
Abstract
In many whole genome studies of gene expression or modified cytosines, data from probes localized to the X-chromosome are removed from analyses due to gender bias. Previously, we observed population differences in cytosine modifications between Caucasian and African lymphoblastoid cell lines (LCLs) on the autosomes using whole genome arrays to measure modified cytosines. DNA methylation plays a critical role in establishment and maintenance of X-chromosome inactivation in females. Therefore, we reasoned that by investigating cytosine modification patterns specifically on the X-chromosome, we could obtain valuable information about a chromosome that is often disregarded in genome-wide analyses. We investigated population differences in cytosine modification patterns along the X-chromosome between Caucasian and African LCLs and identified novel sites that escape methylation on the inactive X-chromosome (Xi) in females. We characterized the chromatin state of these loci by incorporating the extensive histone modification ChIP-seq data generated by ENCODE. To explore the relationship between DNA and histone modifications further, we hypothesized that BRD4, a protein that binds acetylated histones, could be preventing some sites from becoming de novo methylated. To test this, we treated 4 female LCLs with JQ1, a small molecule inhibitor of BRD4, but found that JQ1 treatment induced minor changes in cytosine modification levels, and the majority of sites escaping methylation on the Xi remained unmethylated. This suggests that other epigenetic mechanisms or transcription factors are likely playing a larger role in protecting these sites from de novo methylation on the Xi.
Abbreviations
| 450K array | = | Illumina Infinium HumanMethylation450 BeadChip array |
| BRD4 | = | Bromodomain containing 4 |
| CEU | = | Caucasians of European descent residing in Utah, USA |
| ChIP | = | chromatin immunoprecipitation |
| ENCODE | = | Encyclopedia of DNA Elements |
| LCL | = | lymphoblastoid cell lines |
| Xi | = | inactive X-chromosome |
| YRI | = | Yoruba individuals from Ibadan, Nigeria. |
Introduction
Whole genome genetic, epigenetic, and gene expression studies in cell lines and preclinical studies often neglect sex-based considerations in their analyses.Citation1 However, female and male cells have different responses to chemical and microbial stressors, and several journals are now requiring specification of sex- and gender-related information in their publications.Citation1 Most animal studies use male mice, which can neglect important sex differences that may impact clinical studies. This preponderance of male dominated preclinical studies has led the National Institutes of Health to require more rigorous studies evaluating the effects of sex differences.Citation1
X-chromosome biology is an important difference between male and female cells. Studying cytosine modification patterns on the X-chromosome is especially interesting because de novo DNA methylation is an essential step during X-chromosome inactivation in females.Citation2,3 X-chromosome inactivation involves a sporadic choice to silence one of the 2 X-chromosomes along with transcription of the XIST long non-coding RNA.Citation4-6 XIST spreads along the chromosome, coating and condensing the chromosome in cis.Citation7 DNA methylation is thought to stabilize and lock the inactivated X-chromosome (Xi) in its condensed conformation.Citation8 X-chromosome inactivation is an excellent example of long-range changes in chromatin structure, requiring interplay between long-noncoding RNA, histone modifications, and DNA methylation to convert the entire chromosome to heterochromatin. Therefore, observing specific epigenetic alterations within the context of other epigenetic marks will give a more comprehensive view of the chromatin state.
Epigenetic patterns are capable of predicting genes that escape inactivation on the Xi, because both lack of DNA methylation and presence of activating histone modifications, such as H3 acetylation and H3K4 methylation, are associated with sites that escape inactivation.Citation9-13 In addition, SNP allelic dosage has been used previously to characterize genes that have expression of 2 copies of X-linked genes in female cells.Citation14 Although the majority of sites that escape inactivation are common across tissues, methylation of a subset of genes showed different patterns in different tissues.Citation11
The Illumina Infinium HumanMethylation450 BeadChip (450K) array is a widely used technique for measuring cytosine modification levels genome-wide. The 450K array measures over 480,000 CpGs across all chromosomes. The X-chromosome contains 11,232 probes and the Y-chromosome contains 416 probes. Pseudoautosomal regions are homologous nucleotide sequences on the X- and Y-chromosomes and are expressed by both copies of the X-chromosome in females. Only 13 probes on the 450K array are located in a pseudoautosomal region on the X-chromosome, and they are all in pseudoautosomal region 1 (Xp22). A scan of recent articles that published 450K array data showed that approximately half of the studies removed data from the X- and Y-chromosomes from downstream analyses. Those studies that do retain data from the X- and Y-chromosomes generally deal with them by correcting for gender in their analyses. However, very few studies use the 450K array data to look specifically at the X-chromosome cytosine modification patterns in their samples.
Recently, we generated 450K array data for 60 Caucasian (CEU: Caucasians of Northern/Western European descent residing in Utah) and 73 African (YRI: Yoruba people from Ibadan, Nigeria) lymphoblastoid cell lines (LCLs) and identified population-specific cytosine modification patterns, the genetic architecture of modified cytosines, and their contribution to gene expression patterns.Citation15,16 Similar to previous research by many groups, we had removed the X- and Y-chromosome probes due to the gender bias in cytosine modification patterns. However, we reasoned that analyzing the sex chromosomes separately could provide useful information regarding the biology of these chromosomes, which is frequently disregarded. Therefore, we aimed to investigate cytosine modifications specifically on the X-chromosomes of our Caucasian and African LCL samples. Although we started with cytosine modification data, we sought to incorporate histone modification and DNA binding protein data available from ENCODECitation17,18 to obtain a more comprehensive understanding of the chromatin state across the entire chromosome and put our observations regarding cytosine modification patterns in a more inclusive epigenetic context.
Results
Population-specific X-chromosome cytosine modification patterns
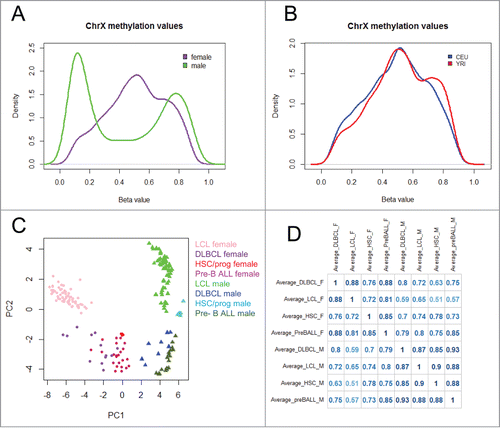
We hypothesized that the CEU and YRI female and male samples would show some population-specific cytosine modification patterns on the X-chromosome. We analyzed female and male samples separately due to the gender differences in cytosine modification distributions (). We found that 763 CpGs were differentially modified between CEU and YRI females at a false discovery rate (FDR) < 0.01 (10.8% of total CpGs analyzed), and 119 of those CpGs were also significant at Bonferroni corrected P < 0.05 using the Wilcoxon Rank-Sum test. The 119 CpGs account for 1.7% of all CpGs on the X-chromosome and correspond to 82 unique genes ( and Table S1). The male samples showed much less variation in X-chromosome cytosine modification between populations, as 344 CpGs were differentially modified between CEU and YRI males at FDR < 0.01 (4.9% of total CpGs analyzed) and only 17 CpGs were significant at Bonferroni corrected P < 0.05 using the Wilcoxon Rank-sum test, accounting for 0.2% of the total number of CpGs on the X-chromosome (Table S2). The observation of females showing more variation is perhaps to be expected, as female LCLs have higher median cytosine modification values on the X-chromosome compared with male LCLs, and that allows more room for variation. Of the 82 genes showing differential modification between CEU and YRI females, there were 4 genes that had greater than 2 probes that showed differential modification: acyl-CoA thioesterase 9 (ACOT9), lysosomal-associated membrane protein 2 (LAMP2), O-linked N-acetylglucosamine transferase (OGT), and phosphatidylinositol glycan anchor biosynthesis, class A (PIGA). PIGA also showed a population difference in males. Furthermore, we found that PIGA, ACOT9, and LAMP2 were differentially expressed between CEU and YRI females (Fig. S1), demonstrating that the variation of cytosine modification patterns on the X-chromosome corresponds with differences in gene expression phenotypes between these 2 populations. At the cut-off of FDR < 0.01, there were 80 CpGs that were different between CEU and YRI among both genders. Overall, the number of differentially modified CpGs on the X-chromosome in both genders is less than the proportion of population-specific CpGs on the autosomes reported in our previous work, where we found 13% of CpGs on autosomes to be differentially modified between CEU and YRI at FDR<0.01.Citation15
Figure 1. Overview of X-chromosome cytosine modification data (A) Distribution of X-chromosome cytosine modification levels in females (purple) and males (green). (B) Distribution of X-chromosome cytosine modification levels in CEU (blue) and YRI (red) female LCLs. (C) Principal Component Analysis of X-chromosome cytosine modification levels in LCLs, diffuse large B cell lymphomas (DLBCL), haematopoietic stem/progenitor cells (HSC/prog), and pre-B cell acute lymphoblastic leukemia (pre-B ALL) samples. (D) Correlation matrix of X-chromosome cytosine modification levels in LCLs, DLBCL, HSC/progenitor cells, and pre-B cell ALL samples.
Comparison of X-chromosome cytosine modification patterns in LCLs with other haematopoietic cell types
To demonstrate the robustness of LCLs as a model for haematopoietic cell types, we compared the LCL 450K array data with 450K array data from diffuse large B cell lymphomas (DLBCL), renal cell carcinomas, and breast carcinomas available from The Cancer Genome Atlas (TCGA) and found they are most closely clustered with DLBCLs compared with the other 2 cell types (Fig. S2). Furthermore, there is not a separation between the cell lines and the primary tissue, which demonstrates that the LCL immortalization by EBV transformation did not significantly alter cytosine modification patterns to the point where it did not closely resemble tissue-specific patterns. Next, we compared the cytosine modification data from the X-chromosome to publicly available 450K array data from haematopoietic stem/progenitor cells (GSE40799),Citation19 pediatric pre-B cell acute lymphoblastic leukemias (pre-BALL) (GSE38235),Citation20 and DLBCLs (TCGA) to investigate the extent to which the X-chromosome cytosine modification patterns in LCLs resembled other haematopoietic cell types. Principal Component Analysis demonstrated a clear separation by gender along the first principle component in all cell types (). We found that the X-chromosome cytosine modification patterns from LCLs were significantly positively correlated to the 3 other cell types in both genders (). Female LCLs showed the highest correlation with DLBCL (r = 0.88) (). It has been reported previously that methylation patterns change over the course of cellular differentiation, aging, and neoplastic transformation, so the marginal differences between these cell types was expected.
Identification of CpGs that escape X-chromosome DNA methylation
In addition to extending the population-specific work we had performed on the autosomes, we were interested in using these data to investigate cytosine modification variation on the X-chromosome and its relation with other epigenetic marks in these 2 human populations. When plotting the frequency distribution of the averaged β-values across all females, we observed a small portion of CpGs that remained lowly methylated (). We hypothesized that we could use this dataset to identify sites that escape methylation on the Xi. The probes located within pseudo-autosomal regions were not included in our analyses because of the propensity of those probes to bind other regions of the genome (specifically, the Y-chromosome) (Table S3). Furthermore, these sites are already known to escape inactivation in females. Cotton et al. previously published a study that investigated sites that escape methylation on the Xi in females.Citation11 Their decision tree for predicting which sites escaped methylation required both males and females to have an average β-value less than 0.15 (<15% methylated). We used a model for prediction of CpGs that escape methylation on the Xi similar to that used by Cotton et al., therefore including CpGs that have an average β-value less than 0.15 in males and females.
Figure 2. Identification of CpGs that escape methylation on the Xi (A) Location of CpGs that escape methylation (top) or are subjected to methylation (bottom) on the Xi. The location of the non-coding RNA XIST is denoted with an arrow. (B–C) Tet-assisted bisulfite sequencing of 2 genes, COL4A6 (B) and PRKX (C). Each pie chart represents a CpG captured by the bisulfite PCR. The proportion of 5mC is shown in black, the proportion of 5hmC is shown in gray, and the proportion of unmodified cytosine is shown in white.

Due to the LCLs being oligoclonal, the silenced X-chromosome may be mosaic within each sample. Using this decision tree for identifying loci that escape methylation, we are able to detect CpGs that are lowly methylated on both X-chromosomes, regardless of whether the cell line is mono- or oligo-clonal (Fig. S3). We identified 225 CpGs that showed less that 15% methylation in female and male samples, suggesting that these sites are lowly methylated on both chromosomes in the female samples. As a comparison, 452 CpGs had an average of less than 20% methylation in females and males. We kept our cut-off at the more stringent level of 15% to minimize false positives. The CpGs showing less than 15% methylation correspond to 104 unique genes, and are located across the length of the X-chromosome, aligned using the hg19 version of the human genome reference ( and Table S4).
To evaluate the validity of our approach for correctly identifying genes that are escaping inactivation on the Xi, we compared our results to those from other studies that have identified genes as escaping inactivation on the Xi using cytosine modification data. We compared the 104 genes found to escape inactivation in our study with 2 studies that used the Illumina 27K arrayCitation11 and the 450K array,Citation21 and found that 12 genes overlapped with the results from the 27K array study and 48 genes overlapped with the 450K array study.
Some genes that have previously been predicted to escape did not meet our criteria for escaping methylation on the Xi. The 450K array does not distinguish between 5-hydroxymethylcytosine (5hmC) and 5-methylcytosine (5mC). Therefore, we hypothesized that some of the CpGs at these genes may be hydroxymethylated instead of methylated, resulting in protection from bisulfite conversion and exclusion from our list of CpGs that escape methylation. We analyzed 2 genes by Tet-assisted bisulfite sequencing (TAB-Seq)Citation22 that had been shown to escape inactivation in previous studies, Collagen, type IV, α 6 (COL4A6) and Protein kinase X-linked (PRKX),Citation11,14,23 to detect 5hmC near the promoter regions of these genes in 2 female LCLs. We found low levels of 5hmC in COL4A6 in 3 of the 10 CpGs analyzed by TAB-Seq (). However, we did not observe any 5hmC in the PRKX promoter region. Therefore, 5hmC may contribute to the cytosine modification levels of some genes on the X-chromosome, but other genes, such as PRKX, are likely among the subset of genes that are more variable in their inactivation status between tissues.
Genes predicted to escape X-chromosome inactivation show higher levels of expression
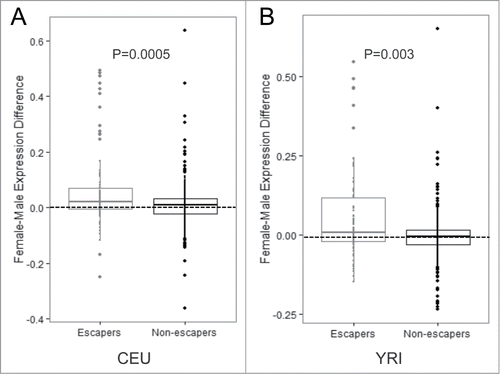
We hypothesized that the genes containing lowly methylated CpGs on the Xi would show higher levels of expression in females compared with males. Using Affymetrix exon array data generated previously for these LCL populations,Citation24 we found that genes that were lowly methylated were more highly expressed in females compared with males, whereas there was no gender difference among genes that were methylated on the Xi (). We next compared our set of genes predicted to escape inactivation to studies that used expression-level data to predict escape from X-chromosome inactivation. Cotton et al. Citation14 used SNP allelic dosage in a similar cohort of HapMap LCLs to identify genes as escaping inactivation, and found 43 genes to be expressed on the Xi. Twenty-seven of those genes contained a CpG measured by the 450K array that passed our quality control criteria, and among those 27 genes, we found 25 of them escape methylation on the Xi (93% overlap) (Table S4). Therefore, our prediction for genes that escape inactivation at the level of DNA methylation is highly correlated with results from a similar cohort of LCLs that used a measure at the level of transcription. In addition, Carrel et al.Citation23 identified 94 genes as escaping inactivation by employing a human/rodent somatic cell hybrid system. Among the genes they investigated, 41 genes were included in our analyses, and 23 of those were also predicted to escape methylation on the Xi in our data set. This higher discordance with this study may be due to the difference in tissues used.
Figure 3. Genes with CpGs predicted to escape inactivation have increased levels of expression (A–B) Expression values for genes containing CpGs that escape methylation compared with genes containing CpGs that are subject to methylation on the Xi in the CEU population (A) and the YRI population (B). P-values were calculated using the Student's t-test.
Overall, by comparing our results to other publicly available datasets, we demonstrated that our analyses identified 46 genes that are likely escaping inactivation that were not found in the studies that used DNA methylation or expression-level data. Of those genes, 10 of them contained more than one CpG that was predicted to escape inactivation, including Forkhead box O4 (FOXO4), Ephrin-B1 (EFNB1), and cysteine-rich hydrophobic domain 1 (CHIC1) (Table S4).
CpGs that escape DNA methylation on the Xi show distinct chromosomal features
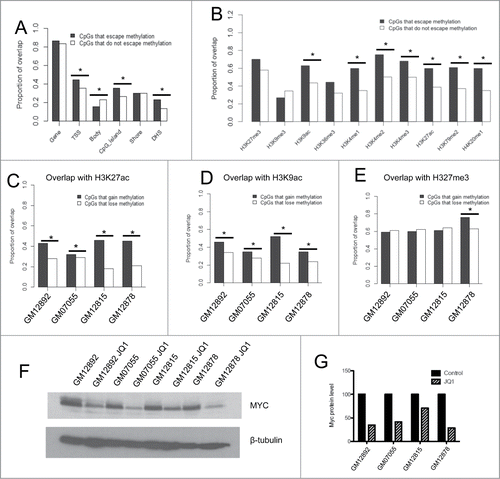
Next, we aimed to determine whether CpGs that escape DNA methylation on the Xi were enriched in certain chromosomal features. First, we investigated whether these CpGs were more likely to be found in distinct genic contexts. We found by Chi-squared tests that the CpGs that escape methylation were more likely to be located near the transcriptional start site of a gene (P = 0.007) and within a CpG island (P = 0.003). They were significantly less likely to be localized in a gene body (P = 0.008) ().
Figure 4. Investigation of the relationship between cytosine modification and histone marks on the Xi (A) Proportion of overlap of CpGs that escape methylation and those that do not with certain genic contexts. (B) Proportion of overlap of CpGs within TSSs that escape methylation and those that do not with histone modifications and transcription factor binding sites obtained from the ENCODE Tier 1 LCL GM12878. (C–E) Proportion of overlap of CpGs that gain methylation or lose methylation following JQ1 treatment with H3K27ac (C), H3K9ac (D), and H3K27me3 (E). Asterisks denote P < 0.05 calculated by the Chi-squared test. (F–G) Western blot of Myc protein levels in 4 LCLs treated with DMSO vehicle control or JQ1 (F). Densitometry quantification analyses performed with ImageJ software (G). (*) denotes P < 0.05 by Chi-squared tests.
We were interested in investigating possible mechanisms that may be promoting an open chromatin state and inhibiting DNA methylation from being established at these loci. Histone modifications are known to play a critical role in X-chromosome inactivation, as hypoacetylation and methylation of H3K9 both occur immediately after Xist RNA coating and before transcriptional silencing of the Xi.Citation25,26 Therefore, we hypothesized that CpGs escaping DNA methylation would be located in regions associated with activating histone marks, including H3 acetylation.
We specifically analyzed the 97 CpGs located in TSSs that escape methylation on the Xi and tested for co-localization with histone modification ChIP-seq data from the ENCODE Tier 1 female LCL GM12878. We found that CpGs within TSSs that escape DNA methylation on the Xi were associated with activating histone marks after correcting for multiple testing, including H3K9ac (P = 1.7 × 10−4), H3K4me-1, −2, −3 (P = 5.9 × 10−7, P = 1.33 × 10−6, and P = 4.9 × 10−4, respectively), and H3K27ac (P = 3.6 × 10−5) (). They were not more likely to be associated with the repressive histone modifications H3K27me3 and H3K9me3 (). We also tested the co-localization for CpGs that escape methylation outside of TSSs with histone marks and found results to be similar (Fig. S4). Furthermore, all CpGs that escaped methylation were enriched in DNase hypersensitive regions, which is consistent with their localization to regions of open chromatin (P = 0.0001) ().
We hypothesized that, in addition to activating histone marks, proteins that bind DNA may be inhibiting DNA methylation from occurring at these sites. CTCF has been shown to protect DNA from becoming methylated on the X-chromosome during development, and this protection is associated with H3 acetylation.Citation13 Therefore, we compared our data to CTCF ChIP-Seq data, including CTCF, for the GM12878 female cell line from ENCODE. CTCF showed higher overlap among all sites that escape DNA methylation on the Xi compared with those that do not, both inside and outside of TSSs, suggesting that CTCF may play a role in protecting sites from inactivation (Fig. S5). Furthermore, we found that 67 of the 96 CpGs (72%) that escape DNA methylation at CTCF binding sites co-localize with H3K9ac and H3K27ac, whereas none of the methylated cytosines at CTCF binding sites co-localize with H3 acetylation. This suggests that CTCF binding along with H3 acetylation is a better predictor of CpGs that escape DNA methylation on the Xi than CTCF binding alone.
Relationship between histone acetylation and DNA methylation on the Xi
The interaction between histone modifications and DNA methylation is thought to be important in mediating chromatin accessibility and transcription regulation. Previous studies have investigated methylation and gene expression changes on the Xi in response to epigenetic therapies, such as histone deacetylase inhibitors and DNA hypomethylating agents.Citation25,26 Considering the evidence that proteins that bind DNA, such as CTCF, can protect DNA on the Xi from becoming methylated, we hypothesized that proteins that bind acetylated histones may be protecting CpGs from becoming methylated on the Xi. Bromodomain containing 4 (BRD4) binds to acetylated histones to mediate transcription. JQ1 is a small molecule inhibitor of BET bromodomains with the highest affinity for the first bromodomain of BRD4.Citation27,28 To determine whether inhibition of BRD4 could result in changes to DNA methylation on the Xi, we treated 4 female LCLs with either 250 nM JQ1 or vehicle control (DMSO) for 48 h and assayed cytosine modification levels using the 450K array. We anticipated that sites that are lowly methylated may gain methylation in the absence of BRD4. The LCLs express DNMT1, DNMT3A, and DNMT3B (Fig. S6); therefore, regaining methylation after exposure to JQ1 is possible through both maintenance and de novo DNA methylation pathways.
First, we found the array results to be robust, because the experimental variation (within the same individual) between the original run and this second run was smaller than the inter-individual variation (). We found that JQ1 treatment did not change the genome-wide cytosine modification levels substantially, as all 4 cell lines had a very strong correlation between DMSO control and JQ1-treated β-values on all autosomes (r > 0.99). We looked specifically at CpGs on the X-chromosome, and found that JQ1 resulted in modest gains and losses of methylation across the X-chromosome in all 4 cell lines, where approximately 600 CpGs gained at least 3% methylation in each of the cell lines, but few achieved greater than a 10% gain in methylation (Table S6). CpGs that did gain methylation (an increase in β-value of at least 0.03) were enriched in H3K27ac and H3K9ac compared with sites that lost methylation (a decrease in β-value of at least 0.03) in each of the cell lines (). We found no enrichment in sites that gain methylation that overlap with H3K27me3, a histone modification that should remain unaffected by JQ1 treatment, in 3 of the 4 cell lines (). We did observe downregulation of MYC in all 4 cell lines at the dose and time point used in this experiment, confirming that JQ1 treatment resulted in transcriptional repression of a known target gene. (). Therefore, there may be some interaction between BRD4 loss at acetylated histones and subsequent gain of DNA methylation, but it is not sufficient to fully restore DNA methylation levels at CpGs that escape methylation on the Xi.
Table 1. Within-individual variation is smaller than inter-individual variation in cytosine modification levels across arrays.We compared β-values from the original 450K array to the more recent 450K array results and found that the 3 samples included in both arrays showed a higher level of intra-individual (across different runs) correlation, measured by the correlation coefficient (r) compared with inter-individual correlations of different samples run on the same chip
Discussion
In this work, we demonstrated how analyses of cytosine modification variation could be performed for the X-chromosome using the Illumina 450K array. We focused on the X-chromosome in particular because of the importance of DNA methylation in X-chromosome biology. We identified a small percentage of CpGs that showed differential modification between CEU and YRI female and male LCLs. Interestingly, males showed less variation between populations, and this could be because females have a higher median methylation level, which may increase variation, and males only have one X-chromosome, which may reduce variation. We investigated CpG loci that escape methylation on the Xi and compared our results with several other studies investigating escape from X-chromosome inactivation.Citation11,14,21,23 The correlation between our study and the other studies ranged from over 90% overlap to approximately 50% overlap. This may be attributable to differences in the CpGs included in the analyses, such as whether the analysis was limited only to CpGs in the promoters of genes, or the variety of tissue types examined, as it has been reported that there is some tissue-specificity in X-chromosome inactivation.Citation11 Overall, we identified 46 novel genes containing CpGs that escape methylation on the Xi, and we correlated these sites with H3 acetylation and H3K4 methylation, suggesting an open chromatin formation.
The interaction between epigenetic modifications on the Xi is complex, and we are still working toward understanding how each epigenetic modification is regulated to establish and maintain the Xi. It has been shown that azacitidine, a hypomethylating agent, can induce re-expression of several X-linked genes, providing experimental evidence for the essential role of DNA methylation in X-chromosome inactivation.Citation2,3 In addition, azacitidine and decitabine have also been shown to cause the Xi to decondense and shift its replication from later to earlier in the cell cycle.Citation29,30 DNA methylation is believed to work synergistically with histone hypoacetylation in maintaining X-chromosome inactivation,Citation26 but additional work is needed to understand the unique and overlapping functions of these marks in establishing and maintaining the Xi.
We were interested in testing whether a protein such as BRD4, which binds acetylated histones, blocks DNA methylation from occurring at these sites that escape methylation on the Xi. JQ1 is a small molecular inhibitor of BRD4, and it has important therapeutic potential in MYC-driven cancers because it causes downregulation of MYC.Citation31,32 However, the extent to which JQ1 influences DNA methylation has not been studied previously. Here, we demonstrated that JQ1 caused only minor changes in DNA methylation patterns on the X-chromosome. The majority of the CpGs that escape methylation on the Xi remained unaffected, suggesting that they are being protected from de novo methylation by other mechanisms. It will be interesting to uncover whether there are other transcription factors or histone modifying proteins that play a role in keeping these loci in a euchromatic state. Other groups that generate genome-wide data, especially 450K array data, could take a similar approach as demonstrated here and analyze the X-chromosome separately to learn more about cytosine modification patterns specifically on the X-chromosome.
To address the matter of accounting for sex differences in biomedical research, the National Institutes of Health is calling for changes in program oversight, review and policy, and publishing criteria.Citation1 This call to action is the result of minimal progress that has been made in balancing male and female models in cell and animal studies. Instead of disregarding information that can shed light on the differences between males and females, biomedical researchers need to account for these differences and utilize all relevant information in their studies. X-chromosome biology is an important difference between male and female cells, and our work has demonstrated the utility of including the X-chromosome in analyses of human epigenetic variation.
Materials and Methods
Identification of population-specific X-chromosome cytosine modification patterns
The cytosine modification data generated from the 450K array on 60 CEU and 73 YRI LCLs (GSE39672) was processed as described in our previous publication.Citation15 Cytosine modification levels were summarized with the Illumina GenomeStudio-Modification Module v1.9 (GenomeStudio). The β-value was calculated as the proportion of modified probe intensity over the sum of the modified and unmodified probe intensities. Similar to the approach taken in our previous work, the M-value was used to detect population-specific cytosine modifications, which is defined as the log2 ratio of the intensities of the methylated probe vs. the unmethylated probe. The M-value reduces the heteroscedastic distribution observed in the β-value distributions and has been shown to provide better detection sensitivity at extreme modification levels.Citation33 After removing probes with common SNPs and that map ambiguously to the genome, as described in our previous publication,Citation15,34 we included 7,033 CpGs on the X-chromosome in our analyses. Cytosines that were differentially modified between the CEU and YRI females and males, separately, were detected by 2 statistical approaches. The first was a linear model: cytosine modification level ˜ population + gender + error. FDR was estimated by 100 permutations across samples using an approximate test.Citation35 Positive betas indicate CEU>YRI and negative betas indicate CEU<YRI. Second, a Wilcoxon Rank-Sum test was performed and CpGs that met Bonferroni corrected P<0.05 were considered differentially modified between the 2 populations to correct for multiple comparisons (Table S1–2).
Comparison of LCL data with other publicly available data sets
We obtained publicly available 450K array data from haematopoietic stem/progenitor cells (GSE40799),Citation19 pediatric pre-B cell acute lymphoblastic leukemias (pre-BALL) (GSE38235),Citation20 diffuse large B cell lymphomas, renal carcinomas, and breast carcinomas (The Cancer Genome Atlas) to compare with our LCL data. We performed quantile normalization across all datasets followed by Principal Components Analysis (PCA).
Analysis of CpGs that escape methylation on the Xi, gene expression levels, and ChIP-seq data sets
We modeled our methods of identifying CpGs that escape methylation on the Xi after the decision-tree of Cotton et al.Citation11 We considered CpGs that showed an average of <15 % methylation in both female and male samples to be unmethylated on the Xi (β-value < 0.15). Theoretically, CpGs subject to X-chromosome inactivation should be at least 50% methylated in females. We used a stringent cut-off of 15% to limit false positives. A graph showing the number of CpGs predicted to escape methylation at several cut-offs ranging from 10-30% methylation is depicted in Fig. S7.
The gene expression data used in this study were previously generated by our group using the Affymetrix Human Exon 1.0ST Array (GSE7851).Citation24 The difference between the average expression level in females and males was calculated for genes that are predicted to escape methylation and genes predicted to be subject to methylation on the Xi, and the Student's t-test was used to test for statistically significant differences between the 2 groups. We used the 450K array annotation file to characterize the genic context of CpGs escaping methylation on the Xi. As denoted in the annotation file, “near the transcriptional start site” (TSS) is defined as within 1500 bp of the TSS. In order to compare the 450K data to transcription factor and histone modification data, we downloaded the data for GM12878, a Tier 1 female cell line in ENCODE. A description of all proteins and histone modifications analyzed is in Table S6. We specifically analyzed the CpGs that were within TSSs and outside of TSSs separately, and Chi-squared tests were used to determine a significant enrichment of CpGs that escape methylation with specific DNA binding proteins and histone modifications.
TAB-Seq
TAB-Seq was performed using the WiseGene 5hmC TAB-Seq kit (cat. no. K001). Five µg of DNA was sonicated to 400 bp and conjugated to UDP-glucose with β-glycosyltransferase. After reaction cleanup, conjugated DNA was oxidized using mTet1, and bisulfite treated. Glycosylated DNA without mTet1 oxidation was used for quantification of modified and unmodified cytosines. After PCR amplification with ZymoTaq polymerase (Zymo Research Co., cat. no. E2001), products were TOPO TA cloned (Life Technologies, cat. no. K4575-02) and sequenced at The University of Chicago DNA Sequencing and Genotyping Facility. Primers used for amplification are listed in Table S7.
Cell lines and JQ1 treatment
Four female LCLs (GM07055, GM12815, GM12878 and GM12892) were cultured in RPMI supplemented with 15% fetal bovine serum and 1% L-glutamine at 37°C. Cells were passaged every 2-3 d JQ1 was generously provided by Dr. James Bradner's Laboratory. For JQ1 treatment, cells were split at 350,000 cells/mL in T25 flasks and treated with 250 nM JQ1 24 h after splitting. Cells were pelleted 48 h after treatment.
450K array profiling and data processing
DNA was isolated from cell pellets using phenol-chloroform extraction and quantified using the Qubit fluorometer (Life technologies). The University of Chicago Genomics Core Facility was provided with 1 µg of DNA in 50 µL of water for bisulfite conversion with the Zymo EZ DNA methylation Kit™ (Zymo Research Co.). Cytosine modification levels were profiled using the Illumina Infinium HumanMethylation450 BeadChip (Illumina, Inc.), according to manufacturer's protocols using an Illumina HiScan System. Approximately 150 ng of bisulfite-converted DNA from each samples was used for array hybridization. Quantile normalized cytosine modification data for the X-chromosome of the 4 LCLs are in Table S5. The raw 450K data were processed as described in our previous publication.Citation15 The raw and processed 450K data are available on Gene Expression Omnibus with accession number GSE62111.
Western blot
Western blot was performed to confirm that c-Myc protein levels decreased following JQ1 treatment in the LCLs. Protein lysates were run on a 12% polyacrylamide gel, transferred to a nitrocellulose membrane, and incubated in primary antibody for MGMT (cat. no: ab69629, Abcam) and rabbit secondary antibody (cat. no: 401393, Millipore). Anti-β tubulin (cat. no: 05-661, Millipore) with mouse secondary antibody (cat. no: sc-2005, Santa Cruz Biotechnology) was used as a loading control.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Material.zip
Download Zip (1.7 MB)Acknowledgments
The authors thank Dr. R. Stephanie Huang, Bonnie LaCroix, and Jamie Meyers of the Pharmacogenomics of Anticancer Agents Research LCL core for maintaining, distributing, and authenticating LCLs. The authors also thank Dr. Pieter Faber from the Genomics Core of The University of Chicago for his help with the 450K arrays and data processing.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was partially supported by grants from the NIH: R21HG006367 (to WZ, LAG, MED), R21CA187869 (to WZ), and NIH/NIGMS Pharmacogenomics of Anticancer Agents Research Grant U01GM61393 (to MED). The funding agency does not have a role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature 2014; 509:282-3; PMID:24834516; http://dx.doi.org/10.1038/509282a.
- Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science 1981; 211:393-6; PMID:6164095; http://dx.doi.org/10.1126/science.6164095
- Graves JA. Five-azacytidine-induced re-expression of alleles on the inactive X chromosome in a hybrid mouse cell line. Exp Cell Res 1982; 141:99-105; PMID:6180921; http://dx.doi.org/10.1016/0014-4827(82)90072-6
- Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, Grompe M, Pizzuti A, Muzny D, Lawrence C, et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991; 351:325-9; PMID:2034278; http://dx.doi.org/10.1038/351325a0
- Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D, Rastan S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991; 351:329-31; PMID:2034279; http://dx.doi.org/10.1038/351329a0
- Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991; 349:38-44; PMID:1985261; http://dx.doi.org/10.1038/349038a0
- Clemson CM, McNeil JA, Willard HF, Lawrence JB. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol 1996; 132:259-75; PMID:8636206; http://dx.doi.org/10.1083/jcb.132.3.259
- Lock LF, Takagi N, Martin GR. Methylation of the Hprt gene on the inactive X occurs after chromosome inactivation. Cell 1987; 48:39-46; PMID:3791414; http://dx.doi.org/10.1016/0092-8674(87)90353-9
- Boggs BA, Cheung P, Heard E, Spector DL, Chinault AC, Allis CD. Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat Genet 2002; 30:73-6; PMID:11740495; http://dx.doi.org/10.1038/ng787
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457-66; PMID:17334365; http://dx.doi.org/10.1038/ng1990
- Cotton AM, Lam L, Affleck JG, Wilson IM, Peñaherrera MS, McFadden DE, Kobor MS, Lam WL, Robinson WP, Brown CJ. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum Genet 2011; 130:187-201; PMID:21597963; http://dx.doi.org/10.1007/s00439-011-1007-8
- Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, Antonarakis SE. DNA methylation profiles of human active and inactive X chromosomes. Genome Res 2011; 21:1592-600; PMID:21862626; http://dx.doi.org/10.1101/gr.112680.110
- Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, Tsuchiya KD, Disteche CM. Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell 2005; 8:31-42; PMID:15669143; http://dx.doi.org/10.1016/j.devcel.2004.10.018
- Cotton AM, Ge B, Light N, Adoue V, Pastinen T, Brown CJ. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol 2013; 14:R122; PMID:24176135; http://dx.doi.org/10.1186/gb-2013-14-11-r122
- Moen EL, Zhang X, Mu W, Delaney SM, Wing C, McQuade J, Myers J, Godley LA, Dolan ME, Zhang W. Genome-wide variation of Cytosine modifications between European and african populations and the implications for complex traits. Genetics 2013; 194:987-96; PMID:23792949; http://dx.doi.org/10.1534/genetics.113.151381
- Zhang X, Moen EL, Liu C, Mu W, Gamazon ER, Delaney SM, Wing C, Godley LA, Dolan ME, Zhang W. Linking the genetic architecture of cytosine modifications with human complex traits. Hum Mol Genet 2014; 23(22):5893-905.
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/10.1038/nature11247
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013; 41:D56-63; PMID:23193274; http://dx.doi.org/10.1093/nar/gks1172
- Weidner CI, Walenda T, Lin Q, Wölfler MM, Denecke B, Costa IG, Zenke M, Wagner W. Hematopoietic stem and progenitor cells acquire distinct DNA-hypermethylation during in vitro culture. Sci Rep 2013; 3:3372; PMID:24284763; http://dx.doi.org/10.1038/srep03372
- Busche S, Ge B, Vidal R, Spinella JF, Saillour V, Richer C, Healy J, Chen SH, Droit A, Sinnett D, et al. Integration of high-resolution methylome and transcriptome analyses to dissect epigenomic changes in childhood acute lymphoblastic leukemia. Cancer Res 2013; 73:4323-36; PMID:23722552; http://dx.doi.org/10.1158/0008-5472.CAN-12-4367
- Cotton AM, Price EM, Jones MJ, Balaton BP, Kobor MS, Brown CJ. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum Mol Genet 2014; 24(6):1528-39.
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012; 149:1368-80; PMID:22608086; http://dx.doi.org/10.1016/j.cell.2012.04.027
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434:400-4; PMID:15772666; http://dx.doi.org/10.1038/nature03479
- Zhang W, Duan S, Kistner EO, Bleibel WK, Huang RS, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, et al. Evaluation of genetic variation contributing to differences in gene expression between populations. Am J Hum Genet 2008; 82:631-40; PMID:18313023; http://dx.doi.org/10.1016/j.ajhg.2007.12.015
- Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL. Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 2001; 107:727-38; PMID:11747809; http://dx.doi.org/10.1016/S0092-8674(01)00598-0
- Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol 2001; 153:773-84; PMID:11352938; http://dx.doi.org/10.1083/jcb.153.4.773
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature 2010; 468:1067-73; PMID:20871596; http://dx.doi.org/10.1038/nature09504
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013; 153:320-34; http://dx.doi.org/10.1016/j.cell.2013.03.036
- Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: implications for methylation-associated cellular processes. Pharmacol Ther 1995; 65:19-46; PMID:7536332; http://dx.doi.org/10.1016/0163-7258(94)00053-6
- Jablonka E, Goitein R, Marcus M, Cedar H. DNA hypomethylation causes an increase in DNase-I sensitivity and an advance in the time of replication of the entire inactive X chromosome. Chromosoma 1985; 93:152-6; PMID:4085302; http://dx.doi.org/10.1007/BF00293162
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 2011; 108:16669-74; PMID:21949397; http://dx.doi.org/10.1073/pnas.1108190108
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146:904-17; PMID:21889194; http://dx.doi.org/10.1016/j.cell.2011.08.017
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587; PMID:21118553; http://dx.doi.org/10.1186/1471-2105-11-587
- Zhang X, Mu W, Zhang W. On the analysis of the illumina 450k array data: probes ambiguously mapped to the human genome. Front Genet 2012; 3:73; PMID:22586432
- Anderson MJ RJ. Permutation tests for linear models. Australian N Zealand J Stat 2002; 43:75-88; http://dx.doi.org/10.1111/1467-842X.00156