
Abstract
DNA methylation can be affected by systemic exposures, such as cigarette smoking and genetic sequence variation; however, the relative impact of each on the epigenome is unknown. We aimed to assess if cigarette smoking and genetic variation are associated with overlapping or distinct sets of DNA methylation marks and pathways. We selected 85 Caucasian current and former smokers with genome-wide single nucleotide polymorphism (SNP) genotyping available from the COPDGene study. Genome-wide methylation was obtained on DNA from whole blood using the Illumina HumanMethylation27 platform. To determine the impact of local sequence variation on DNA methylation (mQTL), we examined the association between methylation and SNPs within 50 kb of each CpG site. To examine the impact of cigarette smoking on DNA methylation, we examined the differences in methylation by current cigarette smoking status. We detected 770 CpG sites annotated to 708 genes associated at an FDR < 0.05 in the cis-mQTL analysis and 1,287 CpG sites annotated to 1,242 genes, which were nominally associated in the smoking-CpG association analysis (Punadjusted < 0.05). Forty-three CpG sites annotated to 40 genes were associated with both SNP variation and current smoking; this overlap was not greater than that expected by chance. Our results suggest that cigarette smoking and genetic variants impact distinct sets of DNA methylation marks, the further elucidation of which may partially explain the variable susceptibility to the health effects of cigarette smoking. Ascertaining how genetic variation and systemic exposures differentially impact the human epigenome has relevance for both biomarker identification and therapeutic target development for smoking-related diseases.
Introduction
Cigarette smoking is a major risk factor for cardiovascular, pulmonary, and neoplastic diseases and contributes to the leading causes of morbidity and mortality globally. While the prevalence of cigarette smoking is declining, due to population growth, the absolute number of smokers is increasing world-wide and the burden of smoking-related diseases is projected to grow.Citation1 Genetic variation is known to play an important role in the risk for many smoking-related complex diseases, but the variable and prolonged susceptibility to the health effects of cigarette smoking are incompletely explained by genetic sequence variation alone. Several recent studies have suggested a role for epigenetic mediations, such as DNA methylation in smoking-related diseases.Citation2-4
DNA methylation involves the addition of a methyl group to DNA, typically in CpG dinucleotide sites. There has been considerable interest in how environmental and personal exposures modulate the establishment and maintenance of the epigenome, including DNA methylation.Citation5-12 In this context, many researchers have investigated the association of variable methylation of DNA from blood with various smoking metrics across the life course.Citation13-26 In cohorts of smokers, differential methylation has been linked to current smoking status and time since smoking cessation, chronic obstructive pulmonary disease (COPD), asthma, and lung cancer.Citation19 It has been reported that genetic sequence variation can also influence DNA methylation patterns.Citation27-36 However, the relative impact of genetic variants and environmental exposures on DNA methylation are incompletely understood.
In this manuscript, we investigate both cigarette smoking (as an environmental factor) and common genetic sequence variations associated with site-specific methylation across the genome. Previous studiesCitation30,37-39 have defined methylation quantitative trait loci (mQTL), but comparisons of the genetic and exposure contexts of methylation in smokers have not been performed simultaneously. We hypothesized that genetic variation and current smoking would demonstrate a subset of overlapping associations with methylation marks. Identifying genetic and exposure factors which differentially impact the plasticity of the human epigenome represents a fertile landscape to investigate both DNA methylation as a biomarker and for future development of pharmacoepigenetic targets for neoplastic and non-neoplastic smoking-related disease.
Results
Cohort description
Demographic and clinical characteristics of the 85 subjects by current smoking status are summarized in . Female subjects accounted for 61.2% of the total cohort; mean age was 65.1 y and mean pack-years smoked was 47.2. Current smokers were significantly younger than former smokers (p=0.003).
Table 1. Cohort characteristics.
cis-mQTL analysis
There were 503,411 individual CpG-SNP pairs tested in our cis-mQTL analysis. Among these, we detected a significant excess of quantitative trait loci for DNA CpG methylation; 3,002 CpG-SNP pairs had an FDR-adjusted P < 0.05. The 3,002 significant tests were comprised of 2,757 unique SNPs associated with 770 unique CpG sites near or in 708 unique genes.
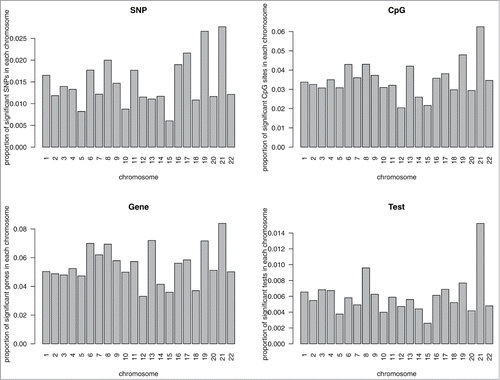
A volcano plot of the –log10 of the P-value (y-axis) relative to the regression coefficient is shown in . A Manhattan plot of the –log10(P-value) vs. physical position of SNPs at each chromosome is shown in ; significant cis-mQTL SNPs are abundant throughout the genome. The proportion of significant mQTLs in this 27K survey is variably distributed across the 22 chromosomes. Chromosome 21 has the largest proportion of significant mQTLs (1.5%), while chromosome 15 has the smallest proportion (0.3%). Chromosome 21 also has the largest proportion (6.3%) of significant CpG sites, the largest proportion (8.4%) of genes corresponding to significant CpG sites, and the largest proportion (2.8%) of SNPs ().
Figure 1. Volcano plot of the cis-mQTL analysis. Dashed red line represents an FDR<0 .05. Gene symbols for the top 20 cis-mQTL tests are shown in the volcano plot.
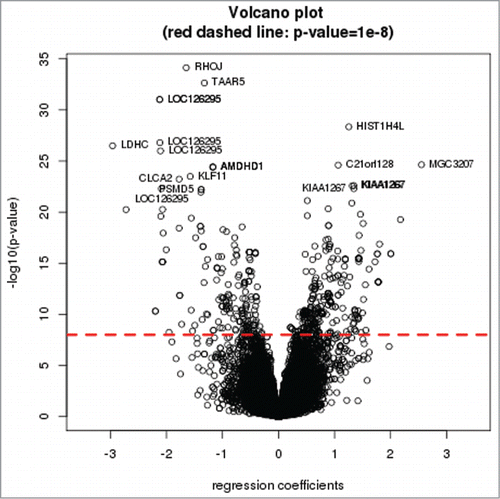
Figure 2. Manhattan plot of cis-mQTL analysis.
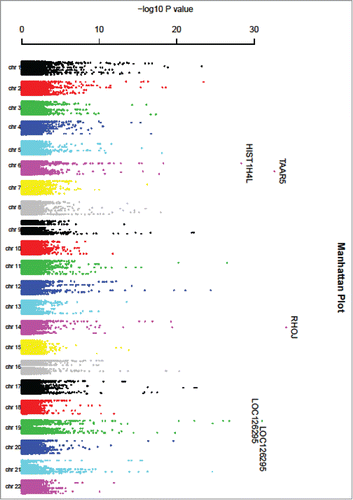
Figure 3. Top-left panel: Proportions of unique significant SNPs (i.e., SNPs in significant cis-mQTL tests) across the 22 chromosomes; Top-right panel: Proportions of unique significant CpG sites (i.e., CpG sites in significant cis-mQTL tests) across the 22 chromosomes; Bottom-left panel: Proportions of unique significant genes (i.e., genes corresponding to CpG sites in significant cis-mQTL tests) across the 22 chromosomes; Bottom-right panel: Proportions of significant cis-mQTL tests across the 22 chromosomes.
We investigated whether the physical distance between CpG sites and SNPs impacted the likelihood of being a mQTL. We binned the 100 kb region surrounding each CpG site (50 kb upstream, 50 kb downstream) into 20 5 kb regions. Within each region, we calculated the percent of association tests with P < 0.001. illustrates the distribution of the mQTLs with a P < 0.001 in the 100 kb region; proximity to the CpG site is associated with an increased likelihood of being a significant mQTL. illustrates the distribution of the mQTLs with a P < 0.001 within 5 kb of the CpG site, and shows a similar pattern to . Among the 3,002 CpG-SNP pairs tested with an FDR <0.05, the mean absolute distances between CpG and SNPs in the first, median, and third quartiles were 5.3 kb, 14.3 kb, and 27.6 kb respectively.
Figure 4. Distribution of mQTLs with p-value < 0.001 by distance from CpG site. For panel (a), each bin has a width of 5KB [range ≤ +45KB to ≥ −45KB from the CpG site]. For panel (b), each bin has a width of 0.5KB [range ≤ +5KB to ≥ −5KB from the CpG site].
![Figure 4. Distribution of mQTLs with p-value < 0.001 by distance from CpG site. For panel (a), each bin has a width of 5KB [range ≤ +45KB to ≥ −45KB from the CpG site]. For panel (b), each bin has a width of 0.5KB [range ≤ +5KB to ≥ −5KB from the CpG site].](/cms/asset/17491e1a-ba1c-4748-930b-f072daca796d/kepi_a_1106672_f0004_oc.gif)
The top 10 statistically significant mQTL tests are shown in , and include 7 CpG sites annotated to 7 genes. The CpG site-SNP pair with the most significant P =7.41×10−35 (cg18771300-rs4902214) is located near the gene ras homolog family member J (RHOJ) on chromosome 14. The parallel boxplots of DNA methylation levels vs. SNP genotype for the top 2 mQTL tests are shown (). Four SNPs were annotated to LOC126295; these 4 were all found to be in high linkage disequilibrium with each other (minimum R2≥ 0.94).
Figure 5. Parallel boxplots of CpG site methylation level vs. SNP genotype for the top 2 significant cis-mQTL tests.
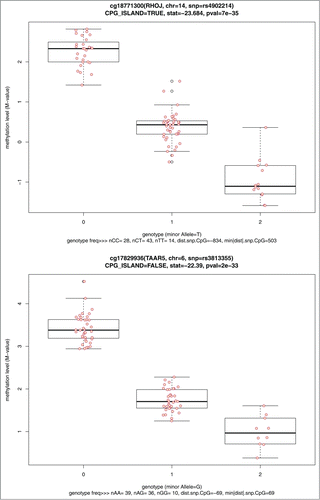
Table 2. Top 10 CpG sites associated with common single nucleotide polymorphisms (cis-mQTLs).
Among the 770 unique CpG sites identified in our mQTL analysis, 63.0% were annotated to CpG islands (as annotated by the R Bioconductor package IlluminaHumanMethylation27k.db). The median (minimum, maximum) distance in base pairs from CpG site to the transcription start site (TSS) was 339 (0, 1482). Two CpG sites (cg10660256 (BHMT, chr5); cg05521696 (SLC2A14, chr12)) were at the reputed TSS; both sites were in CpG islands. The information about these 2 CpG sites is shown in Table S1. At 1,311 (43.7%) of the 3,002 significant CpG-SNP pairs tested, the minor allele was associated with lower percent methylation.
Association of CpG sites with current smoking status
Among the 22,375 CpG sites, 1,287 CpG sites (near or in 1,242 genes) were associated with current smoking status at a nominal P < 0.05. Results for the top 10 associations of current smoking status to DNA methylation are shown in . Although none of our associations met the FDR threshold of < 0.05, several CpG sites, including the top site cg03636183 [annotated to the coagulation factor II receptor-like 3 (F2RL3)] have been previously reported and validated in the literature.Citation19,40 The parallel boxplots of DNA methylation levels by current smoking status for the top 2 CpG sites (in the F2RL3 and CSDE1 genes) are shown in .
Figure 6. Parallel boxplots of CpG site methylation level vs. smoking status for the top 2 significant smoking-CpG-association tests.
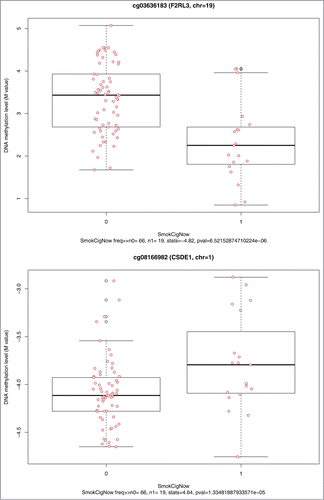
Table 3. Top 10 associations between CpG sites and current smoking status.
The proportion of associated tests was not uniformly distributed across the 22 autosomes. Chromosome 9 had the largest proportion (6.8%) of significant CpGs to total CpGs, while chromosome 8 had the smallest proportion (4.3%). Chromosome 10 has the largest proportion (11.1%) of genes corresponding to significant CpG sites (Fig. S1). The majority (71.0%) of the 1,287 CpG sites associated with current smoking are located in CpG islands. The median (minimum, maximum) distance to the TSS was 276 (0, 1495). There was one CpG island site, cg16944093, annotated to the LIM and senescent cell antigen-like domains 2 (LIMS2) with 0 distance to transcription start site. Approximately 40.2% of the CpG sites associated with current smoking status demonstrated relative hypomethylation in current smokers (data not shown).
Overlap between CpG sites impacted by genetic variants vs. cigarette smoking
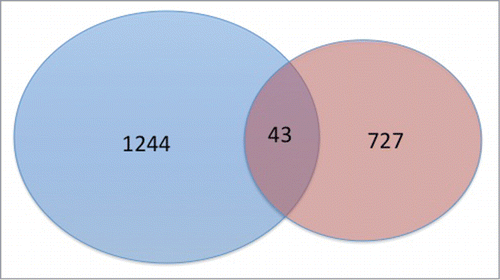
To investigate whether CpG sites associated with mQTLs were also impacted by current smoking, we examined the overlap between sites identified in each of the analyses above. For mQTL analysis, we included CpG sites with an FDR adjusted P < 0.05. For the CpG-smoking association analysis, we included sites with an unadjusted P < 0.05. The intersection of the associated CpG sites is summarized in . There were 727 CpG sites near 675 genes identified only in the cis-mQTL analysis (Table S2). There were 1,244 CpG sites near 1,203 genes significant only in the general linear regression analysis for current smoking (Table S3). There were only 43 CpG sites near 40 genes significant in both analyses (Table S4). By using a binomial test and by assuming the probability of overlapping by chance is at most 5%, the number 43 was not significantly greater than the expected number of overlapping by chance (P = 0.25). Correlations of ranks, P-values, and effect sizes between mQTL results (if multiple SNPs were associated with a CpG, only the result for the SNP having the smallest P-value was used) and the results of smoke-CpG association are −0.018 (P = 0.91), −0.038 (P = 0.81), and −0.056 (P = 0.72), respectively. These findings suggest that the impact of smoking on CpG site methylation may be largely independent of the impact of genetic variation on methylation of CpG sites in current and former smokers.
Figure 7. Proportional Venn diagram of CpG sites associated with cigarette smoking (blue) at an unadjusted P < 0.05 and genetic variants (red) at a FDR < 0.05. A total of 43 sites were significant in both analyses.
Gene set enrichment analysis
We investigated the biologic processes annotated to the CpGs identified in the mQTL-only, current smoking-only, and overlap groups above. Official gene names annotated to each of the CpG sites were used as input for DAVID (The Database for Annotation, Visualization and Integrated Discovery). The 675 genes annotated to CpG sites significant in the cis-mQTL-only group were enriched for 116 biological process categories including membrane depolarization, response to oxidative stress, and immune response (Table S5). The 1,203 genes annotated to CpG sites in the current smoking-only group were enriched for 252 biological process categories including response to metal ion, response to hormone stimulus, and response to endogenous stimulus (Table S6). The 40 genes annotated to CpG sites associated with both cis-mQTLs and current smoking status were enriched in 2 biological process categories related to regulation of foam cell differentiation (Table S7). Of the 116 biological processes enriched in mQTL-only CpG sites and the 252 biological processes were enriched in the smoking-only CpG sites, 14 biological processes common to both sets (Table S8 and Fig. S2).
Discussion
Associations between environmental exposures such as smoking and genetic variation have been reported in various types of diseases. However, it is not clear yet if the exposure and genetic factors associate with the same or distinct sets of DNA methylation marks. This question is particularly timely with regards to smoking, as numerous studies have demonstrated associations between cigarette smoking and variable DNA methylation that persists long after smoking cessation. In the current study, we report that smoking and genetic factors associate with the methylation levels of largely distinct sets of CpG sites. Notably, while individual sites that were impacted by genetic variants and current smoking were distinct, functional annotation analysis suggests that these distinct marks may actually impact a subset of similar biological pathways and processes including response to wounding, cellular proliferation and phospholipid metabolic processes.
Variability of DNA methylation by genotype (mQTL) has been identified in previous studies.Citation30,37-39,41,42 More recently, investigators have suggested that the spatial clustering of variably methylated regions is driven by underlying DNA sequence.Citation43 Because DNA methylation patterns are tissue specific, we compared the overlap between mQTLs identified in our study with mQTLs reported from other tissue samples. Many of the CpG-SNP associations reported in our manuscript have been identified by other groups in other tissue samples (). In addition to independent replication of mQTL loci, the mQTLs observed in multiple tissue types support the importance of the integrative capacity of future studies across genome-wide SNP and epigenetics platforms.
Table 4. cis-mQTL results compared to results in literature.
Liu et al.Citation43 identified smoking-related differentially methylated positions (DMPs) using publicly available methylation data generated on whole blood using the Illumina HumanMethylation450 array. A total of 93.8% of the CpG sites were within 5 Mb of the SNP, supporting the general finding that most mQTL are likely in cis. They identified 97,658 CpG-SNP pairs (6,211 unique CpG sites, 54,828 unique SNPs) that represented cis-mQTL at a P < 1×10−13. We compared our results with those of Liu et al. There are 495 CpG-SNP pairs (143 unique CpG sites and 480 unique SNPs) identified in both our results and Liu et al. In our analyses, there are 727 CpG sites significant in mQTL analysis, but not in the CpG-smoking association analysis. These 727 CpG sites correspond to 2,802 significant CpG-SNP pairs (727 unique CpG sites, 2,596 unique SNPs) in our cis-mQTL analysis. Among the 2,802 CpG-SNP pairs identified in our analysis, 446 (16%) CpG-SNP pairs (130 unique CpG sites and 431 unique SNPs) were identified in both our results and those of Liu et al. There are 1,244 CpG sites only significantly associated with smoking, but not with nearby SNPs in our data analyses. Only 4 (0.32%) of the 1,244 CpG sites appeared in the significant CpG-SNP pairs detected by Liu et al. In our data analyses, there are 43 CpG sites significantly associated with both smoking and nearby SNPs. These 43 CpG sites correspond to 200 significant CpG-SNP pairs (43 unique CpG sites and 190 unique SNPs) in our mQTL analysis. Among these 200 CpG-SNP pairs, 49 (25%) CpG-SNP pairs (13 unique CpG sites and 49 unique SNPs) were also detected by Liu et al.The top CpG-SNP association in our analysis was between cg18771300 and rs4902214, both of which are near the RHOJ gene on chromosome 14. The association between cg18771300 and rs4902214 was also detected by Liu et al.Citation43 The gene RHOJ encodes a small GTP-binding protein associated with focal adhesion in endothelial cells. The encoded protein is activated by vascular endothelial growth factor and may regulate angiogenesis. RhoJ demonstrates endothelial-cell-restricted expression pattern across tissues, including in the lungs.Citation44
Although none of the sites identified in our smoking analysis were significant following correction for multiple testing, the top association between current smoking status and DNA methylation was observed at CpG site cg03636183 near F2RL3 on chromosome 19, which is consistent with the findings in the literature.Citation2,19,25,40,45,46 Differential methylation at this exact site cg03636183 was first reported by Breitling et al.Citation25 and has also been reported by Wan et al.Citation19 and Shenker et al.Citation46 Although initial studies were conducted in largely Caucasian cohorts, Sun et al.Citation47 (2013) have also reported variable methylation of this site in African Americans as well, supporting the generalizability of this finding across races. The F2RL3 gene codes for a protein relevant for cardiovascular physiology and involved in various aspects of blood clotting.Citation2 In addition to exploring the overlap between sites associated with genetic variants and environmental exposures, our work suggests the impact of cigarette smoking is relatively modest compared to the impact of genetic variants on site-specific methylation in blood. This finding should not, however, be misinterpreted as a mitigation of the impact of cigarette smoking on the development of human disease or on the epigenome; future investigations should explore the mechanisms which contribute to these differences as well as the overlapping biological processes impacted by both processes.
The goal of this study was to explore whether differential methylation associated with genetic variation and cigarette smoking impacted overlapping or distinct CpG associations. The strengths of our study include the use of a well-characterized cohort and the systematic, unbiased interrogation of both genetic sequence variations and site-specific methylation throughout the genome. We acknowledge the following limitations to our study. While peripheral blood profiling is a clinically useful endeavor, the generalizability of our findings to organ-specific methylation profiles may be limited. Additionally, we acknowledge that peripheral blood is comprised of a mixture of cell populations and that cell type heterogeneity may impact our findings. In our analysis, we adjusted for this and additional confounders through the inclusion of principal components as covariates in our analyses; reassuringly, our results were similar when we applied established algorithms such as those published by Houseman et al.Citation48 Second, our study was limited to Caucasian subjects—future studies examining mQTLs in other ancestries are needed. We further acknowledge that our modest sample size limited our power to detect both cigarette smoking-associated CpG sites as well as our ability to include imputed SNPs in our analysis. We plan to include imputed SNPs in future analyses conducted in larger cohorts. Despite these limitations, we provide evidence supportive of the utility of integrative genomics profiling including in the development of molecular signatures that may be relevant for the diagnosis, staging, and treatment of smoking-related diseases.
Patients and Methods
Cohort and samples
Ninety four subjects from the COPDGene study (clinicaltrials.gov identifier: NCT 000608764) were profiled for the current analysis.Citation37 COPDGene is a study population initially enrolled from 21 clinical centers throughout the United States between January 2008 and June 2011.Citation37 Subjects were current and former smokers between the ages of 45 to 80 years, with at least 10 pack-years of smoking. All subjects completed questionnaire data and post-bronchodilator spirometry. Current smoking was defined as an affirmative answer to the question “Do you currently smoke cigarettes?” A blood sample for DNA extraction was obtained at the time of enrollment. This study was approved by the Institutional Review Boards of the participating centers, and informed consent was obtained from all subjects.
Methylation profiling with the Illumina HumanMethylation27 BeadChip array
One microgram of DNA from whole blood was bisulfite treated.Citation3 Genome-wide DNA methylation data was generated using the Illumina Infinium HumanMethylation27 BeadChip (Illumina Inc., San Diego, CA). For each locus an intensity value for methylated (Methylated) and unmethylated (Unmethylated) alleles is generated. Percent methylation was expressed as the Illumina β value, which represents a ratio of the Methylated to Unmethylated fluorescence signals, such that β = Max (Methylated, 0)/[Max(Methylated,0)+Max(Unmethylated, 0)+100]. Using this metric, DNA methylation is represented by a variable between 0 (no methylation) and 1 (complete methylation). Percent methylation values were calculated using BeadStudio software (Illumina), then exported for analysis into the statistical software R for further processing.
Data pre-processing and annotation
The annotations for each CpG site were extrapolated using the R Bioconductor packages IlluminaHumanMethylation27k.db and FDb.Infinium Methylation.hg19. Using the method outlined by Du et al, we performed color balance adjustment and quantile normalization. M-values [log2(Methylated/Unmethylated)] were generated for analysis to reduce the effects of severe heteroscedasticity. Scatter plots of the first 2 principal components identified 2 outliers who were removed from further analysis. Four subjects were excluded due to non-Caucasian race, 1 subject was excluded due to missing spirometry data, and 2 subjects were excluded due to unclassified spirometry status.Citation38 Eighty-five subjects were included in the final cohort for analysis. Based on criteria outlined by Christensen et al.,Citation49 we excluded one CpG site having median detection P > 0.05. We did not detect any arrays having detection P-values > 10−5 at more than 25% of CpG loci. Three CpG sites were removed due to lack of adequate annotations. CpG sites annotated to the X- or Y-chromosome were also excluded (1,092 sites). Based on R BioConductor packages SNPlocs.Hsapiens.dbSNP.20120608 and BSgenome.Hsapiens.UCSC.hg19, we excluded 4,107 CpG sites that had SNPs with MAF ≥ 0.05 within 5 base pairs of the interrogated CpG sites or overlapped with a repetitive element. A total of 5,789 CpG sites had SNPs with MAF < 0.05 within 5 base pairs; these were retained in the analysis results. In summary, the cleaned data consisted of 22,375 CpG marks and 85 arrays for analysis.
Genotyping data
Genotyping data were obtained using the Illumina OmniExpress platform and were cleaned as described.Citation50 for the whole COPDGene cohort. For the 85 subjects included in our analysis, we further performed subset-specific quality control using PLINK.Citation51 on the non-imputed genotype data (imputed data were excluded). A total of 156 SNPs were excluded based on Hardy-Weinberg P<0.001. Another 10 SNPs were excluded due to missingness (i.e., the genotyping call rate ≤ 0.1); 48,898 SNPs with a minor allele frequency < 0.05 were also excluded. After frequency and genotyping pruning, there were 581,796 SNPs remaining for analysis. The physical locations for the SNPs were annotated using hg19.
Statistical analysis
To evaluate the association between genetic factors and DNA methylation, we performed a cis-methylation quantitative trait (cis-mQTL) analysis as follows. For each CpG site, we first identified SNPs within 50 kb upstream and downstream from the CpG site.Citation33 We performed general linear regression analysis with methylation M-value as the dependent variable and each regional SNP as an independent variable under an additive model, adjusting for age, sex, pack-years of cigarette smoking, current smoking status, methylation array batch number, the top 5 principal components of the genotype data, and the top 4 principal components of the methylation data. Associations with a false discovery rate (FDR)-adjusted P < 0.05 were considered significant.
To evaluate the association of current smoking status with DNA methylation, we performed general linear regression analysis with DNA methylation M-value as the outcome variable and current smoking status as a binary predictor, adjusting for age at enrollment, sex, pack years of cigarette smoking, DNA methylation array batch number, and the top 4 principal components of the methylation data. Tests with an association P < 0.05 were considered significant.
To investigate the overlap between CpG sites associated with genetic variants and CpG sites associated with smoking, we examined the lists of unique CpG sites identified in each of the analyses above and tested for enrichment in the number of overlapping CpG sites using a binomial test. We also evaluated the correlations of ranks, P-values, and effect sizes between mQTL results and the results of smoking-CpG associations to evaluate if the overlap was more significant than the expected number of overlaps by chance. Lastly, we performed a functional enrichment analysis using Citation (The Data-base for Annotation, Visualization and Integrated Discovery) DAVID52on the set of overlapping CpG sites significant in both the mQTL and current smoking analyses.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1106672_Supplemental_Material.docx
Download MS Word (458.1 KB)References
- Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, Thomson B, Wollum A, Sanman E, Wulf S, Lopez AD, et al. Smoking prevalence and cigarette consumption in 187 countries, 1980-2012. Jama 2014; 311:183-92; PMID:24399557; http://dx.doi.org/10.1001/jama.2013.284692
- Breitling LP, Salzmann K, Rothenbacher D, Burwinkel B, Brenner H. Smoking, F2RL3 methylation, and prognosis in stable coronary heart disease. Eur Heart J 2012; 33:2841-8; PMID:22511653; http://dx.doi.org/10.1093/eurheartj/ehs091
- Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, Rennard S, Agusti A, Anderson W, Lomas DA, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med 2012; 185:373-81; PMID:22161163; http://dx.doi.org/10.1164/rccm.201108-1382OC
- Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, Agusti A, Anderson WH, Lomas DA, DeMeo DL. Systemic steroid exposure is associated with differential methylation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:1248-55; PMID:23065012; http://dx.doi.org/10.1164/rccm.201207-1280OC
- Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int J Epidemiol 2012; 41:79-105; PMID:22253299; http://dx.doi.org/10.1093/ije/dyr154
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 2012; 13:97-109; PMID:22215131
- Bollati V, Baccarelli A. Environmental epigenetics. Heredity (Edinb) 2010; 105:105-12; PMID:20179736; http://dx.doi.org/10.1038/hdy.2010.2
- Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, Haile RW, Laird PW. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships. Hum Genet 2012; 131:1565-89; PMID:22740325; http://dx.doi.org/10.1007/s00439-012-1189-8
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr 2009; 21:243-51; PMID:19663042; http://dx.doi.org/10.1097/MOP.0b013e32832925cc
- Collotta M, Bertazzi PA, Bollati V. Epigenetics and pesticides. Toxicology 2013; 307:35-41; PMID:23380243; http://dx.doi.org/10.1016/j.tox.2013.01.017
- Reamon-Buettner SM, Mutschler V, Borlak J. The next innovation cycle in toxicogenomics: environmental epigenetics. Mutat Res 2008; 659:158-65; PMID:18342568; http://dx.doi.org/10.1016/j.mrrev.2008.01.003
- Guerrero-Bosagna CM, Skinner MK. Environmental epigenetics and phytoestrogen/phytochemical exposures. J Steroid Biochem Mol Biol 2014; 139:270-6; PMID:23274117; http://dx.doi.org/10.1016/j.jsbmb.2012.12.011
- Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 2009; 180:462-7; PMID:19498054; http://dx.doi.org/10.1164/rccm.200901-0135OC
- Knopik VS, Maccani MA, Francazio S, McGeary JE. The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev Psychopathol 2012; 24:1377-90; PMID:23062304; http://dx.doi.org/10.1017/S0954579412000776
- Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet 2013; 4:132; PMID:23882278
- Suter MA, Anders AM, Aagaard KM. Maternal smoking as a model for environmental epigenetic changes affecting birthweight and fetal programming. Mol Hum Reprod 2013; 19:1-6; PMID:23139402; http://dx.doi.org/10.1093/molehr/gas050
- Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120:1425-31; PMID:22851337; http://dx.doi.org/10.1289/ehp.1205412
- Hillemacher T, Frieling H, Moskau S, Muschler MA, Semmler A, Kornhuber J, Klockgether T, Bleich S, Linnebank M. Global DNA methylation is influenced by smoking behaviour. Eur Neuropsychopharmacol 2008; 18:295-8; PMID:18242065; http://dx.doi.org/10.1016/j.euroneuro.2007.12.005
- Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, Agusti A, Anderson W, Lomas DA, Demeo DL. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet 2012; 21:3073-82; PMID:22492999; http://dx.doi.org/10.1093/hmg/dds135
- Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, Davey Smith G, Hughes AD, Chaturvedi N, Relton CL. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics 2014; 6:4; PMID:24485148; http://dx.doi.org/10.1186/1868-7083-6-4
- Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics 2014; 15:151; PMID:24559495; http://dx.doi.org/10.1186/1471-2164-15-151
- Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 2013; 8:e63812; PMID:23691101; http://dx.doi.org/10.1371/journal.pone.0063812
- Philibert RA, Beach SR, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics 2013; 5:19; PMID:24120260; http://dx.doi.org/10.1186/1868-7083-5-19
- Siedlinski M, Klanderman B, Sandhaus RA, Barker AF, Brantly ML, Eden E, McElvaney NG, Rennard SI, Stocks JM, Stoller JK, et al. Association of cigarette smoking and CRP levels with DNA methylation in α-1 antitrypsin deficiency. Epigenetics 2012; 7:720-8; PMID:22617718; http://dx.doi.org/10.4161/epi.20319
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450-7; PMID:21457905; http://dx.doi.org/10.1016/j.ajhg.2011.03.003
- Maccani JZ, Koestler DC, Houseman EA, Marsit CJ, Kelsey KT. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics 2013; 5:619-30; PMID:24283877; http://dx.doi.org/10.2217/epi.13.63
- Eichten SR, Briskine R, Song J, Li Q, Swanson-Wagner R, Hermanson PJ, Waters AJ, Starr E, West PT, Tiffin P, et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell 2013; 25:2783-97; PMID:23922207; http://dx.doi.org/10.1105/tpc.113.114793
- Oakes CC, Claus R, Gu L, Assenov Y, Hullein J, Zucknick M, Bieg M, Brocks D, Bogatyrova O, Schmidt CR, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov 2014; 4:348-61; PMID:24356097; http://dx.doi.org/10.1158/2159-8290.CD-13-0349
- Carless MA, Kulkarni H, Kos MZ, Charlesworth J, Peralta JM, Goring HH, Curran JE, Almasy L, Dyer TD, Comuzzie AG, et al. Genetic effects on DNA methylation and its potential relevance for obesity in Mexican Americans. PLoS One 2013; 8:e73950; PMID:24058506; http://dx.doi.org/10.1371/journal.pone.0073950
- Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, Craig DW, Redman M, Gershon ES, Liu C. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet 2010; 86:411-9; PMID:20215007; http://dx.doi.org/10.1016/j.ajhg.2010.02.005
- Drong AW, Nicholson G, Hedman AK, Meduri E, Grundberg E, Small KS, Shin SY, Bell JT, Karpe F, Soranzo N, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS One 2013; 8:e55923; PMID:23431366; http://dx.doi.org/10.1371/journal.pone.0055923
- van Eijk KR, de Jong S, Boks MP, Langeveld T, Colas F, Veldink JH, de Kovel CG, Janson E, Strengman E, Langfelder P, et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics 2012; 13:636; PMID:23157493; http://dx.doi.org/10.1186/1471-2164-13-636
- Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 2011; 12:R10; PMID:21251332; http://dx.doi.org/10.1186/gb-2011-12-1-r10
- Zhi D, Aslibekyan S, Irvin MR, Claas SA, Borecki IB, Ordovas JM, Absher DM, Arnett DK. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 2013; 8:802-6; PMID:23811543; http://dx.doi.org/10.4161/epi.25501
- Quon G, Lippert C, Heckerman D, Listgarten J. Patterns of methylation heritability in a genome-wide analysis of four brain regions. Nucleic Acids Res 2013; 41:2095-104; PMID:23303775; http://dx.doi.org/10.1093/nar/gks1449
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 2010; 6:e1000952; PMID:20485568; http://dx.doi.org/10.1371/journal.pgen.1000952
- Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, Curran-Everett D, Silverman EK, Crapo JD. Genetic epidemiology of COPD (COPDGene) study design. Copd 2010; 7:32-43; PMID:20214461; http://dx.doi.org/10.3109/15412550903499522
- Wan ES, Castaldi PJ, Cho MH, Hokanson JE, Regan EA, Make BJ, Beaty TH, Han MK, Curtis JL, Curran-Everett D, et al. Epidemiology, genetics, and subtyping of preserved ratio impaired spirometry (PRISm) in COPDGene. Respir Res 2014; 15:89; PMID:25096860
- Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet 2013; 93:876-90; PMID:24183450; http://dx.doi.org/10.1016/j.ajhg.2013.10.004
- Zhang Y, Yang R, Burwinkel B, Breitling LP, Brenner H. F2RL3 Methylation as a Biomarker of Current and Lifetime Smoking Exposures. Environ Health Perspect 2014; 122:131-7; PMID:24273234
- Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013; 31:142-7; PMID:23334450; http://dx.doi.org/10.1038/nbt.2487
- Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, et al. Patterns of population epigenomic diversity. Nature 2013; 495:193-8; PMID:23467092; http://dx.doi.org/10.1038/nature11968
- Liu Y, Li X, Aryee MJ, Ekstrom TJ, Padyukov L, Klareskog L, Vandiver A, Moore AZ, Tanaka T, Ferrucci L, et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet 2014; 94:485-95; PMID:24656863; http://dx.doi.org/10.1016/j.ajhg.2014.02.011
- Yuan L, Sacharidou A, Stratman AN, Le Bras A, Zwiers PJ, Spokes K, Bhasin M, Shih SC, Nagy JA, Molema G, et al. RhoJ is an endothelial cell-restricted Rho GTPase that mediates vascular morphogenesis and is regulated by the transcription factor ERG. Blood 2011; 118:1145-53; PMID:21628409; http://dx.doi.org/10.1182/blood-2010-10-315275
- Breitling LP. Current genetics and epigenetics of smoking/tobacco-related cardiovascular disease. Arterioscler Thromb Vasc Biol 2013; 33:1468-72; PMID:23640490; http://dx.doi.org/10.1161/ATVBAHA.112.300157
- Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 2012; 22:843-51; PMID:23175441; http://dx.doi.org/10.1093/hmg/dds488
- Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, Smith JA, Almli LM, Binder EB, Klengel T, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet 2013; 132:1027-37; PMID:23657504; http://dx.doi.org/10.1007/s00439-013-1311-6
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13:86; PMID:22568884; http://dx.doi.org/10.1186/1471-2105-13-86
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:e1000602; PMID:19680444; http://dx.doi.org/10.1371/journal.pgen.1000602
- Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, DeMeo DL, Hunninghake GM, Litonjua AA, Sparrow D, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet 2010; 42:200-2; PMID:20173748; http://dx.doi.org/10.1038/ng.535
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559-75; PMID:17701901; http://dx.doi.org/10.1086/519795
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003; 4:P3; PMID:12734009; http://dx.doi.org/10.1186/gb-2003-4-5-p3