
Abstract
Neurofilament heavy polypeptide (NEFH) has recently been identified as a candidate DNA hypermethylated gene within the functional breast cancer hypermethylome. NEFH exists in a complex with neurofilament medium polypeptide (NEFM) and neurofilament light polypeptide (NEFL) to form neurofilaments, which are structural components of the cytoskeleton in mature neurons. Recent studies reported the deregulation of these proteins in several malignancies, suggesting that neurofilaments may have a role in other cell types as well. Using a comprehensive approach, we studied the epigenetic inactivation of neurofilament genes in breast cancer and the functional significance of this event. We report that DNA methylation-associated silencing of NEFH, NEFL, and NEFM in breast cancer is frequent, cancer-specific, and correlates with clinical features of disease progression. DNA methylation-mediated inactivation of these genes occurs also in multiple other cancer histologies including pancreas, gastric, and colon. Restoration of NEFH function, the major subunit of the neurofilament complex, reduces proliferation and growth of breast cancer cells and arrests them in Go/G1 phase of the cell cycle along with a reduction in migration and invasion. These findings suggest that DNA methylation-mediated silencing of the neurofilament genes NEFH, NEFM, and NEFL are frequent events that may contribute to the progression of breast cancer and possibly other malignancies.
Introduction
The acquisition of DNA methylation in gene promoters is a hallmark of tumorigenesis that leads to the functional inactivation of genes.Citation1 Many candidate DNA hypermethylated genes have been identified in cancers through genome-wide screens, but the biological significance for the majority of these newly identified DNA methylated loci remains to be elucidated. To characterize the vast amount of DNA methylation data generated, comprehensive functional approaches have been initiated and novel tumor suppressor genes and pathways as well as biomarkers for early detection and prognosis prediction of cancer have been discovered.Citation2-11
Neurofilament heavy polypeptide (NEFH) has been identified as a candidate DNA hypermethylated gene within the functional breast cancer hypermethylome, a comprehensive approach to define genome-wide functional methylation changes in breast cancer that is based on whole transcriptome expression arrays on breast cancer cell lines after pharmacological inhibition of DNA methylation.Citation6 NEFH encodes the neurofilament heavy polypeptide and assembles along with neurofilament medium polypeptide (NEFM) and neurofilament light polypeptide (NEFL) into 10 nm filamentous structures known as neurofilaments.Citation12 Neurofilaments are type IV intermediate filaments that are the main structural component of the cytoskeleton in mature neuronsCitation13 determining axon caliber and conductivity for proper neuronal function.Citation14,15 Neurofilaments have been implicated in the pathology of neurological diseases,Citation16-18 but recent data also point to a role in the pathology of cancer.Citation19-21 For example, NEFL and NEFM are both located within the 8p21 chromosome region and LOH of this region has been described in several cancers including breast cancer.Citation22-24 Furthermore, aberrant expression and methylation of neurofilament genes were recently detected in esophageal squamous cell, renal cell and hepatocellular carcinoma as well as in Ewing sarcoma.Citation19,21,25,26
In the present study we used a comprehensive approach to study the epigenetic inactivation of neurofilament genes in breast cancer and the functional significance of these events. We show that DNA methylation-associated silencing of NEFH, NEFL, and NEFM in breast cancer is frequent, cancer-specific and correlates with clinical features of disease progression. Furthermore, DNA methylation-mediated inactivation of these genes occurs in other types of cancer including pancreas, gastric, and colon cancers. Restoration of NEFH expression, the main subunits of the neurofilament complex, reduces proliferation and growth of breast cancer cells and arrests them in Go/G1 phase of the cell cycle. We further observed a reduction in migration and invasion in breast cancer cells with restored NEFH expression. Together, these results suggest that DNA methylation-mediated silencing of the neurofilament genes NEFH, NEFM, and NEFL are frequent events that contribute to the progression of breast cancer and possibly many other types of cancer.
Results
Epigenetic alteration of NEFH is associated with loss of gene function in breast cancer cells
NEFH was identified as a potential DNA hypermethylated gene by the genome-wide screen for functionally DNA hypermethylated genes in breast cancer.Citation6 NEFH was transcriptionally silent in MCF7 and MDA-MB-231 cells and re-expressed ≥ 2 log-fold on the transcriptional level after treatment with the DNMT inhibitor 5-aza-2′-deoxycytidine (DAC) (33.78 log-fold re-expression for MCF-7 and 8.46 log-fold for MDA-MB-231, respectively) but not with the histone deacetylase (HDAC) inhibitor Trichostatin A (TSA) (). We validated the expression array results by RT-PCR in Mock-, DAC-, and TSA-treated MCF7 and MDA-MB-231 cells and included non-transformed and non-tumorigenic MCF10A breast cells as well as non-immortalized human mammary epithelial cells (HMEC) into our assay. We detected expression of NEFH after DAC treatment in MCF7 and MDA-MB-231 cells but not at basal level or after TSA treatment (). Interestingly, while NEFH is strongly expressed in HMECs, we did not detect expression in MCF10A cells, a spontaneously immortalized breast epithelial cell line. NEFH DNA methylation status of these samples correlated with the expression results for MCF7, MDA-MB-231, and HMECs showing complete methylation of the NEFH promoter in MCF7 and MDA-MB-231 cells but no methylation in HMECs (). Interestingly, MCF10A cells showed an unmethylated promoter status for NEFH despite the loss of NEFH expression suggesting alternative modes of inactivation.
Figure 1. Silencing of NEFH is associated with DNA promoter hypermethylation or repressive chromatin structure. (A) Appearance of NEFH within the breast cancer hypermethylome. Cell lines were treated with either 5 μmol/L DAC for 96 hours or 300 nmol/L TSA for 18 hours. Gene expression changes (analyzed on 4 × 44 K Agilent platform) are plotted by fold change (log scale) after DAC (y-axis) or TSA (x-axis) treatment. (B) Expression (mRNA) of NEFH in untreated (Mock), DAC (5 μmol/L 96 hours) or TSA (300 nmol/L18 hours) treated MDA-MB-231 and MCF7 cells as well as in untreated MCF10A and HMEC cells or normal breast (NB). (C) Methylation status of the NEFH region from +41 to +198 relative to transcription start site (TSS) in cells without treatment (Mock) or cells treated with DAC or TSA assessed by MSP. U and M marking unmethylated and methylated bands, respectively. DKO and IVD were used as control for unmethylated and methylated DNA, respectively. (D) ChIP at the NEFH promoter region from +125 bp to +296 bp relative to TSS for α-H3k4me2 and α-H3k27me3. All values are corrected for IgG background and are normalized to the relative amount of Input obtained by real-time PCR from 2 independent experiments ± SEM. Group comparisons were carried out using Student t test, P***< 0.001 and P*< 0.05.
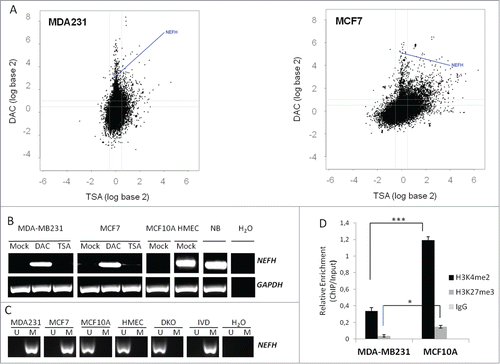
To further investigate whether the loss of NEFH expression in MCF10A cells could be due to histone modifications, we performed ChIP for the H3K4me2 and the H3K27me3 mark at the NEFH promoter in MCF10A and MDA-MB-231 cells. MCF10A cells showed a significant enrichment of the active H3K4me2 and the repressive H3K27me3 mark as compared to MDA-MB-231 cells () that have low levels of both of these marks, as this is typical for DNA methylated promoters. These results suggest that a bivalent chromatin structure at the NEFH promoter in MCF10A cells can adequately suppress the expression of NEFH, similar to the poised state of embryonic stem cells, or as previously shown for other genes.Citation7
Promoter methylation of neurofilament genes NEFH, NEFL, and NEFM is frequent in breast primary tumors and correlates with adverse clinical features
To investigate whether the association between loss of NEFH expression and NEFH promoter methylation is present in patients, we analyzed the DNA methylation and mRNA expression status of NEFH in primary tumors. Since neurofilaments are obligate heteropolymers that are composed of 3 subunits, NEFH, NEFL, and NEFM,Citation27 we also investigated the methylation and expression status of NEFL and NEFM in the 10 primary breast tumors (breast tumor samples 1T to 10T). We observed downregulation in gene expression (gene expression of tumors was normalized to normal breast tissue) and a methylated gene promoter for NEFH in 50% of samples (breast tumor samples 1T, 4T, 7T, 8T and 9T), for NEFL in 80% (breast tumor samples 1T, 2T, 3T, 4T, 5T, 6T, 7T and 9T), and for NEFM in 50% of samples (4T, 6T, 7T, 8T and 9T) (). This data suggest that methylation associated inactivation of NEFH, NEFL, and NEFM is a frequent event (50% to 80%) in breast primary tumors but not in normal breast tissues (Fig. 2B). However, some samples (i.e., sample 2 for NEFH and NEFM) showed downregulation in gene expression despite having an unmethylated gene promoter, suggesting regulation by alternative transcriptional silencing mechanisms other than DNA promoter methylation, similar to what we demonstrated for MCF10A cells above.
Figure 2. Methylation of neurofilament genes NEFH, NEFL and NEFM is frequent in breast primary tumors. (A) Methylation and quantitative mRNA expression of NEFH, NEFL and NEFM in 10 breast primary tumors (from 1T to 10T). Methylation status was assayed by MSP for the NEFH region from +41 to +198, for the NEFL region from +86 to +167 and for the NEFM region from +60 to +150 relative to TSS. U and M marking unmethylated and methylated bands, respectively. Quantitative mRNA expression in tumors is shown in fold change (log2) relative to expression in normal breast tissue. (B) Expression (mRNA) of NEFH, NEFL, NEFM and GAPDH in 5 normal breast tissues from non-cancer patients. (C) DNA methylation frequency (in %) of NEFH, NEFL, and NEFM were assessed in a cohort of 14 normal breast tissues from non-cancer patients and in a cohort of 185 primary breast cancers (BC) of stages 0 (DCIS) to 4. DNA methylation frequencies are plotted by tumor stage and are shown as an overall result. (D) Bisulfite sequencing of the NEFH region from −20 bp to +320, the NEFL region from −10 bp to +290 bp and the NEFM region from −140 bp to +240 bp relative to the TSS. Five sequencing reactions are shown for each sample representing clonal plasmids. White and black circles represent unmethylated and methylated CpG dinucleotides, respectively. NB presented the lowest methylation frequency ranging from 3.6% to 16.5% among the 3 genes on bisulfite sequencing. MCF-7 showed a high methylation frequency with almost every CpG methylated for the 3 genes while HMECs showed low methylation frequencies ranging from 11% to 25%. Breast primary tumor sample BC73 showed a methylation frequency ranging from 48% to 60% among the 3 genes.
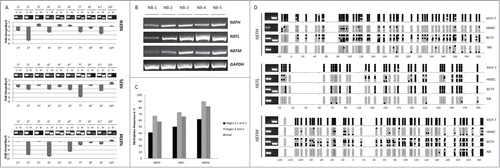
To test for tumor specific methylation of NEFH, NEFL, and NEFM and to gain a broader estimate of their methylation frequencies, we utilized a cohort of 185 primary breast cancer specimens of different clinical stages (Stages 0–4) along with 14 normal breast specimens from non-cancerous patients. Promoter methylation of the 3 neurofilament genes was not observed in the 14 normal breast specimens but in 58% of tumor samples for NEFH (108 out of 185 samples), 66% for NEFL (123 out of 185 samples) and 82% for NEFM (152 out of 185 samples) (see total frequency in ). NEFH, NEFL, and NEFM, thus, show high frequency of tumor specific methylation in vivo. We further estimated the methylation frequency in early versus late stage tumors and observed that the methylation frequency increases with tumor stage for all 3 genes [, compare stages 0, 1 and 2 (early stage) with stages 3 and 4 (late stage)]. Next, we confirmed the promoter methylation status obtained through MSP with bisulfite sequencing on selected samples. For NEFH we sequenced a region from −20 bp to +320, for NEFL from −10 bp to +290 bp and for NEFM from −140 bp to +240 bp relative to the TSS. The methylation status obtained by MSP correlated with bisulfite sequencing results for NEFH, NEFL and NEFM in MCF7 and HMEC cells, the breast primary tumor sample BC73 and normal breast (NB) (). NB, unmethylated by MSP, presented the lowest methylation frequency ranging from 3.6% to 16.5% among the 3 genes by bisulfite sequencing. MCF-7, methylated by MSP, showed a high methylation frequency with almost every CpG methylated for the 3 genes while HMECs, unmethylated by MSP, showed low methylation frequencies ranging from 11% to 25%. Breast primary tumor sample BC73, partially methylated by MSP, showed a methylation frequency ranging from 48% to 60% among the 3 genes.
We next tested for correlations between common clinicopathologic patient characteristics and gene methylation status in our cohort of 185 breast cancer patients for which well-annotated clinical follow-up data were available. Clinicopathological characteristics of the cohort are summarized in Table S1. Promoter methylation of NEFH, NEFL and NEFM was significantly associated with adverse prognostic parameters such as tumor stage, perinodal invasion, and HER2 receptor overexpression (). We further tested whether these genes were associated with differences in overall survival of breast cancer patients using a univariate and multivariate Cox regression model. Methylation of NEFL (HR = 1.81, P = 0.042) predicted for poor survival but when including age and stage into the model, the significant correlation between methylation status and survival was lost ( and Fig. S1).
Table 1. Statistical association between clinicopathological characteristics and methylation frequencies (%) of genes
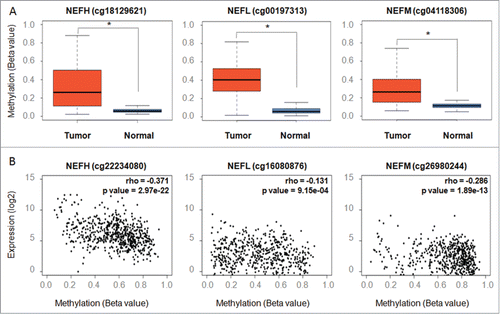
Finally, we extracted data for NEFH, NEFL, and NEFM from genome-wide DNA methylation (Illumina Infinium HumanMethylation450 array) and gene expression profiles (Illumina HiSeq RNA-SeqV2) of breast tissues from The Cancer Genome Atlas (TCGA) to validate our findings. We compared methylation levels of NEFH, NEFL, and NEFM based on 9, 11, and 20 Infinium probes within the promoter region (Table S2 for probe information), respectively, in 641 tumors vs. 96 normal breast tissues. We thereby observed significant differences in methylation for all 3 genes between tumor and normal tissues with the tumors harboring higher levels of DNA methylation ( and Table S3). We further identified modest yet significant differences in methylation between high (stage 3 and 4) and low (stage 1 and 2) stage tumors for NEFH, NEFL, and NEFM (Table S4). Integrated analysis of DNA methylation and gene expression for NEFH, NEFL, and NEFM in 638 TCGA breast tumors confirmed the inverse relationship we observed in our cell line models and our small cohort of 10 breast tumors ( and Table S5).
Figure 3. Validating methylation of NEFH, NEFL, and NEFM and its inverse correlation with gene expression in breast tissues from The Cancer Genome Atlas (TCGA). (A) Methylation levels (β values) of representative probes from the Infinium HumanMethylation450 platform located within the NEFH, NEFL and NEFM promoter show a significant difference between normal breast tissues (n = 96 ) and primary breast tumors (n = 641 ). (B) Scatter plots depicting the correlation between the gene expression (y-axis; log2 values; analyzed on IlluminaHiSeq_RNA-SeqV2 platform) and DNA methylation (x-axis; β values; analyzed on Infinium HumanMethylation450 platform) of NEFH, NEFL and NEFM in 641 primary breast cancers from the TCGA data portal. Spearman rank correlation coefficients (rho) are displayed. A P value < 0.05 was considered statistically significant.
Restoration of NEFH expression suppresses tumor cell proliferation and growth by arresting MDA-MB-231 cells in G0/G1 phase
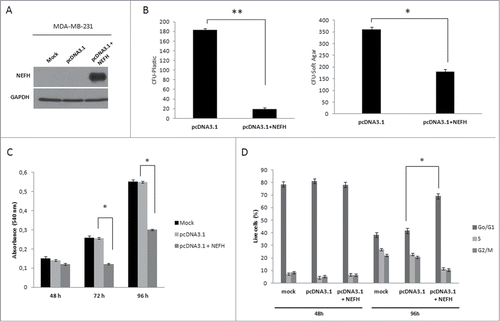
Given the tumor specific and highly frequent inactivation of neurofilament genes in breast cancer, we functionally investigated the restoration of NEFH protein, the main subunits of the neurofilament complex, in breast cancer cells. To test for a potential tumor suppressor function of NEFH in breast cancer, we transiently transfected MDA-MB-231 cells with empty vector (pcDNA3.1/V5-HisB) or NEFH expression vector (pcDNA3.1/V5-HisB+NEFH). Ninety-six hours post transfection, expression of NEFH protein was confirmed by western blot (). NEFH expression in MDA-MB-231 cells showed a growth suppressive effect in colony formation and soft agar assays (). NEFH transfected cells formed significantly less colonies than cells transfected with empty vector in both assays (colony formation: P < 0.01, soft agar: P < 0.05). To explore the observed growth suppressive effect in NEFH expressing cells further, we performed MTT assays in MDA-MB-231 cells 48, 72, and 96 hours post transfection. Cells with restored NEFH presented a significant lower proliferation rate than cells lacking NEFH expression after 72 and 96 hours (P < 0.05) () but not at 48 hours. We further performed cell cycle analysis and found an increase in number of NEFH transfected cells in Go/G1 phase and a decrease of cells in S phase 96 hours post transfection (P < 0.01) ( and Fig. S2). No difference in cell numbers between Go/G1 and S phase was observed 48 hours post transfection. These results suggest that restoration of NEFH expression suppresses tumor cell proliferation and growth by arresting cells in Go/G1 phase.
Figure 4. Restoration of NEFH function suppresses tumor cell proliferation and growth by arresting MDA-MB-231 cells in Go/G1 phase. (A) MDA-MB-231 cells were transiently transfected with pcDNA3.1 (empty vector) or pcDNA3.1-NEFH and re-expression of NEFH was confirmed 96 hours post transfection by Western blot using α-NEFH antibody and α-GAPDH as a control. (B) Tumor cell clonogenicity was assessed on plastic and in soft-agar. Cells were transiently transfected with pcDNA3.1 (empty vector) or pcDNA3.1-NEFH and replated 24 hours post transfection for selection with Geneticin/G418. After 14 d of selection, colonies were stained with Giemsa and counted. MDA-MB-231 Mock cells (not transfected but treated with Lipofectamine 2000) were used as control for Geneticin selection. After 14 d of selection all MDA-MB-231 Mock cells were dead. Data presented are the mean of 2 independent experiments ± SEM. Group comparisons were carried out using Student t test, P**< 0.01 and P*< 0.05. (C) Proliferation rates of MDA-MB-231 cells 48, 72, and 96 hours post transfection with pcDNA3.1 (empty vector) and pcDNA3.1-NEFH and MDA-MB-231 mock transfected was measured by MTT assay. Data presented are the mean of 2 independent experiments ±SEM . Group comparisons were carried out using Student t test, P < 0.05. (D) Cell cycle distribution measured with Hoechst 33258 by flow cytometry of MDA-MB-231 cells 48 and 96 hours post transfection with pcDNA3.1 (empty vector) or pcDNA3.1-NEFH. MDA-MB-231 Mock cells (untransfected but treated with Lipofectamine 2000) were used as control of the experiment. Data represent 2 independent experiments. Group comparisons were carried out using Student t test, P < 0.01.
Restoration of NEFH expression in MDA-MB-231 breast cancer cells slows tumor progression
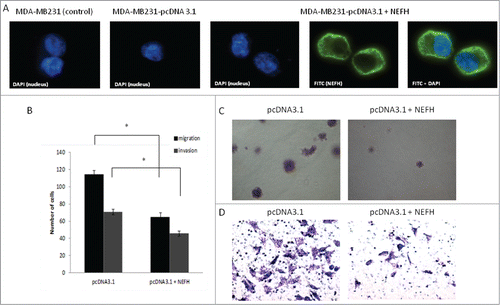
Given the functional involvement of neurofilaments in the organization of the cytoskeleton, we examined whether the ectopic overexpression of NEFH in the MDA-MB-231 cells, which do not express this protein, could affect the motility and thus migration and invasion behavior of MDA-MB-231 breast cancer cells. First, we analyzed the cellular localization of NEFH in these cells. Immunofluorescence staining of NEFH revealed a cytoplasmic localization in MDA-MB- 231 cells, which complies with a potential role of this protein in the cytoskeleton of breast cancer cells (). Next, we restored NEFH expression in MDA-MB-231 cells and performed migration and invasion assays. We observed a significant reduction in migration and invasion of NEFH expressing MDA-MB-231 cells as compared to cells lacking NEFH expression (P < 0.05 for both assays) ().
Figure 5. Restoration of NEFH expression in MDA-MB-231 breast cancer cells slows tumor progression. (A) The cellular localization of NEFH protein in MDA-MB-231 cells transiently transfected with pcDNA3.1 and pcDNA3.1-NEFH was determined by immunofluorescence with anti-NEFH (FITC). Nuclei were visualized by staining with DAPI. The NEFH protein was not expressed in MDA-MB-231 cells transfected with pcDNA3.1 and just the nuclei was visualized in these cells. MDA-MB-231 wild type cells were also stained for NEFH to determine endogenous levels of NEFH expression and the specificity of the NEFH antibody and as NEFH protein was not expressed in MDA-MB-231 wild type cells just the nuclei was visualized in these cells. (B) Invasion and migration assays in MDA-MB-231 cells transfected with pcDNA3.1 (empty vector) or pcDNA3.1-NEFH. Twenty-four hours post transfection, cells were harvested, suspended in serum-free media and added either directly to the top chamber of transwell plates (migration assay) or to the top chamber of transwell plates that were layered with 2 mg/ml matrigel (invasion assay). Growth media containing 10% serum was added to the bottom chamber and cells were allowed to migrate or invade for 24 and 48 hours respectively. Cells attached to the membrane were fixed and subsequently stained with giemsa or crystal violet. For quantification of migrated or invaded cells X fields per well were randomly selected and counted. Data presented are the mean of 2 independent experiments § SEM. Group comparisons were carried out using Student t test, P < 0.05. (C) Representative images of the invasion assay in MDA-MB-231 cells transfected with either pcDNA3.1 (empty vector) or pcDNA3.1-NEFH. (D) Representative images of the migration assay in MDA-MB-231 cells transfected with pcDNA3.1 (empty vector) or pcDNA3.1-NEFH.
Neurofilament genes NEFH, NEFL, and NEFM are frequently DNA methylated in various types of cancer
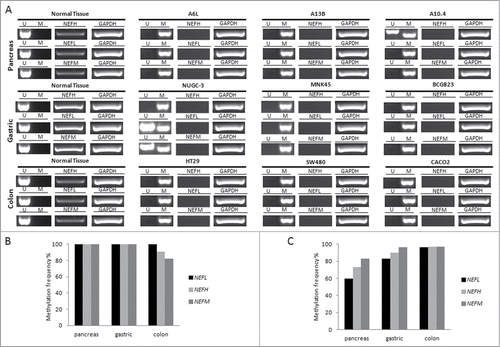
In order to analyze whether DNA methylation associated silencing of NEFH, NEFL, and NEFM occurs in other tumor types than breast cancer, we assayed the NEFH, NEFL, and NEFM expressions status in various cancer cells (pancreas, gastric and colon) and their corresponding normal cells. Furthermore, we determined the methylation status of NEFH, NEFL, and NEFM in the various cancer cells and correlated expression and methylation status. NEFH, NEFL, and NEFM were unmethylated and expressed in all normal tissues but silenced in association with a methylated gene promoter in 66.7% to 100% of the corresponding cancer cell lines (). To test whether the observed methylation induced gene silencing reflects also an event in vivo, we assayed the methylation status of NEFH, NEFL, and NEFM in primary tumor specimens of these tumor types [pancreas (n = 30), gastric (n = 30), colon (n = 128)]. We found promoter methylation of NEFH in 73%, NEFL in 60%, and NEFM in 83% of pancreas cancer specimen. In gastric and colon cancer specimen we observed even higher methylation frequencies for all 3 genes (90% for NEFH, 93% for NEFL and 97% for NEFM in gastric cancer; 97% for NEFH, 96% for NEFL and 97% for NEFM in colon cancer) (). These data indicate that methylation of the neurofilament genes is a highly frequent event in various human malignancies.
Figure 6. Promoter methylation and mRNA expression of NEFH, NEFL and NEFM in various types of cancer. (A) Promoter methylation and mRNA expression of NEFH, NEFL and NEFM was assayed in 3 representative cell lines of each cancer type by MSP and RT-PCR, respectively. U and M marking unmethylated and methylated bands, respectively. Basal expression of NEFH, NEFL, and NEFM was confirmed in normal tissue of each cancer type. GAPDH expression was assayed as control. (B) Overall DNA methylation frequency (in %) of NEFH, NEFL, and NEFM in pancreas, gastric and colon cancer cell lines. (C) Overall DNA methylation frequency (in %) of NEFH, NEFL and NEFM in primary tissue of pancreas (n = 30), gastric (n = 30), and colon cancer (n = 128).
Discussion
In the present study, we discovered the epigenetic inactivation of the neurofilament gene family in breast cancer and show that neurofilaments have functions in other cell types than neurons. Promoter methylation of NEFH, NEFL, and NEFM in breast cancer occurs in a tumor-specific manner, is frequent and associated with loss of gene expression. Although methylation appears to be the predominant mechanism for inactivation of neurofilament genes in breast cancer, we show for NEFH in MCF10A cells an alternative epigenetic silencing mechanism. Transcriptional inactivation of NEFH in those cells is associated with a bivalent chromatin pattern at the NEFH promoter, enriched for the repressive H3K27me3 and the active H3K4me2 histone marks. These results suggest that in MCF10A cells repressive chromatin at the NEFH promoter, similar to the poised state of embryonic stem cells,Citation28 can suppress the expression of NEFH. Further, we speculate that silencing of NEFH in non-tumorigenic MCF10A cells by repressive chromatin could be an early event that predisposes DNA methylation.Citation29-31 Previous studies suggested that CpG island-containing gene promoters marked with a bivalent chromatin are prone to undergo an epigenetic switch and become de novo DNA methylated in cancer.Citation30,32 The quantitative replacement of flexible, polycomb-mediated gene silencing with the more stable silencing by DNA methylationCitation33 may lead to further or more permanent repression of gene expression in cancers.Citation34
We further show that DNA methylation of neurofilament genes is not specific to breast cancers but occurs in other types of cancers as well, i.e., in pancreas, colon, and gastric cancers. These results and the recent findings that showed NEFH inactivation in association with promoter methylation in esophageal squamous cell and hepatocellular carcinomas as well as in Ewing sarcomasCitation19,21,25 suggest that neurofilament genes may be epigenetically downregulated in a wide range of malignancies and that these gene family may have importance in tumorigenesis. Using proliferation and colony formation assays, we show that NEFH possesses tumor cell growth suppressive activity in breast cancer cells. Further in-depth studies revealed that the observed growth-repressive effect is associated with an arrest of NEFH expressing cells in Go/G1 phase of the cell cycle. These findings agree with a recent study by Kim et al. that showed that esophageal cancer cells with restored NEFH expression display decreased growth and proliferation and an increased number of cells arrested in G2 phase of the cell cycle.Citation25 Kim et al. further showed that lack of NEFH expression due to epigenetic inactivation was associated with increased β-catenin expression and a subsequent elevated activation of oncogenic β-catenin downstream effectors.Citation25 Interestingly, oncogenic β-catenin downstream effectors involve genes that have a role in cell cycle progression.Citation35 Deregulation of the Wnt/β-catenin pathway and accumulation of β-catenin have been observed in breast cancerCitation36 but mutations of β-catenin have not been reported yet.Citation37
Given the importance of neurofilaments for cytoskeletal organization in neurons, we questioned whether the loss of neurofilament protein NEFH in breast cancer cells might affect the process of EMT that depends on re-organization of the cytoskeleton. Indeed, we found that breast cancer cells with restored NEFH expression behave less invasive than cells with epigenetically lost NEFH expression as assessed by migration and invasion assays. An increased invasiveness upon epigenetic loss of NEFH has as well been described in esophageal cancers.Citation25 These results suggest that silencing of NEFH affects the invasive behavior of breast cancer cells, possibly through participation of neurofilaments in cytoskeletal re-organization. Lack of NEFH in breast cancer cells might destabilize intermediate filaments and cause the disarrangement of the cytoskeleton and the subsequent dynamic deformation of cells leading to higher motility and ability to migrate and invade into neighboring tissues. Further support for this hypothesis is given by our finding that promoter methylation of neurofilament genes is associated with adverse clinical parameters and poor overall survival. The association of NEFH methylation with prognostic factors such as advanced tumor stage, perinodal invasion, and Her2 receptor expression supports a potential role for NEFH in disease progression. NEFL, however, is the most promising candidate for validation as a marker for disease progression since methylation of NEFL is predictive for survival outcome of breast cancer patients. Expression of NEFL has been established as a prognostic marker in breast cancer;Citation38 however, methylation of NEFL as marker for disease progression needs to be validated in an independent cohort. In summary, the present study describes the epigenetic downregulation of the neurofilament genes NEFH, NEFM, and NEFL in breast cancer and highlights their significance for disease progression as well as their potential implication in other types of cancer.
Materials and Methods
Cell culture and drug treatments
Cell lines were purchased from American Type Culture Collection (ATCC, Virginia, USA) and cultured in appropriate media (Mediatech, Virginia, USA) supplemented with 10% FBS (Atlanta Biologicals, Georgia, USA) and 1x Penicillin-Streptomycin (Mediatech, Virginia, USA) at 37°C in 5% CO2 atmosphere. The HCT116 derivative cell line lacking the major DNA methyltransferases DNMT1 and 3b (DNMT1-/- and DNMT3b-/-; Double Knockout or DKO) was maintained as previously described.Citation2 Drug treatment with 5-aza-2'-deoxycytidine (DAC) or Trichostatin A (TSA) was carried out as previously described.Citation6
Human tissue samples
Primary tumor specimen and normal breast tissues from cancer-free donors were obtained from the archives of the Department of Pathology, Johns Hopkins University (Baltimore, MD) with Johns Hopkins Institutional Review Board approval (IRB-X) and Health Insurance Portability and Accountability Art compliance. The use of patient-derived material was approved by the Johns Hopkins University's Committee Research Ethics Board and written consent was obtained from all patients. All paraffin-embedded samples underwent independent review to confirm diagnosis by a pathologist (EG). Patient characteristics are shown in Table S1.
TCGA breast cancer data
DNA methylation profiles (Infinium HumanMethylation450) of 737 breast tissues (96 normal and 641 tumor samples) and gene expression profiles (IlluminaHiSeq_RNA-SeqV2) of 974 breast tumors as well as clinical data for each patient were downloaded from the TCGA data repository (https://tcga-data.nci.nih.gov/tcga/). Level 1 data (raw signal intensities of probes) were downloaded for Infinium HumanMethylation450 arrays and probes of low quality based on a detection p-value threshold of 0.05, cross-reactive probes (i.e., targeting several genomic locations) as well as probes containing SNPs based on the extended annotation of Price et al.Citation39,40 were removed. Additionally, probes associated to X and Y-chromosomes were removed from the analysis. β-values were computed using the formula: β-value = M/[U+M] where M and U are the raw “methylated” and “unmethylated” signals, respectively. The β-values were corrected for type I and type II bias using the peak-based correction.Citation40,41 Finally, probes associated with NEFH, NEFL and NEFM within the following genomic locations were selected: TSS1500, TSS200, 1stExon or 5′UTR. For NEFH we identified 9 probes were, for NEFL 11 and for NEFM 20 probes (Table S2). For IlluminaHiSeq_RNA-SeqV2, level 3 data were downloaded, which contain the calculated expression signal of a particular composite exon of a gene per sample. For 638 tumors both DNA methylation and gene expression profiles were available and could be integrated.
Gene expression, methylation analysis, and ChIP
Total RNA was extracted using RNeasy Mini Kit (Qiagen, California, USA) or purchased from Stratagene (Stratagene, California, USA) (normal tissues), treated with DNase 1 (Qiagen, California, USA) and reverse transcriped (RT) into cDNA using Ready-To-Go You-Prime First-Strands Beads (GE Healthcare, New Jersey, USA) with addition of pd(N)6 Random Hexamers (GE Healthcare, New Jersey, USA) according to the manufacturer's instructions. Genomic DNA was extracted using the standard phenol-chloroform extraction method. Bisulfite modification of genomic DNA was performed with EZ DNA Methylation Kit (Zymo Research, California, USA). Methylation-Specific PCR (MSP) was performed as previously described.Citation42 PCR products for bisulfite sequencing were cloned using the TOPO TA Cloning Kit (Life technologies, California, USA), purified from single colonies using QIAprep Spin Miniprep Kit (Qiagen, California, USA), and sequenced with M13 reverse primer by Johns Hopkins Medical Institutions Synthesis & Sequencing Facility. For ChIP, cells were crosslinked in 1% formaldehyde as previously described.Citation43 Nuclear extraction using CEBN and CEB, and ChIP on 1 × 106 cells per IP was performed as previously described.Citation44 α-H3-K4me2, α-H3-K27me3 and α-IgG antibodies from Millipore were used. IP specific products were amplified using real-time PCR. Relative quantities of histone modifications were determined by calculating the fold enrichment relative to the IgG background and normalize them to input: (CtIgG − Ctmodification) / (CtIgG − CtInput).Citation45 Table S6 lists primer sequences used in this study.
Expression vectors
Wild type NEFH (NM_021076) was amplified from normal brain by RT-PCR and cloned into pcDNA3.1/V5-His B expression vector (Life technologies, California, USA) via KpnI and EcoRI cutting sites.
Immunofluorescence
MDA-MB-231 cells (1 × 10Citation5) transfected with empty pcDNA3.1 vector or pcDNA3.1+NEFH, were grown on coverslips overnight. Cells were fixed in 3.7% formalin, permeabilized with 0.25% Triton X-100 in PBS and blocked in 0.2% fish skin gelatin solution in PBS for 30 minutes at 37 degree. NEFH protein expression was detected with anti-NEFH (Sigma, Missouri, USA), anti-mouse DAPI and anti-goat IgG-AF647. MDA-MB-231 wild type cells (mock cells; not transfected with the NEFH expression plasmid) were also stained for NEFH to determine endogenous levels of NEFH expression.
Colony formation and soft agar assay
Cells were transfected with 4 μg of either empty control vector or NEFH expression vector using Lipofectamine 2000 reagent (Life technologies, California, USA). For colony formation assays, transiently transfected cells were harvested 24 hours post transfection, re-plated in 10 cm2 dishes in triplicates and selected with 0.4 mg/ml G418 for 14 d Colonies were stained with Giemsa and counted. For soft agar assays, 1 × 104 transfected cells in complete media containing 0.3% agar were layered on top of 0.6% agar in 24-well plates in triplicates. Colonies were selected with 0.8 mg/ml Geneticin/G418 (Life technologies, California, USA) for 14 d and counted after staining with 0.005% crystal violet. In these assays, MDA-MB-231 cells upon mock transfection (just with Lipofectamine 2000) were used as control.
Analysis of proliferation rate and cell cycle
MDA-MB-231 cells were transfected with 4 μg of either empty control vector or NEFH expression vector using Lipofectamine 2000 reagent (Life technologies, California, USA) to perform analysis of proliferation rate and cell cycle. Additionally, mock-transfected MDA-MB-231 cells (just treated with Lipofectamine 2000) were included as control into both assays. Proliferation rates were measured at 48, 72, and 96 hours post transfection using the MTT assay (Life technologies, California, USA). Cells were washed and covered with 100 μL complete growth medium. MTT stock solution (10 μL) was added to each well and incubated for 4 hours at 37°C in a CO2 incubator. Then, 100 μL of SDS-HCl solution was added and incubated for 4 hours at 37°C in a CO2 incubator. The absorbance was measured at 570 nm. Cell cycle analysis was performed 48 and 96 hours post transfection. MDA-MB-231 cells were washed with phosphate buffered saline (PBS) (Life Technologies, California USA), trypsinized, washed with PBS and fixed for 10 min at room temperature in a solution containing 300 uL 1X PBS, 0.55% NP-40 (Sigma-Aldrich, St. Louis, USA), 3.7% Formaldehyde (Sigma-Aldrich, St. Louis, USA) and 0.011 mg/mL Hoechst 33258 (Life Technologies, California, USA). The solution was aspirated and cells were washed 2 times with PBS and analyzed using the BD LSR flow cytometer.
Transwell migration and invasion assay
To measure cell migration, 2.5 × 104 cells were suspended in serum-free media and added to the top chamber of 24-well transwell plates (8 μm pore-size) (Corning, Massachusetts, USA). Growth media containing 10% serum was added to the bottom chamber. After 24 hours of incubation, cells attached to the membrane were fixed and stained with crystal violet. Cells on top of the membrane were removed by wiping with cotton swabs and migrated cells were counted. To measure cell invasion, transwell plates (8 μm pore-size) (Corning, Massachusetts, USA) were layered with 2 mg/ml matrigel (Corning, Massachusetts, USA) and 1 × 105 cells suspended in serum-free media were added to the top chamber of the transwell plates. Growth media containing 10% serum was added to the bottom chamber. After 48 hours of incubation, cells attached to the membrane were fixed and stained with giemsa. Cells on top of the membrane were removed. Cells were counted in the light microscope (3 random fields).
Western blot
NEFH and GAPDH protein expression was detected in whole cell protein extracts with α-NEFH (Convance, New Jersey, USA) and α-GAPDH (Millipore, Massachusetts, USA), respectively.
Statistical analysis
Group comparisons were performed with 2-tailed paired Student's t test. The relationships between gene methylation and clinicopathological characteristics was tested using Pearson's χ2. Survival curves were plotted by Kaplan-Meier method and compared by long-rank test. Hazard ratio (HR) for prognostic value of gene methylation status was calculated using univariate and multivariate Cox regression analysis. All statistical analyses were performed using the STATA 9.2 software package (STATA Corp., Texas, USA). TCGA data were analyzed using the R statistical package. Differences between the median methylation of tumor and the median methylation of normal tissues as well as between the median methylation of high and the median methylation of low stage tumors per probe were assessed using a 2-tailed paired Student's t test. A Spearman correlation coefficient was computed to test for correlation between DNA methylation and gene expression for each probe. P values of less than 0.05 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the Johns Hopkins Tumor Registry for their help with clinicopathologic patient information as well as Kathy Bender and Joann Murphy for administrative support.
Supplemental_Material.zip
Download Zip (1.3 MB)Funding
This study was supported by the NIH, Susan G. Komen Foundation, Mary Kay Foundation, American College of Surgeons/Society of University Surgeons, German Academic Exchange Service (DAAD), Dr. Jost Henkel Stiftung and the Mentored Patient-Oriented Research Career Development Award from NCI (K23).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011; 11:726-34; PMID:21941284; http://dx.doi.org/10.1038/nrc3130.
- Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den Bosch S, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3:1709-23; PMID:17892325; http://dx.doi.org/10.1371/journal.pgen.0030157.
- Glockner SC, Dhir M, Yi JM, McGarvey KE, Van Neste L, Louwagie J, Chan TA, Kleeberger W, de Bruine AP, Smits KM, et al. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res 2009; 69:4691-9; PMID:19435926; http://dx.doi.org/10.1158/0008-5472.CAN-08-0142.
- Zhang W, Glockner SC, Guo M, Machida EO, Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins DN, et al. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res 2008; 68:2764-72; PMID:18413743; http://dx.doi.org/10.1158/0008-5472.CAN-07-6349.
- Yi JM, Dhir M, Van Neste L, Downing SR, Jeschke J, Glockner SC, de Freitas Calmon M, Hooker CM, Funes JM, Boshoff C, et al. Genomic and epigenomic integration identifies a prognostic signature in colon cancer. Clin Cancer Res 2011; 17:1535-45; PMID:21278247; http://dx.doi.org/10.1158/1078-0432.CCR-10-2509.
- Jeschke J, Van Neste L, Glockner SC, Dhir M, Calmon MF, Deregowski V, Van Criekinge W, Vlassenbroeck I, Koch A, Chan TA, et al. Biomarkers for detection and prognosis of breast cancer identified by a functional hypermethylome screen. Epigenet 2012; 7:701-9; PMID:22647880; http://dx.doi.org/10.4161/epi.20445.
- Jeschke J, O'Hagan HM, Zhang W, Vatapalli R, Calmon MF, Danilova L, Nelkenbrecher C, Van Neste L, Bijsmans IT, Van Engeland M, et al. Frequent inactivation of cysteine dioxygenase type 1 contributes to survival of breast cancer cells and resistance to anthracyclines. Clin Cancer Res 2013; 19:3201-11; PMID:23630167; http://dx.doi.org/10.1158/1078-0432.CCR-12-3751.
- Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Translat Med 2011; 3:75ra25; PMID:21430268; http://dx.doi.org/10.1126/scitranslmed.3001875.
- Fackler MJ, Umbricht CB, Williams D, Argani P, Cruz LA, Merino VF, Teo WW, Zhang Z, Huang P, Visvananthan K, et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res 2011; 71:6195-207; PMID:21825015; http://dx.doi.org/10.1158/0008-5472.CAN-11-1630.
- Bediaga NG, Acha-Sagredo A, Guerra I, Viguri A, Albaina C, Ruiz Diaz I, Rezola R, Alberdi MJ, Dopazo J, Montaner D, et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res 2010; 12:R77; PMID:20920229; http://dx.doi.org/10.1186/bcr2721.
- Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, Maher ER, Latif F. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res 2011; 71:2988-99; PMID:21363912; http://dx.doi.org/10.1158/0008-5472.CAN-10-4026.
- Liem RK, Yen SH, Salomon GD, Shelanski ML. Intermediate filaments in nervous tissues. J Cell Biol 1978; 79:637-45; PMID:83322; http://dx.doi.org/10.1083/jcb.79.3.637.
- Steinert PM, Roop DR. Molecular and cellular biology of intermediate filaments. Annu Rev Biochem 1988; 57:593-625; PMID:3052284; http://dx.doi.org/10.1146/annurev.bi.57.070188.003113.
- Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec 1970; 167:379-87; PMID:5454590; http://dx.doi.org/10.1002/ar.1091670402.
- Hoffman PN, Griffin JW, Price DL. Control of axonal caliber by neurofilament transport. J Cell Biol 1984; 99:705-14; PMID:6204997; http://dx.doi.org/10.1083/jcb.99.2.705.
- Kudo LC, Parfenova L, Vi N, Lau K, Pomakian J, Valdmanis P, Rouleau GA, Vinters HV, Wiedau-Pazos M, Karsten SL. Integrative gene-tissue microarray-based approach for identification of human disease biomarkers: application to amyotrophic lateral sclerosis. Hum Mol Genet 2010; 19:3233-53; PMID:20530642; http://dx.doi.org/10.1093/hmg/ddq232.
- Skvortsova V, Shadrina M, Slominsky P, Levitsky G, Kondratieva E, Zherebtsova A, Levitskaya N, Alekhin A, Serdyuk A, Limborska S. Analysis of heavy neurofilament subunit gene polymorphism in Russian patients with sporadic motor neuron disease (MND). Eur J Hum Genet 2004; 12:241-4; PMID:14722583; http://dx.doi.org/10.1038/sj.ejhg.5201144.
- Mersiyanova IV, Perepelov AV, Polyakov AV, Sitnikov VF, Dadali EL, Oparin RB, Petrin AN, Evgrafov OV. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet 2000; 67:37-46; PMID:10841809; http://dx.doi.org/10.1086/302962.
- Alholle A, Brini AT, Gharanei S, Vaiyapuri S, Arrigoni E, Dallol A, Gentle D, Kishida T, Hiruma T, Avigad S, et al. Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics 2013; 8:1198-204; PMID:24005033; http://dx.doi.org/10.4161/epi.26266.
- Huang Z, Zhuo Y, Shen Z, Wang Y, Wang L, Li H, Chen J, Chen W. The role of NEFL in cell growth and invasion in head and neck squamous cell carcinoma cell lines. J Oral Pathol Med 2013; 43(3):191-8.
- Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, Hoshida Y, Llovet JM, Powers S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 2013; 145:1424-35 e1-25; PMID:24012984; http://dx.doi.org/10.1053/j.gastro.2013.08.055.
- Emi M, Fujiwara Y, Nakajima T, Tsuchiya E, Tsuda H, Hirohashi S, Maeda Y, Tsuruta K, Miyaki M, Nakamura Y. Frequent loss of heterozygosity for loci on chromosome 8p in hepatocellular carcinoma, colorectal cancer, and lung cancer. Cancer Res 1992; 52:5368-72; PMID:1356616.
- Vogelstein B, Fearon ER, Kern SE, Hamilton SR, Preisinger AC, Nakamura Y, White R. Allelotype of colorectal carcinomas. Science 1989; 244:207-11; PMID:2565047; http://dx.doi.org/10.1126/science.2565047.
- Yaremko ML, Kutza C, Lyzak J, Mick R, Recant WM, Westbrook CA. Loss of heterozygosity from the short arm of chromosome 8 is associated with invasive behavior in breast cancer. Genes Chromosomes Cancer 1996; 16:189-95; http://dx.doi.org/10.1002/(SICI)1098-2264(199607)16:3%3c189::AID-GCC6%3e3.0.CO;2-V.
- Kim MS, Chang X, LeBron C, Nagpal JK, Lee J, Huang Y, Yamashita K, Trink B, Ratovitski EA, Sidransky D. Neurofilament heavy polypeptide regulates the Akt-β-catenin pathway in human esophageal squamous cell carcinoma. PloS One 2010; 5:e9003; PMID:20140245; http://dx.doi.org/10.1371/journal.pone.0009003.
- Dubrowinskaja N, Gebauer K, Peters I, Hennenlotter J, Abbas M, Scherer R, Tezval H, Merseburger AS, Stenzl A, Grunwald V, et al. Neurofilament Heavy polypeptide CpG island methylation associates with prognosis of renal cell carcinoma and prediction of antivascular endothelial growth factor therapy response. Cancer Med 2014; 3(2):300-9; PMID:24464810.
- Petzold A. Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss. J Neurol Sci 2005; 233:183-98; PMID:15896809; http://dx.doi.org/10.1016/j.jns.2005.03.015.
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553-60; PMID:17603471; http://dx.doi.org/10.1038/nature06008.
- Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008; 105:12979-84; PMID:18753622; http://dx.doi.org/10.1073/pnas.0806437105.
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007; 39:232-6; PMID:17200670; http://dx.doi.org/10.1038/ng1950.
- Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW, Vogelstein B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 2003; 3:89-95; PMID:12559178; http://dx.doi.org/10.1016/S1535-6108(02)00234-9.
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39:237-42; PMID:17211412; http://dx.doi.org/10.1038/ng1972.
- Ohm JE, Baylin SB. Stem cell chromatin patterns: an instructive mechanism for DNA hypermethylation? Cell Cycle 2007; 6:1040-3; PMID:17457052; http://dx.doi.org/10.4161/cc.6.9.4210.
- Easwaran H, Johnstone SE, Van Neste L, Ohm J, Mosbruger T, Wang Q, Aryee MJ, Joyce P, Ahuja N, Weisenberger D, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res 2012; 22:837-49; PMID:22391556; http://dx.doi.org/10.1101/gr.131169.111.
- Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003; 2:339-45; PMID:12851486; http://dx.doi.org/10.4161/cc.2.4.433.
- Hayes MJ, Thomas D, Emmons A, Giordano TJ, Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res 2008; 14:4038-44; PMID:18593979; http://dx.doi.org/10.1158/1078-0432.CCR-07-4379.
- Kizildag S, Zengel B, Vardar E, Sakizli M. β-catenin gene mutation in invasive ductal breast cancer. J BUON 2008; 13:533-6.
- Li XQ, Li L, Xiao CH, Feng YM. NEFL mRNA expression level is a prognostic factor for early-stage breast cancer patients. PloS One 2012; 7:e31146; PMID:22319610; http://dx.doi.org/10.1371/journal.pone.0031146.
- Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 2013; 6:4; PMID:23452981; http://dx.doi.org/10.1186/1756-8935-6-4.
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011; 3:771-84; http://dx.doi.org/10.2217/epi.11.105.
- Dedeurwaerder S, Defrance M, Bizet M, Calonne E, Bontempi G, Fuks F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief Bioinform 2014; 15:929-41; PMID:23990268; http://dx.doi.org/10.1093/bib/bbt054.
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93:9821-6; PMID:8790415; http://dx.doi.org/10.1073/pnas.93.18.9821.
- Fahrner JA, Eguchi S, Herman JG, Baylin SB. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res 2002; 62:7213-8; PMID:12499261.
- Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, Baylin SB. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res 2012; 40:4334-46; PMID:22278882; http://dx.doi.org/10.1093/nar/gks031.
- Thijssen PE, Tobi EW, Balog J, Schouten SG, Kremer D, El Bouazzaoui F, Henneman P, Putter H, Slagboom PE, Heijmans BT, et al. Chromatin remodeling of human subtelomeres and TERRA promoters upon cellular senescence Commonalities and differences between chromosomes. Epigenetics 2013; 8:512-21; PMID:23644601; http://dx.doi.org/10.4161/epi.24450.