Zaret KS, Weissman IL, Smale ST. Pioneer factor interactions and unmethylated CpG dinucleotides mark silent tissue-specific enhancers in embryonic stem cells. Proc Natl Acad Sci U S A 2007; 104:12377-82; PMID:17640912; http://dx.doi.org/10.1073/pnas.0704579104
- Huang S. Genetic and non-genetic instability in tumor progression: link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev 2013; 32:423-48; PMID:23640024; http://dx.doi.org/10.1007/s10555-013-9435-7
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 2001; 104:365-76; PMID:11239394; http://dx.doi.org/10.1016/S0092-8674(01)00224-0
- Wan M, Liang J, Xiong Y, Shi F, Zhang Y, Lu W, He Q, Yang D, Chen R, Liu D, et al. The trithorax group protein Ash2l is essential for pluripotency and maintaining open chromatin in embryonic stem cells. J Biol Chem 2013; 288:5039-48; PMID:23239880; http://dx.doi.org/10.1074/jbc.M112.424515
- Lim DA, Huang YC, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J, Ernst P, Alvarez-Buylla A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009; 458:529-33; PMID:19212323; http://dx.doi.org/10.1038/nature07726
- Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE, Iliopoulos O, Hjelmeland AB, Rich JN. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ 2012; 19:428-39; PMID:21836617; http://dx.doi.org/10.1038/cdd.2011.109
- Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol 2001; 21:2249-58; PMID:11259575; http://dx.doi.org/10.1128/MCB.21.7.2249-2258.2001
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 2004; 24:5639-49; PMID:15199122; http://dx.doi.org/10.1128/MCB.24.13.5639-5649.2004
- Cierpicki T, Risner LE, Grembecka J, Lukasik SM, Popovic R, Omonkowska M, Shultis DD, Zeleznik-Le NJ, Bushweller JH. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat Struct Mol Biol 2010; 17:62-8; PMID:20010842; http://dx.doi.org/10.1038/nsmb.1714
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007; 318:447-50; PMID:17761849; http://dx.doi.org/10.1126/science.1149042
- Roidl D, Hacker C. Histone methylation during neural development. Cell Tissue Res 2014; 356:539-52; PMID:24817100; http://dx.doi.org/10.1007/s00441-014-1842-8
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009; 63:600-13; PMID:19755104; http://dx.doi.org/10.1016/j.neuron.2009.08.021
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17:98-110; PMID:20129251; http://dx.doi.org/10.1016/j.ccr.2009.12.020
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483:474-8; PMID:22343901; http://dx.doi.org/10.1038/nature10860
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150:12-27; PMID:22770212; http://dx.doi.org/10.1016/j.cell.2012.06.013
- Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nature Med 2011; 17:330-9; PMID:21386836; http://dx.doi.org/10.1038/nm.2305
- Tsankova NM, Canoll P. Advances in genetic and epigenetic analyses of gliomas: a neuropathological perspective. J Neurooncol 2014; 119:481-90; PMID:24962200; http://dx.doi.org/10.1007/s11060-014-1499-x
- Popova SN, Bergqvist M, Dimberg A, Edqvist PH, Ekman S, Hesselager G, Ponten F, Smits A, Sooman L, Alafuzoff I. Subtyping of gliomas of various WHO grades by the application of immunohistochemistry. Histopathology 2014; 64:365-79; PMID:24410805; http://dx.doi.org/10.1111/his.12252
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011; 27:1571-2; PMID:21493656; http://dx.doi.org/10.1093/bioinformatics/btr167
- Dahl JA, Collas P. MicroChIP: chromatin immunoprecipitation for small cell numbers. Methods Mol Biol 2009; 567:59-74; PMID:19588085; http://dx.doi.org/10.1007/978-1-60327-414-2_4
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci 2004; 24:5603-10; PMID:15201333; http://dx.doi.org/10.1523/JNEUROSCI.0589-04.2004
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci 2008; 9:42; PMID:18442397; http://dx.doi.org/10.1186/1471-2202-9-42