
Abstract
African Americans are at increased risk for spontaneous preterm birth (PTB). Though PTB is heritable, genetic studies have not identified variants that account for its intergenerational risk, prompting the hypothesis that epigenetic factors may also contribute. The objective of this study was to evaluate DNA methylation from maternal leukocytes to identify patterns specific to PTB and its intergenerational risk. DNA from peripheral leukocytes from African American women that delivered preterm (24–34 weeks; N = 16) or at term (39–41 weeks; N = 24) was assessed for DNA methylation using the HumanMethylation450 BeadChip. In maternal samples, 17,829 CpG sites associated with PTB, but no CpG site remained associated after correction for multiple comparisons. Examination of paired maternal-fetal samples identified 5,171 CpG sites in which methylation of maternal samples correlated with methylation of her respective fetus (FDR < 0.05). These correlated sites were enriched for association with PTB in maternal leukocytes. The majority of correlated CpG sites could be attributed to one or more genetic variants. They were also significantly more likely to be in genes involved in metabolic, cardiovascular, and immune pathways, suggesting a role for genetic and environmental contributions to PTB risk and chronic disease. The results of this study may provide insight into the factors underlying intergenerational risk for PTB and its consequences.
Abbreviations
| DOHaD | = | developmental origin of health and disease |
| FDR | = | false discovery rate |
| GA | = | gestational age |
| PTB | = | preterm birth |
| SES | = | socioeconomic status |
Introduction
Though the overall rate of preterm birth has slightly decreased in recent years, African Americans have more than 1.5 times the risk of spontaneous preterm birth (PTB; <37 weeks gestation) and more than twice the risk of early PTB (<32 weeks) when compared to Caucasians.Citation1-3 Studies to date have identified numerous maternal risk factors for PTB,Citation1,4-7 such as low socioeconomic status (SES), but less than half of the increased risk in African Americans is explained by SES and other known risk factors.Citation8-10
Personal and family history of PTB are the greatest risk factors for PTB, and studies estimate its heritability at 17–30%.Citation11-15 Citation16,17 However, genetic studies have not identified variants that account for this intergenerational risk,Citation1,11,18-21 prompting the hypothesis that epigenetic factors may also contribute to PTB.Citation17,22-24 Few studies have evaluated the epigenetics of PTB, and those that have focus mostly on those born preterm.Citation25-28 Other studies focus on the short-term and long-term consequences of PTB for the neonate,Citation1,29-32 in part, because of interest in the developmental origin of health and disease (DOHaD) hypothesis.Citation32,33
We recently evaluated DNA methylation in leukocytes from African American umbilical cord blood samples and identified thousands of DNA methylation differences between preterm and term fetuses.Citation25 These DNA methylation differences may underlie some of the risks associated with being born preterm, though longitudinal studies will be more informative for determining whether methylation differences observed at birth have long-term consequences or whether they simply reflect developmental differences. Cruickshank and colleagues performed one such study in 12 PTB cases and 12 matched controls.Citation28 They evaluated DNA methylation from blood at birth and at 18 years and observed substantial overlap with the PTB-associated CpGs we reported at birth. DNA methylation differences observed at birth were no longer associated with PTB status at 18 years for the majority of CpG sites examined, but they report 10 CpGs that continue to differ in methylation at both time points, suggesting the potential for a long-term epigenetic signature of PTB.
No study has examined genome-wide DNA methylation in leukocytes of women who deliver preterm. However, studies have recently begun documenting the long-term implications of delivering preterm for maternal health. Women who deliver preterm are at increased risk to develop cardiovascular and other chronic disorders as they age.Citation34-42 For example, a series of studies demonstrate that mothers who deliver very preterm are at subsequent risk for Type 2 diabetes.Citation38,43 The first, conducted primarily in Caucasian women from the Nurses' Health Study II, reports that women who deliver very early preterm are more likely to be diagnosed with Type 2 diabetes in the decade following pregnancy.Citation43 A second investigation of ∼30,000 women from the Black Women's Health Study also reports that early preterm birth associates with a higher risk for developing Type 2 diabetes even after correcting for age at first birth, family history of diabetes, education, personal history of preterm birth, and body mass index.Citation38 They then demonstrated that the increased diabetes risk was independent of gestational diabetes history, consistent with other studies.Citation39,40
The mechanism underlying the relationship between PTB and the development of chronic disorders later in life is not yet clear, but some suggest that inflammation or immune dysregulation may increase risk for PTB and other chronic disorders.Citation38,44 DNA methylation patterns regulate the functional properties of immune cellsCitation45,46 and associate with inflammatory markers,Citation47 chronic disorders,Citation48-50 and PTB.Citation27,51 We hypothesize that DNA methylation patterns may reveal genes whose regulation is unique to women who deliver preterm or provide insight into its intergenerational risk
Results
The cohort is comprised of African American women that deliver early preterm [gestational age (GA) range: 24.1–34.0 weeks] and at term (39.0–40.9 weeks). As expected, the groups differed by GA and birthweight, but did not differ significantly by any other demographic or clinical factor ().
Table 1. Demographics table for maternal samples
Association between maternal DNA methylation and PTB
First, we examined the association between DNA methylation at each CpG site and PTB. Overall, 17,829 CpG sites associated with PTB (1.83 × 10−6 < P < 0.05). Among the CpG sites with the strongest association were 2 (cg22486214, cg16980736) in regulatory associated protein of MTOR (RPTOR; 2.20 × 10−5 < P < 1.03 × 10−4). However, no CpG site remained associated after correction for multiple testing (FDR < 0.05; Table S1). We have previously reported DNA methylation differences that associate with GA in leukocytes from umbilical cord blood of fetuses born to this cohort of women.Citation25 CpG sites that associate with PTB in maternal leukocytes are more likely to associate with GA in fetuses when compared to those that do not associate with PTB (5.2% vs. 3.5%; P < 2.2 × 10−16), suggesting that there may be epigenetic factors shared between mothers who deliver preterm and their fetuses.
Correlation between maternal and fetal methylation
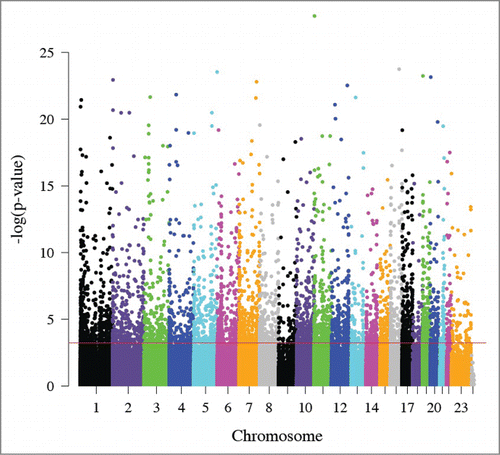
We next evaluated the relationship between maternal and fetal DNA methylation across the genome. We identified 5,171 CpG sites in which maternal methylation associated with fetal methylation (FDR < 0.05; ; Table S2), 98.8% of which occurred in the same direction. These correlated CpG sites were consistent with the “heritable” CpG sites recently reported by McRae and colleaguesCitation52 (Result S1). To empirically assess whether these differences are greater than expected by chance, we repeated the analysis comparing methylation from each mother to methylation from an unrelated fetus that was matched for PTB status and sex. Only 35 CpG sites associated between unrelated pairs (FDR < 0.05), suggesting that the high degree of correlation observed between a mother and her fetus is substantially greater than expected by chance.
Figure 1. Manhattan plot of the relationship between maternal and fetal DNA methylation. The x-axis represents the position of each CpG site by chromosome. The y-axis represents the negative log10 of the P-value for the association between maternal and fetal methylation for each CpG site. The red line indicates experiment-wide significance based on a false discovery rate of 5% such that the 5,171 CpG sites above this line are significantly correlated in leukocytes from a mother and her fetus.
There is a wide range of variation in methylation levels across the 5,171 correlated CpG sites (), that is consistent with distribution of variation in all CpG sites, assessed on the array (). Correlated CpG sites were more likely to occur in regions of low CpG density (i.e., shelves and open seas) and less likely occur in regulatory regions near the transcription start site (i.e., promoters, 1st exon, and 5′UTRs) when compared to uncorrelated CpG sites (1.5 × 10−6 < P < 2.2 × 10−16; ). They were also more likely to be located in genes involved in metabolic (i.e., Type 1 and Type 2 diabetes mellitus), cardiovascular (i.e., viral myocarditis, arrhythmogenic right ventricular cardiomyopathy), and immune (i.e., graft vs. host disease, allograft rejection) pathways (). The majority of correlated CpG sites (3,857; 74.6%) could be attributed to one or more genetic variants, defined as a SNP that either overlapped with the CpG probe sequence (1,393; 26.9%) or associated with the CpG as a methylation quantitative trait locus (meQTL; 2,464; 47.7%). We also found that CpG sites that correlated between a mother and her fetus were enriched for meQTLs at increasingly stringent significance levels (), consistent with the results of McRae et al., who reported that sequence variation accounts for the majority of heritable DNA methylation patterns.Citation52 The remaining 1,314 CpG sites (25.4%) could not be attributed to genetic factors and may reflect the shared environment. Both classes of CpG sites support the pathways identified in the combined analysis when evaluated individually (). Evaluation of the 200 genes containing CpG sites whose methylation levels are both predictive of cord blood methylation and associate with PTB in maternal samples also reveal enrichment for genes in the Type 2 diabetes mellitus pathway (KEGG:04930; P = 0.015).
Figure 2. Distribution of maternal DNA methylation for CpG sites. The x-axis represents the standard deviation (SD) of each CpG site's methylation (β) values in maternal leukocytes. The y-axis indicates the proportion of CpG sites in bins determined by SD. Black represents CpG sites that may be attributed to genetic variation while gray represents CpG sites that cannot be attributed to genetic variation. Graph (A) depicts the distribution of correlated CpG sites (n = 5,171). Graph (B) depicts the distribution of all CpG sites (n = 479,808).
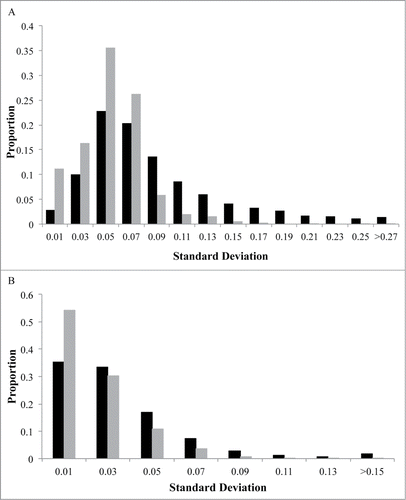
Figure 3. Correlated CpG sites and their enrichment in meQTLs. The x-axis represents increasing statistical significance of genotype-methylation associations (cis-meQTLs). The –log10 is used to represent the statistical threshold (α level) used to define meQTLs (i.e., 1 is equivalent to P < 0.1, and 2 is equivalent to P < 0.01, etc.). The y-axis represents the odds ratios comparing the probability that a correlated CpG site associates with a genotype (i.e., is an meQTL) with greater likelihood than an uncorrelated CpG site. The vertical lines represent the confidence interval for each odds ratio.
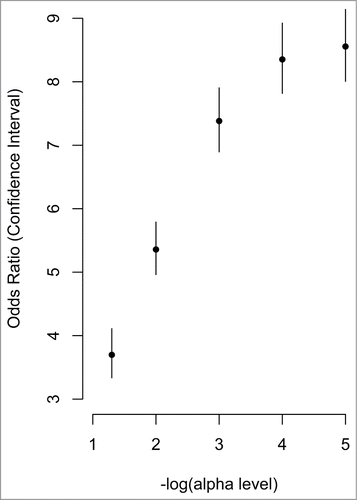
Table 2. Enrichment for correlation analysis of maternal and fetal blood
Table 3. Pathway analysis of CpG sites in genes that correlate in maternal-fetal pairs
Correlated CpG sites were more likely than uncorrelated sites to associate with PTB in maternal samples (; OR = 1.7; P < 2.2 × 10−16), but there was no difference in the rates of CpG sites influenced by genetic versus non-genetic factors among those associated with PTB (P = 0.41). Thus, we re-examined the relationship between maternal and fetal DNA methylation separately in PTB and term birth pairs. There were 79 CpG sites that correlated (FDR < 0.05) in the PTB pairs, 57 (72.2%) of which were unique to the PTB samples. PTB-specific sites were enriched in genes involved in vascular smooth muscle contraction (KEGG:04270; P = 0.037). Consistent with the results of the combined analysis, the majority (70.2%) of the correlated CpG sites specific to PTB were attributable to genetic variation.
Correlation Between Maternal and Fetal Gene Expression
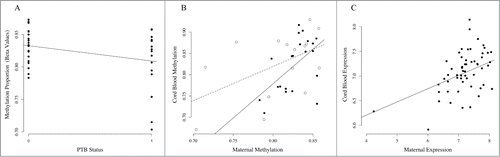
We next examined whether genes containing correlated CpG sites have correlated gene expression levels. Correlated CpG sites (N = 5,171) were located in 3,297 expression probes, representing 2,282 unique genes and 1,015 transcript variants. Maternal expression of 738 transcripts (22.3%; 2.3 × 10−14 < P < 0.05) predicted fetal expression, 357 (10.8%) of which remained associated after correction for multiple tests (FDR < 0.05; Table S3). For example, methylation of CpG sites in MHC class I polypeptide-related sequence B (MICB) associate with PTB in maternal leukocytes and correlate with methylation in fetal samples (). Maternal MICB expression also predicts fetal MICB expression (P = 1.09 × 10−3; ). Interestingly, levels of the protein encoded by MICB fall prior to parturition. Citation53 Pathway analysis of genes with correlated expression levels (FDR < 0.05) were consistent with the results of those performed with correlated CpG sites (data not shown).
Figure 4. Association of MICB methylation and expression in PTB and TB pairs. (A) Association between cg06284756 in maternal leukocytes and PTB. The x-axis represents PTB status where 0 denotes term birth and 1 denotes PTB. The y-axis represents the methylation levels (β values) for cg06284756. (B) Correlation between maternal methylation (x-axis) and fetal methylation (y-axis) for cg06284756. Open circles represent maternal-fetal pairs that are preterm and closed circles represent pairs that are term. The dashed line represents correlation in PTB pairs, and the solid line represents correlation in term birth (TB) pairs. (C) Correlation between maternal leukocyte MICB expression (x-axis) and fetal leukocyte MICB expression (y-axis).
Discussion
In this study, no CpG site of large effect size was associated with PTB in African American women, though thousands of CpG sites were nominally significant. Only 5.2% of the CpG sites that were associated with GA in the leukocytes of umbilical cord blood of fetuses born preterm also associated with PTB in maternal samples, suggesting that the majority of CpG sites associated with GA in fetuses may reflect developmental differences. Nevertheless, our study does provide insight into correlated methylation and expression patterns in maternal-fetal pairs and suggests they are enriched in biological pathways implicated in PTB and chronic disease risk.
One of the biggest risk factors for PTB is a prior history or a family history. We identified 5,171 CpG sites in which maternal methylation predicts fetal methylation. The vast majority of correlated CpG sites (98.8%) occurred in the same direction, consistent with a high degree of genetic and environmental similarity in these pairs. These correlated CpG sites were enriched in areas of low CpG density, regions of high inter-individual variation that are more likely to associate with environmental factors and complex diseases.Citation54 Though this study did not specifically evaluate sequence variation, methylation of almost 75% of correlated CpG sites could be attributed to genetic variation, such as a SNP or meQTL. The results of this study were consistent with those reported in a large multigenerational cohort of Caucasians,Citation52 which determined that heritable CpG sites were primarily under genetic influence. African Americans and Caucasians have distinct patterns of genetic-epigenetic correlationCitation55 that may contribute to the increased risk for PTB and other disorders more common in African Americans.
Correlated CpG sites were also enriched among genes whose expression levels were correlated in maternal-fetal pairs, providing a potential mechanism linking correlated methylation in women who deliver preterm to biological differences. For example, MICB is part of the MHC class I chain and is induced by cellular stress to initiate an immune response.Citation53 Activation of inflammatory pathways has been implicated in the timing of parturition specifically in PTB,Citation11 and we observed lower methylation of CpGs in MICB in PTB as well as correlated methylation and expression patterns in maternal-fetal pairs. These results suggest a complex relationship between sequence variation, DNA methylation, and gene expression that should be considered in future studies of PTB.
Correlated CpG sites were enriched in genes involved in chronic disorders that are common in African Americans,Citation1,34-36,56-59 suggesting an epigenetic link between PTB and metabolic, cardiovascular, and immune dysregulation across generations. Though we made every effort to limit inclusion of clinical factors that could influence these results, such as gestational diabetes or preeclampsia, the implications of these findings are difficult to interpret. However, the results suggest that chronic disorders diagnosed subsequent to preterm delivery may not be limited to those indicated by gestational diabetes or hypertension. On the contrary, spontaneous PTB with unknown etiology may also increase maternal risk for chronic disorders during her lifetime. In general, African American women have higher levels of inflammation when compared to Caucasian women, and chronic inflammation has been presented as a potential mechanism through which disparities in the rates of PTB and other chronic conditions occur.Citation11,17,24,38,44,57,60 For example, Liu and colleagues examined DNA methylation of 8 imprinted genes in umbilical cord blood samples for association with PTB and infection status.Citation26 Though they did not find any association with PTB, they reported that pleiomorphic adenoma gene-like 1 (PLAGL1) DNA methylation associates with chorioamnionitis. Consistent with these results, correlated CpG sites exclusive to PTB pairs were also identified in genes involved in immune regulation. Other environmental factors, such as high BMI, psychosocial stress, smoking, and infection also increase inflammation and PTB risk. It will be important for future studies to consider these factors, as DNA methylation may mediate these relationships. For example, in an animal model, Yao and colleagues demonstrated that prenatal stress results in neuroendocrine and metabolic differences as well as shorter gestational length in subsequent generations. They linked their findings to epigenetic regulation of the placenta and other key tissues, providing a potential mechanism for inter-generational transmission of stress.Citation61
This study has a number of strengths and limitations. The primary limitation is the sample size, which is, in part, due to the fact that we restricted the design to only African Americans with spontaneous PTB prior to 34 weeks gestation and uncomplicated controls. However, our cases span different categories of PTB based on GA, extremely preterm, very preterm, and moderate preterm, based on the World Health Organization classification, and it would have been ideal if there was sufficient power to examine each group individually. In this study, we did not identify any individual CpG sites that associated with PTB in maternal samples. Thus, it is reasonable to conclude that there are no CpG sites of large effect, though evaluations of larger cohorts may reveal associations of more subtle effect. However, our study was well powered to detect CpG sites whose methylation and expression levels correlated in maternal-fetal pairs. In time, these results may provide a foundation through which individual epigenetic patterns can be used in early pregnancy to predict risk for preterm delivery.
This is the first epigenetic study of maternal-fetal pairs for PTB and the first study of heritable CpG sites in African Americans, an understudied population with an increased risk of PTB. The results of this study support a complex genetic and environmental relationship underlying the intergenerational risk for PTB and are consistent with the hypothesis that pregnancy complications, including spontaneous PTB, may be an early indicator of future risk for mothers as well as their fetuses. Future studies should prospectively examine women who are at high risk for PTB throughout pregnancy and beyond.
Methods
Nashville Birth Cohort
All subjects were recruited at Centennial Women's Hospital and the Perinatal Research Center in Nashville, TN beginning in 2003 as part of the Nashville Birth Cohort that was established to examine biological risk factors that distinguish spontaneous preterm from term labor. Pregnant women were enrolled at the time of admission for labor at preterm or term after obtaining informed consent. Maternal demographic and clinical data (race, socioeconomic education, household income, marital status, cigarette smoking) were recorded from medical records or by interviews during the consenting process. Demographic and clinical data specific to the fetus were collected from clinical records. Gestational age (GA) was determined by maternal reporting of the last menstrual period and corroboration by ultrasound dating. Birth weight percentile was based on GA in accordance with the United States national reference.Citation62 Race was identified by self-reporting that traced back to 3 generations from maternal and paternal side of the fetus. Only African Americans of non-Hispanic ethnicity were included in this study.
Subjects were included in this study if they had contractions (rate of 2 contractions/10 min) leading to delivery either at preterm or at term. Cases delivered preterm with intact membranes between 241/7 weeks and 340/7 weeks. Controls delivered (>390/7 weeks) with spontaneous term labor and delivery and no current or history of pregnancy related complications including preterm birth and preterm or pre-labor rupture of the membranes (pPROM). In addition, controls were excluded if they had any surgical procedures during pregnancy, were treated for preterm labor, or were treated for suspected intra-amniotic infection. Subjects who had multiple gestations, preeclampsia, placenta-previa, fetal anomalies, and/or medical or surgical complications during pregnancy were excluded from the study. This study was conducted in accordance with the Helsinki Declaration of 1975.
Biological Sample Collection and DNA Extraction
Maternal peripheral blood samples were collected in EDTA tubes at time of admission for labor. Blood samples were centrifuged at 3,000 rpm to separate plasma, and buffy coats were aliquoted and stored at −80°C. DNA was extracted using the Autopure automated system (Gentra Systems, Minneapolis, MN).
DNA Methylation Analysis
For each subject, >485,000 CpG sites across the genome were interrogated using the HumanMethylation450 BeadChip (Illumina, San Diego, CA).Citation63,64 Briefly, 1 μg of DNA from maternal leukocytes was converted with sodium bisulfite, amplified, fragmented, and hybridized on the BeadChip according to the manufacturer's instructions. CpGassoc was used to perform quality control and calculate β-values.Citation65 Data points with probe detection P-values >0.001 were set to missing, and CpG sites with missing data for >10% of samples were excluded from analysis; 479,808 CpG sites passed the above criteria. Samples with probe detection call rates <90% and those with an average intensity value of either <50% of the experiment-wide sample mean or <2,000 arbitrary units (AU) were excluded from further analysis. One sample of female DNA from a stable lymphoblast cell line (Coriell) was included on each BeadChip as a technical control throughout the experiment and assessed for reproducibility using the Pearson correlation coefficient, to ensure that Pearson correlation coefficient >0.99 for all pairwise comparisons of technical replicates. For each individual sample and CpG site, the signals from methylated (M) and unmethylated (U) bead types were used to calculate a β value as β = M/(U+M).
Statistical Analysis
MethLAB was used to test for association with PTB via linear regressions that modeled β-values as the outcome and PTB as the independent variable, adjusting for maternal age, cell composition and positional effects of the array as covariates.Citation66 Cell type proportions were estimated using publically available data (GSE36069) as a reference panel for applying the method described by Houseman and colleaguesCitation67,68 to our data. We examined the association between methylation of each CpG site and potential confounding factors including: birth weight percentile, gravidity, parity, infection, and smoking. In a univariate analysis, these factors did not associate with methylation of any CpG site after correction for multiple testing; thus, they were not included as covariates in the final model. Logit transformation of the β-values (i.e., M-values) did not substantially alter the results, so analyses of untransformed β are presented to ease biological interpretation and to make comparisons to our previous study.Citation25 For all genome-wide analyses, the False Discovery Rate (FDR) was controlled at 5% using Storey's q-value.Citation69 For all replication analyses, we set the significance threshold at a nominal p < 0.05.
To evaluate the relationship between DNA methylation in maternal and fetal (leukocytes from umbilical cord blood) samples, linear regressions compared β-values for each maternal sample to those of her fetus for each CpG site while accounting for positional effects on the array and cellular proportions. To compare the observed correlations to what would be observed if maternal and fetal methylation were completely independent, we repeated the analysis comparing each mother to an unrelated fetus that was matched for case status, positional effects, and fetal sex. We also performed an exploratory analysis that evaluated the relationship between methylation in preterm and term pairs separately.
The location of each CpG site was determined using the Illumina array annotation for the HumanMethylation450 BeadChip based on build 37 of the human genome. Chi-square tests were used to compare the number of correlated CpG sites that did or did not occur in a particular gene region (e.g., promoter, 5′UTR, body, 1st exon, 3′UTR, or intragenic regions) to the sites not associated with PTB in that gene region. We performed similar tests of enrichment for regions characterized by CpG density (islands, shores, shelves, and open seas). DAVID was used to evaluate whether groups of CpG sites, were in genes enriched for any specific biological pathways and focused specifically on KEGG pathways.Citation70,71
To determine if genetic variation influenced DNA methylation, methylation quantitative trait loci (meQTL) were identified by applying the approach described previouslyCitation55 to the methylation data from African American subjects in the Grady Trauma Project.Citation47,72-74 Briefly, the relationship between the proportion of methylation at each CpG site and each SNP within 50 kb of that site was examined via linear regression, where methylation was modeled as a linear function of the number of reference alleles (0, 1, or 2). CpG sites were excluded from the meQTL analysis if the probe sequence contained a SNP with a minor allele frequency greater than 1% in any population, as identified from the 1,000 genomes project (TGP). Citation75 In total, 98,741 CpG sites had a TGP SNP within its probe sequence, and an additional 74,712 were meQTLs in an African American cohort (FDR < 0.05). We then plotted the odds ratio of whether correlated CpG sites were enriched in meQTLs at varying significance levels.
Gene expression
Correlation of gene expression was assessed using publicly available data from maternal-fetal pairs (GSE27272).Citation76 Expression of total RNA in umbilical cord blood and maternal peripheral blood was evaluated using the HumanRef-8v3.0 BeadChip (Illumina). The data was extracted using Illumina's BeadStudio Software v3 and then quantile normalized using Lumi.Citation77 For each gene containing a correlated CpG site (Table S2), linear regressions were used to compare expression of each maternal sample to that of her fetus to evaluate whether genes with correlated CpG sites also have correlated expression levels.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Material.zip
Download Zip (578 KB)Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by grants from the National Institutes of Health (MH085806 and MD009064 to AKS) and developmental funds from the Dept. of Obstetrics & Gynecology at The University of Texas Medical Branch (RM). Salary support for SEP was provided, in part, by T32GM008490 and The Burroughs Wellcome Fund's Molecules to Mankind Program (M2M). Research reported in this publication was also supported in part by the Emory Integrated Genomics Core (EIGC).
References
- Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet 2008; 371:75-84; PMID:18177778; http://dx.doi.org/10.1016/S0140-6736(08)60074-4
- Kistka ZA, Palomar L, Lee KA, Boslaugh SE, Wangler MF, Cole FS, DeBaun MR, Muglia LJ. Racial disparity in the frequency of recurrence of preterm birth. Am J Obstet Gynecol 2007; 196:131 e1-6
- Hamilton BE, Martin JA, Ventura SJ. Births: preliminary data for 2011. National vital statistics reports : from the Centers for Disease Control and Prevention, National Center for Health Statistics, National Vital Statistics System 2012; 61:1-18
- Cho G, Min KJ, Hong HR, Kim S, Hong JH, Lee JK, Oh MJ, Kim H. High-risk human papillomavirus infection is associated with premature rupture of membranes. BMC Pregnancy Childbirth 2013; 13:173; PMID:24011340; http://dx.doi.org/10.1186/1471-2393-13-173
- Jeffcoat MK, Geurs NC, Reddy MS, Goldenberg RL, Hauth JC. Current evidence regarding periodontal disease as a risk factor in preterm birth. Ann Periodontol 2001; 6:183-8; PMID:11887462; http://dx.doi.org/10.1902/annals.2001.6.1.183
- Kelly RH, Russo J, Holt VL, Danielsen BH, Zatzick DF, Walker E, Katon W. Psychiatric and substance use disorders as risk factors for low birth weight and preterm delivery. Obstet Gynecol 2002; 100:297-304; PMID:12151153; http://dx.doi.org/10.1016/S0029-7844(02)02014-8
- Spiegler J, Stichtenoth G, Weichert J, Konig IR, Schlaud M, A VDW, Olbertz D, Gurth H, Schiffmann JH, Bohnhorst B, et al. Pregnancy risk factors for very premature delivery: what role do hypertension, obesity and diabetes play? Arch Gynecol Obstet 2013; 288:57-64; PMID:23400353; http://dx.doi.org/10.1007/s00404-013-2739-6
- Marret S, Ancel PY, Marpeau L, Marchand L, Pierrat V, Larroque B, Foix-L'Helias L, Thiriez G, Fresson J, Alberge C, et al. Neonatal and 5-year outcomes after birth at 30–34 weeks of gestation. Obstet Gynecol 2007; 110:72-80; PMID:17601899; http://dx.doi.org/10.1097/01.AOG.0000267498.95402.bd
- Goldenberg RL, Cliver SP, Mulvihill FX, Hickey CA, Hoffman HJ, Klerman LV, Johnson MJ. Medical, psychosocial, and behavioral risk factors do not explain the increased risk for low birth weight among black women. Am J Obstet Gynecol 1996; 175:1317-24; PMID:8942508; http://dx.doi.org/10.1016/S0002-9378(96)70048-0
- McGrady GA, Sung JF, Rowley DL, Hogue CJ. Preterm delivery and low birth weight among first-born infants of black and white college graduates. Am J Epidemiol 1992; 136:266-76; PMID:1415148
- Preterm birth causes, consequences, and prevention. In: Behrman RE BA, ed. Institute of Medicine (US) Understanding premature birth and assuring healthy outcomes. Washington (DC): National Academies Press (US), 2007.
- Ward K, Argyle V, Meade M, Nelson L. The heritability of preterm delivery. Obstet Gynecol 2005; 106(6):1235-9; PMID:16319246; http://dx.doi.org/10.1097/01.AOG.0000189091.35982.85
- Haataja R, Karjalainen MK, Luukkonen A, Teramo K, Puttonen H, Ojaniemi M, Varilo T, Chaudhari BP, Plunkett J, Murray JC, et al. Mapping a new spontaneous preterm birth susceptibility gene, IGF1R, using linkage, haplotype sharing, and association analysis. PLoS Genet 2011; 7:e1001293; PMID:21304894; http://dx.doi.org/10.1371/journal.pgen.1001293
- Treloar SA, Macones GA, Mitchell LE, Martin NG. Genetic influences on premature parturition in an Australian twin sample. Twin Res 2000; 3:80-2; PMID:10918619; http://dx.doi.org/10.1375/136905200320565526
- Clausson B, Lichtenstein P, Cnattingius S. Genetic influence on birthweight and gestational length determined by studies in offspring of twins. BJOG 2000; 107:375-81; PMID:10740335; http://dx.doi.org/10.1111/j.1471-0528.2000.tb13234.x
- Committee on Practice Bulletins-Obstetrics TACoO, Gynecologists. Practice bulletin no. 130: prediction and prevention of preterm birth. Obstet Gynecol 2012; 120:964-73; PMID:22996126; http://dx.doi.org/10.1097/AOG.0b013e3182723b1b
- Parets SE, Bedient CE, Menon R, Smith AK. Preterm birth and its long-term effects: methylation to mechanisms. Biology 2014; 3:498-513; PMID:25256426; http://dx.doi.org/10.3390/biology3030498
- Myking S, Boyd HA, Myhre R, Feenstra B, Jugessur A, Devold Pay AS, Ostensen IH, Morken NH, Busch T, Ryckman KK, et al. X-chromosomal maternal and fetal SNPs and the risk of spontaneous preterm delivery in a Danish/Norwegian genome-wide association study. PloS one 2013; 8:e61781; PMID:23613933; http://dx.doi.org/10.1371/journal.pone.0061781
- Steffen KM, Cooper ME, Shi M, Caprau D, Simhan HN, Dagle JM, Marazita ML, Murray JC. Maternal and fetal variation in genes of cholesterol metabolism is associated with preterm delivery. J Perinatol 2007; 27:672-80; PMID:17855807; http://dx.doi.org/10.1038/sj.jp.7211806
- Ehn NL, Cooper ME, Orr K, Shi M, Johnson MK, Caprau D, Dagle J, Steffen K, Johnson K, Marazita ML, et al. Evaluation of fetal and maternal genetic variation in the progesterone receptor gene for contributions to preterm birth. Pediatric Res 2007; 62:630-5; PMID:17805208; http://dx.doi.org/10.1203/PDR.0b013e3181567bfc
- Plunkett J, Muglia LJ. Genetic contributions to preterm birth: implications from epidemiological and genetic association studies. Ann Med 2008; 40:167-95; PMID:18382883; http://dx.doi.org/10.1080/07853890701806181
- Kaprio J. Twins and the mystery of missing heritability: the contribution of gene-environment interactions. J Int Med 2012; 272:440-8; PMID:22934540; http://dx.doi.org/10.1111/j.1365-2796.2012.02587.x
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012; 109:1193-8; PMID:22223662; http://dx.doi.org/10.1073/pnas.1119675109
- Menon R, Conneely KN, Smith AK. DNA methylation: an epigenetic risk factor in preterm birth. Reprod Sci; 19:6-13; PMID:22228737; http://dx.doi.org/10.1177/1933719111424446
- Parets SE, Conneely KN, Kilaru V, Fortunato SJ, Syed TA, Saade G, Smith AK, Menon R. Fetal DNA Methylation Associates with Early Spontaneous Preterm Birth and Gestational Age. PloS one 2013; 8:e67489; PMID:23826308; http://dx.doi.org/10.1371/journal.pone.0067489
- Liu Y HC, Murphy S, Huang Z, Overcash F, Thompson J, Brown H, Murtha AP. DNA methylation at imprint regulatory regions in preterm birth and infection. Am J Obst Gynecol 2013; 208(5):395.e1-7; pii S0002-9378:00142–7
- Tendl KA, Schulz SM, Mechtler TP, Bohn A, Metz T, Greber-Platzer S, Kasper DC, Herkner KR, Item CB. DNA methylation pattern of CALCA in preterm neonates with bacterial sepsis as a putative epigenetic biomarker. Epigenetics 2013; 8:1261-7; PMID:24135723; http://dx.doi.org/10.4161/epi.26645
- Cruickshank MN, Oshlack A, Theda C, Davis PG, Martino D, Sheehan P, Dai Y, Saffery R, Doyle LW, Craig JM. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Gen Med 2013; 5:96; PMID:24134860; http://dx.doi.org/10.1186/gm500
- Yuan W, Basso O, Sorensen HT, Olsen J. Indicators of fetal growth and infectious disease in childhood–a birth cohort with hospitalization as outcome. Eur J Epidemiol 2001; 17:829-34; PMID:12081101; http://dx.doi.org/10.1023/A:1015626329533
- Bhutta AT, Cleves MA, Casey PH, Cradock MM, Anand KJ. Cognitive and behavioral outcomes of school-aged children who were born preterm: a meta-analysis. JAMA 2002; 288:728-37; http://dx.doi.org/10.1001/jama.288.6.728
- Singhal A, Fewtrell M, Cole TJ, Lucas A. Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet 2003; 361:1089-97; PMID:12672313; http://dx.doi.org/10.1016/S0140-6736(03)12895-4
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 2008; 359:61-73; PMID:18596274; http://dx.doi.org/10.1056/NEJMra0708473
- Barker DJ. The origins of the developmental origins theory. J Int Med 2007; 261:412-7; PMID:17444880; http://dx.doi.org/10.1111/j.1365-2796.2007.01809.x
- Kessous R, Shoham-Vardi I, Pariente G, Holcberg G, Sheiner E. An association between preterm delivery and long-term maternal cardiovascular morbidity. Am J Obstet Gynecol 2013; 209:368 e1-8; PMID:23800639; http://dx.doi.org/10.1016/j.ajog.2013.05.041
- Catov JM, Newman AB, Roberts JM, Kelsey SF, Sutton-Tyrrell K, Harris TB, Colbert L, Rubin SM, Satterfield S, Ness RB, et al. Preterm delivery and later maternal cardiovascular disease risk. Epidemiology 2007; 18:733-9; PMID:17917602; http://dx.doi.org/10.1097/EDE.0b013e3181567f96
- Hastie CE, Smith GC, Mackay DF, Pell JP. Maternal risk of ischaemic heart disease following elective and spontaneous pre-term delivery: retrospective cohort study of 750 350 singleton pregnancies. Int J Epidemiol 2011; 40:914-9; PMID:21278195; http://dx.doi.org/10.1093/ije/dyq270
- Smith GC, Pell JP, Walsh D. Pregnancy complications and maternal risk of ischaemic heart disease: a retrospective cohort study of 129,290 births. Lancet 2001; 357:2002-6; PMID:11438131; http://dx.doi.org/10.1016/S0140-6736(00)05112-6
- James-Todd T, Wise L, Boggs D, Rich-Edwards J, Rosenberg L, Palmer J. Preterm birth and subsequent risk of type 2 diabetes in black women. Epidemiology 2014; 25:805-10; PMID:25166879; http://dx.doi.org/10.1097/EDE.0000000000000167
- Lykke JA, Paidas MJ, Damm P, Triche EW, Kuczynski E, Langhoff-Roos J. Preterm delivery and risk of subsequent cardiovascular morbidity and type-II diabetes in the mother. BJOG 2010; 117:274-81; PMID:20015308; http://dx.doi.org/10.1111/j.1471-0528.2009.02448.x
- Catov JM, Dodge R, Yamal JM, Roberts JM, Piller LB, Ness RB. Prior preterm or small-for-gestational-age birth related to maternal metabolic syndrome. Obst Gynecol 2011; 117:225-32; PMID:21252733; http://dx.doi.org/10.1097/AOG.0b013e3182075626
- Robbins CL, Hutchings Y, Dietz PM, Kuklina EV, Callaghan WM. History of preterm birth and subsequent cardiovascular disease: a systematic review. Am J Obstet Gynecol 2014; 210:285-97; PMID:24055578; http://dx.doi.org/10.1016/j.ajog.2013.09.020
- Melbye M, Wohlfahrt J, Andersen AM, Westergaard T, Andersen PK. Preterm delivery and risk of breast cancer. Br J Cancer 1999; 80:609-13; PMID:10408874; http://dx.doi.org/10.1038/sj.bjc.6690399
- James-Todd TM, Karumanchi SA, Hibert EL, Mason SM, Vadnais MA, Hu FB, Rich-Edwards JW. Gestational age, infant birth weight, and subsequent risk of type 2 diabetes in mothers: Nurses' Health Study II. Preventing chronic disease 2013; 10:E156; PMID:24050526; http://dx.doi.org/10.5888/pcd10.120336
- Dunlop AL, Kramer MR, Hogue CJ, Menon R, Ramakrishan U. Racial disparities in preterm birth: an overview of the potential role of nutrient deficiencies. Acta obstetricia et gynecologica Scandinavica 2011; 90:1332-41; PMID:21910693; http://dx.doi.org/10.1111/j.1600-0412.2011.01274.x
- Backdahl L, Bushell A, Beck S. Inflammatory signalling as mediator of epigenetic modulation in tissue-specific chronic inflammation. Int J Biochem Cell Biol 2009; 41:176-84; PMID:18793748; http://dx.doi.org/10.1016/j.biocel.2008.08.023
- Rodriguez-Cortez VC, Hernando H, de la Rica L, Vento R, Ballestar E. Epigenomic deregulation in the immune system. Epigenomics 2011; 3:697-713; PMID:22126290; http://dx.doi.org/10.2217/epi.11.99
- Sun YV, Lazarus A, Smith JA, Chuang YH, Zhao W, Turner ST, Kardia SL. Gene-specific DNA methylation association with serum levels of C-reactive protein in African Americans. PloS one 2013; 8:e73480; PMID:23977389; http://dx.doi.org/10.1371/journal.pone.0073480
- Barres R, Zierath JR. DNA methylation in metabolic disorders. Am J Clin Nutrition 2011; 93:897S-900; PMID:21289222; http://dx.doi.org/10.3945/ajcn.110.001933
- Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 2009; 5:401-8; PMID:19488075; http://dx.doi.org/10.1038/nrendo.2009.102
- Ordovas JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol 2010; 7:510-9; PMID:20603647; http://dx.doi.org/10.1038/nrcardio.2010.104
- Kim J, Pitlick MM, Christine PJ, Schaefer AR, Saleme C, Comas B, xe Cosentino V, Gadow E, Murray JC. Genome-Wide Analysis of DNA Methylation in Human Amnion. Scientific World J 2013; 2013:11
- McRae AF, Powell JE, Henders AK, Bowdler L, Hemani G, Shah S, Painter JN, Martin NG, Visscher PM, Montgomery GW. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Gen Biol 2014; 15:R73; PMID:24887635; http://dx.doi.org/10.1186/gb-2014-15-5-r73
- Huang SY, Chiang CH, Chen FP, Yu CL. The alteration of placental-derived soluble MHC class I chain-related protein A and B during pregnancy. Acta obstetricia et gynecologica Scandinavica 2011; 90:802-7; PMID:21426309; http://dx.doi.org/10.1111/j.1600-0412.2011.01131.x
- van Dongen J, Ehli EA, Slieker RC, Bartels M, Weber ZM, Davies GE, Slagboom PE, Heijmans BT, Boomsma DI. Epigenetic variation in monozygotic twins: a genome-wide analysis of DNA methylation in buccal cells. Genes 2014; 5:347-65; PMID:24802513; http://dx.doi.org/10.3390/genes5020347
- Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ, Tylavsky FA, Conneely KN. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Gen 2014; 15:145; PMID:24555763; http://dx.doi.org/10.1186/1471-2164-15-145
- Geiss LS, Wang J, Cheng YJ, Thompson TJ, Barker L, Li Y, Albright AL, Gregg EW. Prevalence and incidence trends for diagnosed diabetes among adults aged 20 to 79 years, United States, 1980-2012. JAMA 2014; 312:1218-26; http://dx.doi.org/10.1001/jama.2014.11494
- Gibbs RS, Romero R, Hillier SL, Eschenbach DA, Sweet RL. A review of premature birth and subclinical infection. Am J Obstet Gynecol 1992; 166:1515-28; PMID:1595807; http://dx.doi.org/10.1016/0002-9378(92)91628-N
- Kerkhof GF, Willemsen RH, Leunissen RW, Breukhoven PE, Hokken-Koelega AC. Health profile of young adults born preterm: negative effects of rapid weight gain in early life. J Clin Endocrinol Metab 2012; 97:4498-506; PMID:22993033; http://dx.doi.org/10.1210/jc.2012-1716
- Melville JM, Moss TJ. The immune consequences of preterm birth. Front Neurosci 2013; 7:79; PMID:23734091; http://dx.doi.org/10.3389/fnins.2013.00079
- Corwin EJ, Guo Y, Pajer K, Lowe N, McCarthy D, Schmiege S, Weber M, Pace T, Stafford B. Immune dysregulation and glucocorticoid resistance in minority and low income pregnant women. Psychoneuroendocrinology 2013; 38:1786-96; PMID:23541234; http://dx.doi.org/10.1016/j.psyneuen.2013.02.015
- Yao Y, Robinson AM, Zucchi FC, Robbins JC, Babenko O, Kovalchuk O, Kovalchuk I, Olson DM, Metz GA. Ancestral exposure to stress epigenetically programs preterm birth risk and adverse maternal and newborn outcomes. BMC medicine 2014; 12:121; PMID:25286408; http://dx.doi.org/10.1186/s12916-014-0121-6
- Oken E, Kleinman KP, Rich-Edwards J, Gillman MW. A nearly continuous measure of birth weight for gestational age using a United States national reference. BMC pediatrics 2003; 3:6; PMID:12848901; http://dx.doi.org/10.1186/1471-2431-3-6
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288-95; PMID:21839163; http://dx.doi.org/10.1016/j.ygeno.2011.07.007
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011; 3:771-84; PMID:22126295; http://dx.doi.org/10.2217/epi.11.105
- Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics 2012; 28:1280-1; PMID:22451269; http://dx.doi.org/10.1093/bioinformatics/bts124
- Kilaru V, Barfield RT, Schroeder JW, Smith AK, Conneely KN. MethLAB: a graphical user interface package for the analysis of array-based DNA methylation data. Epigenetics 2012; 7:225-9; PMID:22430798; http://dx.doi.org/10.4161/epi.7.3.19284
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC bioinformatics 2012; 13:86; PMID:22568884; http://dx.doi.org/10.1186/1471-2105-13-86
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PloS one 2012; 7:e41361; PMID:22848472; http://dx.doi.org/10.1371/journal.pone.0041361
- Ahmad QR, Allen RC, Andersen TC, D Anglin J, Barton JC, Beier EW, Bercovitch M, Bigu J, Biller SD, Black RA, et al. Direct evidence for neutrino flavor transformation from neutral-current interactions in the Sudbury Neutrino Observatory. Physical review letters 2002; 89:011301; PMID:12097025; http://dx.doi.org/10.1103/PhysRevLett.89.011301
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 2009; 4:44-57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; http://dx.doi.org/10.1093/nar/gkn923
- Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, Klengel T, Mehta D, Binder EB, Epstein MP, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol 2014; 38:231-41; PMID:24478250; http://dx.doi.org/10.1002/gepi.21789
- Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW, Rex-Haffner M, Loeschner A, Gonik M, Mercer KB, et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci U S A 2013; 110:8302-7; PMID:23630272; http://dx.doi.org/10.1073/pnas.1217750110
- Norrholm SD, Jovanovic T, Smith AK, Binder E, Klengel T, Conneely K, Mercer KB, Davis JS, Kerley K, Winkler J, et al. Differential Genetic and Epigenetic Regulation of catechol-O-methyltransferase is Associated with Impaired Fear Inhibition in Posttraumatic Stress Disorder. Front Behav Neurosci 2013; 7:30; PMID:23596403; http://dx.doi.org/10.3389/fnbeh.2013.00030
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491:56-65; PMID:23128226; http://dx.doi.org/10.1038/nature11632
- Votavova H, Dostalova Merkerova M, Fejglova K, Vasikova A, Krejcik Z, Pastorkova A, Tabashidze N, Topinka J, Veleminsky M, Jr., Sram RJ, et al. Transcriptome alterations in maternal and fetal cells induced by tobacco smoke. Placenta 2011; 32:763-70; PMID:21803418; http://dx.doi.org/10.1016/j.placenta.2011.06.022
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547-8; PMID:18467348; http://dx.doi.org/10.1093/bioinformatics/btn224