
ABSTRACT
A-kinase anchor protein 12 (AKAP12) is a regulator of protein kinase A and protein kinase C signaling, acting downstream of RAS. Epigenetic silencing of AKAP12 has been demonstrated in different cancer entities and this has been linked to the process of tumorigenesis. Here, we used quantitative high-resolution DNA methylation measurement by MassARRAY to investigate epigenetic regulation of all three AKAP12 promoters (i.e., α, β, and γ) within a large cohort of juvenile myelomonocytic leukemia (JMML) patient samples. The AKAP12α promoter shows DNA hypermethylation in JMML samples, which is associated with decreased AKAP12α expression. Promoter methylation of AKAP12α correlates with older age at diagnosis, elevated levels of fetal hemoglobin and poor prognosis. In silico screening for transcription factor binding motifs around the sites of most pronounced methylation changes in the AKAP12α promoter revealed highly significant scores for GATA-2/-1 sequence motifs. Both transcription factors are known to be involved in the haematopoietic differentiation process. Methylation of a reporter construct containing this region resulted in strong suppression of AKAP12 promoter activity, suggesting that DNA methylation might be involved in the aberrant silencing of the AKAP12 promoter in JMML. Exposure to DNMT- and HDAC-inhibitors reactivates AKAP12α expression in vitro, which could potentially be a mechanism underlying clinical treatment responses upon demethylating therapy. Together, these data provide evidence for epigenetic silencing of AKAP12α in JMML and further emphasize the importance of dysregulated RAS signaling in JMML pathogenesis.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare childhood myelodysplastic/ myeloproliferative disorder with an incidence of 1.2 cases per million children per year. The average age at diagnosis is 1.8 y and, if left untreated, this malignancy is rapidly fatal for most affected children. Until now, the only curative treatment for JMML is allogeneic haematopoietic stem cell transplantation (HSCT).Citation1 Still, about 35% of patients relapse after HSCT, which demonstrates a strong need for a better understanding of the molecular mechanisms that are involved in the pathogenesis of JMML.Citation2
Promoter hypermethylation and subsequent gene inactivation are frequent events in myeloid malignancies, including JMML.Citation3-6 Two recent studies linked aberrant DNA methylation patterns to an aggressive phenotype of JMML.Citation7,8 Using a candidate gene approach, a prognostic score based on four frequently hypermethylated genes (BMP4, CALCA, CDKN2B, and RARB) could be identified, which retained prognostic value independently of other clinical risk factors.Citation7 Several studies have shown that aberrant methylation patterns may be governed by deregulated signaling.Citation9,10 In this way, it becomes increasingly clear that abnormal epigenetic marks are closely linked to aberrant cell signaling, a characteristic of various cancer entities.Citation11 A hallmark of JMML is its hypersensitivity to growth stimuli, such as the granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-1 (IL-1), and the tumor necrosis factor α (TNF-α).Citation12 GM-CSF hypersensitivity has been linked to a constitutively active Ras signaling pathway, which is believed to be the major driver of the disease.Citation13,14 Indeed, in >80% of JMML patients different members of the RAS/MAP kinase (MAPK) pathway are targeted by mutations in a mostly mutually exclusive fashion. Gain of function mutations occur in KRAS and NRAS (15–20%), and in the non-receptor tyrosine phosphatase PTPN11 (35%), whereas loss of function mutations occur in NF1 (15–20%) and CBL (15%).Citation15-18
The A-kinase anchor protein 12 (AKAP12) is a key regulator of protein kinase A and C signaling and has been described to suppress the PKC-Raf/MEK/ERK pathway, located downstream of RAS.Citation19 Epigenetic suppression of AKAP12 has been reported in various childhood myeloid leukemias.Citation20 None of the six JMML patients investigated in that study showed aberrant methylation of the AKAP12 promoter. Yet, the suppressive role of AKAP12 on RAS downstream signaling and the dependence of JMML on activated RAS signaling suggested a more detailed study on the potential role of AKAP12 in JMML pathogenesis. In the present study, we investigated the epigenetic regulation of AKAP12 in a cohort of 81 children diagnosed with JMML.
Results and discussion
Hypermethylation of the AKAP12α promoter in JMML
AKAP12 is a known tumor suppressor gene that can be silenced by deletions or by epigenetic mechanisms. Aberrant DNA methylation in the promoter region has been shown to specifically silence AKAP12 in Src- and Ras-transformed fibroblasts.Citation10 Re-expression of AKAP12 in vitro and in vivo suppresses oncogenic growth,Citation21 while overexpression of AKAP12 leads to cell cycle arrest.Citation22 In line with this, cells react with increased AKAP12 expression levels upon mitogenic stimuli.Citation23,24 The AKAP12 gene locus encodes for three isoforms α, β, and γ.Citation25 Recent whole-genome bisulfite studies have shown that the vast majority of CpGs in the adult mammalian genome are methylated. The only exceptions are gene regulatory regions like enhancers and promoters that are associated with (potentially) active genes.Citation26,27 Gene regulatory regions are enriched for CpG islands, which are usually protected from methylation and thus can be found most frequently in the vicinity of constitutively active genes. All three AKAP12 isoforms are under the control of independent promoter regions, each containing one or more CpG islands. In gastric, colorectal, and hepatic cancer one or both major AKAP12 isoforms (α, β) are targeted by promoter DNA hypermethylation and are transcriptionally repressed.Citation23,28-30
We used quantitative DNA methylation analysis at single CpG unit resolution to test if the AKAP12 promoter is epigenetically silenced in JMML. All three promoter regions corresponding to known AKAP12 isoforms were tested using the MassARRAY technology in a cohort of 81 JMML cases and leukocytes from healthy individuals as controls (). It is well known that methylation changes occurring in gene regulatory regions in a physiological or pathological context usually start from the borders of a given regulatory region and that is why one can generally observe the strongest methylation changes in CpG island shores but also in the distal regions of a given promoter or enhancer.Citation27,31 This effect is nicely recapitulated in . The distal region of the AKAP12α promoter covered by amplicon α1 was found to be significantly hypermethylated in a subset of JMML samples as compared to the control samples (P < 0.0001) while the region covered by amplicon α2 showed only a slight increase in DNA methylation levels. In contrast, the regions covered by amplicons β and γ showed no relevant methylation changes and a slight hypomethylation, respectively (). The average DNA methylation of amplicon α1 showed a large spread in JMML (range: 16–78%), as compared to healthy cells (21–32%). We subsequently defined a genomic region as being hyper- or hypo-methylated, respectively, if the average methylation levels exceeded two standard deviations from the mean of normal granulocytes. According to these criteria, 42% of JMML cases showed hypermethylation in the AKAP12α1 promoter region. Additionally, we analyzed the AKAP12α1 promoter methylation status of CD34+ cells from umbilical cord blood of healthy neonates, which are thought to represent the most appropriate healthy counterpart of JMML-initiating cells. These cells displayed an even lower promoter methylation profile than the healthy controls with an average CpG methylation of 16% in amplicon α1.
Figure 1. Quantitative DNA methylation analysis of the AKAP12α, β, and γ promoters in JMML. (A) Schematic representation of the AKAP12 locus showing the promoter regions of AKAP12α-, β- and γ-isoforms with their respective CpG islands. All isoforms share a common N-terminus encoded by 2 exons (C2 and C3). CpG islands are depicted as gray boxes (<300 bp, light gray). The MassARRAY amplicons (gray lines) and their positions relative to their respective TSS are illustrated underneath the CpG islands: α1: −868to −438 bp from TSS, α2: −462 to −131 bp from TSS, β1: −193 to +267 bp from TSS, γ1: −243 to +229 bp from TSS. Methylation heat maps are shown for each amplicon. Each row represents a sample and each column a CpG unit. Percentage of DNA methylation ranges from 0% (light green) to 100% (dark blue). Gray indicates unavailable data. HG: healthy granulocytes. (B) The mean DNA methylation levels for each amplicon are displayed as dot plots. The methylation levels between JMML samples (circles) and healthy granulocytes (diamonds) were compared using an unpaired t test with Welch's correction. (C) Leukemic cells isolated from bone marrow (BM) and peripheral blood (PB) were obtained from three JMML patients. DNA methylation values measured for the AKAP12 locus (amplicons α1, α2, β and γ) show a very good correlation between the two tissues (r = 0.996). (D) The methylation profile of amplicon α1 at CpG-unit resolution is shown for JMML and HG. JMML cases were grouped according to their methylation into 25% quartiles. The lower quartile (LQ) contains hypomethylated JMML samples (dashed black line) and the upper quartile (UQ) represents the hypermethylated ones (black line). The average methylation profile of HG (n = 23) is depicted as a light gray line. (E) Methylation levels for CpG units 12, 13, and 14 in AKAP12 amplicon α1 are depicted for HG and all JMML samples, as well as for hypomethylated (LQ) and hypermethylated (HQ) JMML samples. JMML samples show a statistically significant increase in methylation as compared to HG (P < 0.0001).
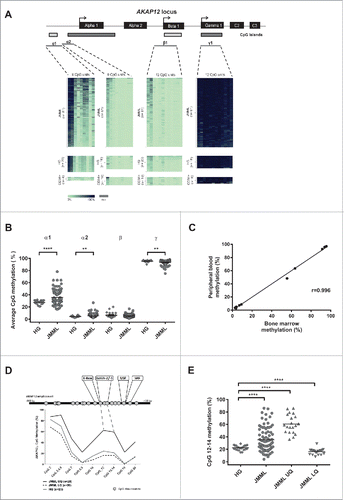
The promoter region of AKAP12β was unmethylated in JMML samples as well as in healthy controls. The promoter of AKAP12γ was strongly methylated in samples from healthy individuals, which correlates with the fact that its expression is known to be testis-specific.Citation25,32 In JMML cases, a slight hypomethylation was observed (P = 0.002; ). Indeed, compared to healthy controls, 23% of the JMML samples showed reduced promoter CpG island methylation within amplicon γ1. We did not observe any correlation between AKAP12γ hypomethylation and AKAP12α hypermethylation (data not shown).
Aberrant AKAP12α methylation in JMML is independent of the cell source
Since the clinical samples from 81 JMML patients were obtained either from bone marrow (BM) or from peripheral blood (PB), we investigated whether the source of the leukemic material had an influence on the observed methylation pattern. Therefore, matched pair analyses of malignant leukocytes isolated from BM and from PB were carried out. Highly similar DNA methylation values within the AKAP12α promoter region (amplicon α1) were observed between cells that originate from different tissue sources of the same patient (r = 0.996; ). Moreover, DNA methylation profiles did not differ between granulocytes and mononuclear cells, derived from BM or PB. Together, these data indicate that the heterogeneity of DNA methylation levels observed in this JMML cohort is most likely explained by differences in clonality levels between samples. Anyhow, we cannot fully rule out the possibility that varying levels of contamination with lymphocytes or inherent epigenetic instability may contribute to the observed heterogeneity of DNA methylation patterns.
Identification of potentially disrupted gene regulatory regions of AKAP12
In order to identify key regulatory regions within the differentially methylated AKAP12α promoter region, a search for highly differentially methylated CpG sites was performed. In the DNA methylation status of AKAP12α is given at single CpG unit resolution. In the JMML samples representing the lower quartile of DNA methylation, the methylation levels almost parallel those of healthy controls. The upper quartile shows most strongly elevated methylation levels in amplicon α1 between CpG 12 and CpG 14 located 614–587 bp upstream of the AKAP12α transcription start site (TSS). The scatter plot depicted in compares CpG methylation levels in this specific region between the upper and lower methylation quartile of JMML samples (JMML UQ and JMML LQ, respectively) and also to healthy individuals. A wide range of methylation values can be observed in this region and these methylation differences might reflect in part the phenotypic diversity among the samples. Overall, the described region shows a highly significant methylation increase as compared to normal controls (P < 0.0001). Applying our criteria for hypermethylation to this subregion of the AKAP12α promoter (CpGs 12–14), 49% of the JMML samples would be categorized as showing significant hypermethylation.
This aberrantly methylated region upstream of the AKAP12α TSS has been shown to be part of a sequence stretch that is highly conserved in mammals.Citation25 We used an in silico prediction tool to search for potential transcription factor binding sites (TFSEARCH; version 1.3) in this region. We found several putative transcription factor (TF) binding sites at and proximal to the hypermethylated region (). The in silico analysis predicted a GATA2/GATA1 binding site that precisely overlaps with the observed hypermethylation hotspot in the AKAP12α promoter region. This is of special interest, since both GATA2 expression and binding are tightly regulated epigenetically during early haematopoietic commitment.Citation26,27 Our motif search further identified an additional E-box predicted to bind the homeobox protein Nkx-2 that is located immediately 5′ of the differentially methylated DNA stretch. An upstream stimulatory factor (USF) binding site was predicted 50 bp downstream of the hypermethylated region. Finally, a serum response factor (SRF) binding site was predicted which is located 502 bp upstream of the transcription start site (TSS). Together, these findings suggest that this differentially methylated region harbors TF binding sites that may be disrupted by DNA hypermethylation. This, in turn, might ultimately result in aberrant silencing of AKAP12α in JMML.
DNA methylation of the AKAP12α promoter correlates with AKAP12α transcription
In order to test if AKAP12α promoter hypermethylation leads to AKAP12α silencing, we investigated AKAP12α mRNA expression levels in JMML. Quantitative RT-PCR of 19 JMML cases and 5 control samples (granulocytes of healthy donors) revealed a 13-fold decreased AKAP12α expression in JMML (P < 0.0001; ).
Figure 2. AKAP12α expression and promoter methylation. (A) AKAP12α mRNA expression levels measured by qRT-PCR in five healthy granulocyte samples and 19 JMML patient samples are shown. Horizontal lines represent mean values. The AKAP12α mRNA expression was significantly lower in JMML patients as compared to HG (P = 0.0002; Mann-Whitney U test). (B) Schematic representation of the AKAP12α promoter sequence with and without GATA-motif cloned into the CpG-free (pCpGL) luciferase reporter vector. K562 cells were transfected either with an unmethylated or with an in vitro methylated pCpGL vector construct. Luciferase activity was measured and normalized to the unmethylated Renilla-luciferase construct. The results are relative to the activity of the unmethylated AKAP12α promoter. The bar chart shows the mean luciferase activity ± SD (n = 4) of the methylated promoter construct and the unmethylated construct. Statistical significance was analyzed by a Student's t-test.
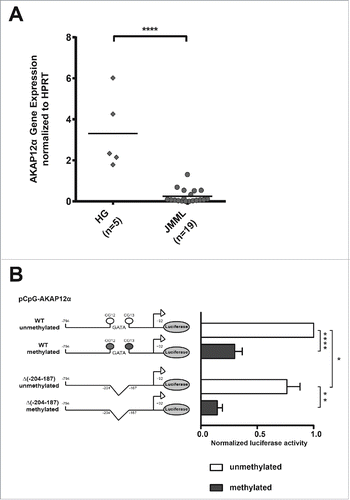
To further investigate the effects of AKAP12α promoter methylation and the impact of GATA binding on promoter activity, we carried out luciferase reporter assays using the pCpGL-basic reporter vector. The genomic region spanning the AKAP12α amplicons α1 and α2 (−794 bp to +92 bp around the AKAP12α TSS) was cloned into the CpG-free vector. In addition, a construct harboring a deletion of the predicted GATA binding motif was generated using site-directed mutagenesis (from now on referred to as AKAP12α_ΔGATA). The luciferase reporter vector containing the AKAP12α insert was either methylated in vitro or left unmethylated. Each construct was transiently transfected into K562 cells, and luciferase activity was measured. The unmethylated AKAP12α_ΔGATA construct reproducibly showed a modest reduction of promoter activity (76% activity as compared to the wild type construct; P = 0.0439) in the luciferase assay (). More importantly, both methylated constructs showed a significant decrease of promoter activity as compared to their unmethylated counterparts (P < 0.0001 for AKAP12 wild type; P = 0.0013 for AKAP12α_ΔGATA). Again, this effect was more pronounced in the AKAP12α_ΔGATA construct which showed only 14% promoter activity while the methylated wild type construct still had 30% promoter activity as compared to the unmethylated wild type construct (). In summary, these findings suggest that GATA binding plays, at least in part, a role in the activation of the AKAP12α promoter. This is further supported by our observation that deletion of the GATA motif has no effect on promoter activity in HEK293T cells, which do not express GATA transcription factors (data not shown). At the same time, our data clearly indicate that additional transcription factor binding events, which are also methylation dependent, crucially contribute to AKAP12α expression.
Next, we tested whether demethylation of the AKAP12α promoter leads to re-expression of AKAP12α. Therefore, we chose two myeloid leukemia cell lines, Kasumi-1 and HL-60, which harbor RAS mutations and thus show a constitutively active MAPK-signaling pathway (Table S4).Citation33 MassARRAY analysis showed high levels of DNA methylation (94%) at the AKAP12α promoter in both cell lines under steady state conditions (). In line with these findings, qRT-PCR revealed low mRNA expression levels of AKAP12α in Kasumi-1 and HL-60 at baseline. A recent publication has demonstrated that depletion of HDAC7 leads to re-expression of AKAP12 in endothelial cells.Citation34 Therefore, we asked the question whether HDAC-inhibition alone or in combination with DNMT-inhibition leads to re-expression of AKAP12α.
Figure 3. Epigenetic silencing of AKAP12α in RAS-mutated AML cell lines can be reverted by DAC treatment. Time course of AKAP12α1 promoter methylation in Kasumi-1 (A), and HL-60 cells (B) upon treatment with either SB939, DAC or a combination of both drugs and subsequent drug withdrawal. Cells were treated with the respective drug(s) for 72 h (day 1–3) and then observed for 11 d upon drug withdrawal (day 6–14). Time course of AKAP12α mRNA expression in Kasumi-1 (C), and HL-60 cells (D) upon drug treatment and subsequent drug withdrawal. Untreated control cells (Ctrl) are shown as light gray bars, time-points under drug treatment are shown in dark gray, and time-points upon drug withdrawal are depicted as black bars.
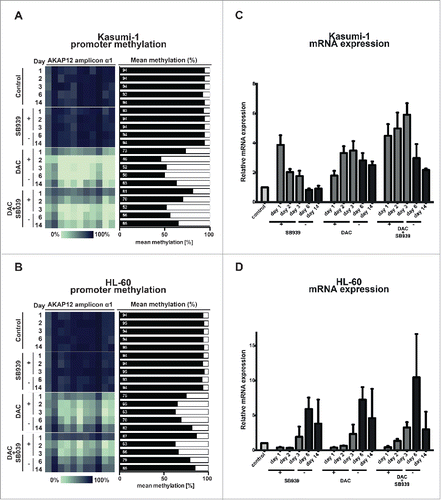
Both cell lines were treated for 72 h either with the DNMT-inhibitor DAC, the pan-HDAC-inhibitor SB939, or a combination of both inhibitors. DNA methylation levels of the AKAP12α promoter, as well as the AKAP12α mRNA expression levels were quantified every 24 h during the treatment period (day 1–3) and 3 d and 11 d after end of drug treatment (day 6 and day 14). Untreated cells were cultured in parallel as a control. DAC treatment led to a time-dependent decrease of DNA methylation in the AKAP12α promoter region. At the end of the 3-day treatment period, we observed an absolute methylation difference, as compared to baseline of 42% and 29% in Kasumi-1 and HL-60, respectively (). This effect was accompanied by a 2.3- to 3.5-fold increase in AKAP12α expression at the end of the treatment period in HL-60 and Kasumi-1 cells, respectively (). HDAC inhibition did not affect DNA methylation levels at the AKAP12α promoter as a single agent nor in combination with DAC. In contrast, HDAC inhibition resulted in a modest transient increase of AKAP12α expression levels in both cell lines and the combination of DAC + SB939 resulted in a slightly stronger reactivation of AKAP12α expression than either agent alone (). Interestingly, the kinetics of AKAP12α reactivation were different in both cell lines, anyhow, the reactivation of AKAP12α was only transient under all conditions tested: AKAP12α gene expression decreased following drug withdrawal and this decrease seemed to be paralleled by increasing levels of AKAP12α promoter methylation ().
Together, these findings demonstrate that high levels of AKAP12α promoter DNA methylation indicate transcriptional repression of AKAP12α and that pharmacological demethylation may lead to re-expression of AKAP12α. These data are well in line with published genome-wide data that show a global inverse correlation of DNA methylation dynamics with gene expression changes in the context of normal haematopoietic differentiation.Citation27 Since hyperactive RAS signaling is a hallmark of JMML, re-expression of the negative RAS signaling regulator AKAP12 could be one potential mechanism that might explain the treatment responses frequently observed in patients upon demethylating therapy.Citation1,35 In addition, HDAC-inhibition was also able to re-express AKAP12a, although this effect even under ongoing treatment was only transient if no DNMT-inhibitor was given concomitantly. This transient effect of HDAC-inhibition on AKAP12α expression is in slight contrast to the data published by Turtoi et al., but this discrepancy might be explained by the fact that Turtoi et al. used an siRNA knock-down approach to abrogate HDAC7 expression, which not only inhibits the function of the enzyme but also depletes the entire protein.Citation34 In addition, our data set is generated in haematopoietic cells while the paper of Turtoi et al. use vascular endothelial cells for their experiments. It is clear that different cell types might show distinct regulatory mechanisms as indicated already by the differential response observed between Kasumi-1 and HL-60.
AKAP12α promoter methylation correlates with clinical prognostic factors and is elevated in relapsed JMML cases
We correlated AKAP12α DNA methylation levels of 81 JMML patients with their clinical and hematological characteristics (). This revealed significant correlation with a number of parameters: Higher levels of AKAP12α promoter methylation were associated with platelet counts < 70 × 10Citation9/l (P = 0.02), bone marrow blasts ≥ 5% (P = 0.03), and aberrant karyotype (P = 0.03). The mutational status was also associated with significant differences in AKAP12α promoter methylation (P = 0.04). In a post-hoc test the presence of a PTPN11 mutation was associated with significantly higher methylation levels as compared to CBL-mutated (P = 0.007), and KRAS-mutated (P = 0.017) cases and showed a tendency for higher methylation as compared to samples with no known mutation (P = 0.048).
Table 1 Correlation of AKAP12 promoter methylation with clinical parameters in JMML patients (N = 81).
Interestingly, the strongest correlation of AKAP12α DNA methylation levels was observed with two well-established prognostic parameters, namely increased age at diagnosis (P < 0.001), and elevated fetal hemoglobin levels (HbF; P < 0.001).Citation2,36 In line with this, patients who would experience a relapse after HSCT showed a higher methylation level at diagnosis as compared to those who did not relapse (P = 0.03). No differences were found with respect to overall survival and event-free survival.
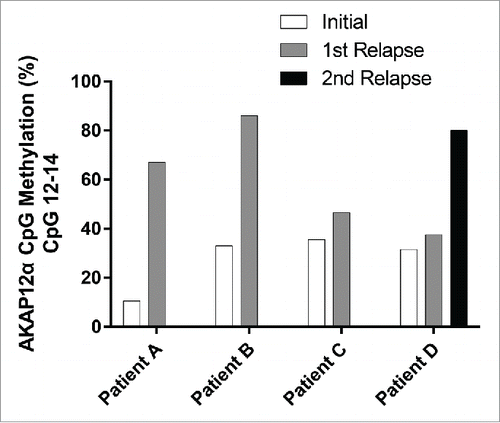
In order to investigate whether the levels of AKAP12α promoter methylation are not only associated with the risk of relapse but also increase during the course of disease, we analyzed DNA samples from four JMML patients of whom we had material, both at the time of diagnosis and at relapse after one or more transplants. Samples taken at the time of relapse displayed elevated AKAP12α promoter CpG methylation levels as compared to the diagnostic samples (). In the patient who experienced a second relapse, the AKAP12α promoter methylation seemed to increase during the course of disease with the highest level of AKAP12α promoter methylation detected at the time of the second relapse.
Figure 4. Longitudinal comparison of AKAP12α DNA methylation levels at diagnosis and at relapse. The mean AKAP12α promoter methylation level measured at CpGs 12–14 is depicted for JMML patients (n=4) who experienced relapse after treatment.
In conclusion, we demonstrate that the AKAP12α promoter shows high DNA methylation levels in JMML, which go along with reduced AKAP12α mRNA expression levels. Our data further suggest that differential binding of myeloid transcription factors might contribute to AKAP12α silencing. Importantly, AKAP12α promoter methylation levels are associated with known prognostic factors such as age at diagnosis and elevated HbF levels, which suggests involvement of AKAP12α in disease biology. Alternatively, since recent literature described several differentially methylated loci in JMML, which all correlated with clinical outcome, it might be tempting to speculate that a global epigenetic re-patterning occurs during the onset of JMML pathogenesis.Citation7,8 Further studies will be needed to elucidate the exact molecular mechanisms leading to AKAP12α transcriptional repression and the impact of aberrant signaling in establishing aberrant methylation patterns within the AKAP12α promoter region.
Material and methods
Patient samples
Mononuclear cells and granulocytes were isolated via density gradient centrifugation from peripheral blood or bone marrow of 81 unselected German JMML patients. Informed consent was obtained from the parents or legal guardians in accordance with the Declaration of Helsinki and approval was granted from the institutional review committees of each participating center. Patients had been registered in the European Working Group of MDS in Childhood (EWOG-MDS) studies EWOG-MDS 98 and EWOG-MDS 2006 (www.clinicaltrials.gov identifiers NCT00047268 and NCT00662090). Peripheral blood granulocytes (n = 23) from healthy donors were used as controls. CD34+ cells (n = 10) from bone marrow of healthy donors were obtained from Lonza (Basel, Switzerland). Genomic DNA was isolated using either the Puregene (Qiagen, Hilden, Germany) or the Transfast (Peqlab, Erlangen, Germany) isolation kit. Trizol reagent (Invitrogen, Karlsruhe, Germany) was used to isolate total RNA. For a subset of JMML samples (n = 23) DNA and RNA was extracted simultaneously using the AllPrep kit (Qiagen, Hilden, Germany).
Cell lines
The human myeloid leukemia cell lines Kasumi-1, HL-60, and K562 were grown in RPMI 1640 medium (PAA Laboratories, Cölbe, Germany) supplemented with 10% FCS, 1% sodium-pyruvate, and 1% penicillin/streptomycin (Sigma, St. Louis, MO, USA). Cells were kept at 37°C in an atmosphere containing 5% CO2. DNA and RNA were isolated using the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) and Trizol reagent, respectively.
5-aza-2′-deoxycytidine (DAC) and SB939 treatment of cell lines
Cell lines (Kasumi-1 & HL-60) were seeded at a density of 0.8×106 cells/ml. Cell lines were treated with the DNMT inhibitor 5-aza-2′-deoxycytidine (DAC) (Sigma-Aldrich, St. Louis, MO, USA), and the histone deacetylase inhibitor SB939 (Cayman Chemical, Ann Arbor, MI, USA) either alone or in combination for 72 h. Cell culture medium and inhibitors were renewed every 24 h. After completion of the 72 h treatment, cells were grown for additional 72 h in cell culture medium without addition of inhibitors. Samples for isolation of genomic DNA and total RNA were taken at 24, 48, 72, and 144 h from treated and control cells.
Functional studies of the AKAP12α promoter
Functional promoter CpG methylation analysis was carried out using a CpG-free vector backbone (pCpGL-basic) provided by Dr. Michael Rehli (Department of Internal Medicine III, University Hospital, Regensburg, Germany). Table S1 depicts the AKAP12α promoter construct (−794 bp to +92 bp relative to TSS) covering the promoter CpG-island used for luciferase reporter assay. The construct was amplified with Phusion High-Fidelity DNA polymerase (Fermentas, St. Leon-Rot, Germany), and purified using the Qiaquick PCR purification kit (Qiagen, Hilden, Germany). Subsequently, the PCR-product was cloned into the pCpGL-basic vector using the BglII and HindIII restriction sites. To test the impact of GATA binding on AKAP12α promoter activity, a 17-nt fragment containing a GATA motif was deleted using QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions. Briefly, two oligonucleotide primers complementary to the vector were extended using PfuUltra HF DNA polymerase. Following temperature cycling, the reaction mixture was placed on ice for 2 min, and then treated with DpnI (1h, 37°C) to digest the parental DNA template and to select for deletion-containing synthesized DNA. The mutated plasmid was then ethanol precipitated and transformed into PIR electrocompetent cells. The deletion was verified by Sanger sequencing.
Primer sequences for site-directed mutagenesis:
GATA-CG-Δ(26–42)-FW cctgacatcacttgttctgggcctgcgtggc
GATA-CG-Δ(26–42)-RV gccacgcaggcccagaacaagtgatgtcagg
In vitro methylation of plasmid DNA was carried out as described.Citation37 Briefly, the plasmid was incubated with M.SssI (2.5 U/μg DNA) in the presence of 160 μM SAM for 4 h at 37°C. Another 160 μM SAM was added after 2 h to boost the reaction. A control plasmid was treated equally but without the addition of methyltransferases or SAM. All plasmids were treated with proteinase K to remove bound proteins. The completeness of methylation was tested via digestion of the plasmid with the methylation sensitive restriction enzyme HpaII and subsequent agarose gel separation.
Transient transfection assays
Transient transfection of K562 cells was performed using the Amaxa Cell Line Nucleofector Kit V, according to the manufacturer's instructions. Briefly, for each transfection experiment, 1×106 cells/sample were used, pelleted by centrifugation (200 g, 10 min, RT) and resuspended in 100 µL Nucleofector solution. One microgram of Firefly luciferase reporter vector (pCpGL-basic) and 3 µg of Renilla luciferase control vector (pRL-Actin) were added and nucleofection was performed using “T-016.” Pre-warmed cell culture medium (RPMI1640 + 10% FCS; 500 µl) was added to the cuvette and the cells were subsequently plated into a 96-well plate in quintuplicates (technical replicates) and cultured at 37°C in 5% CO2. Luciferase activity was measured 24 h later. Firefly luciferase activity was normalized to Renilla luciferase activity, and the average of technical replicates was calculated. Each experiment was performed in biological quadruplicates.
Bisulfite treatment and quantitative DNA methylation analysis
Genomic DNA was bisulfite treated using the EZ DNA methylation kit (Zymo Research, Irvine, CA). Bisulfite-converted DNA was PCR amplified using Hotstar Taq (Qiagen, Hilden, Germany). Amplified regions ranged 400–500 bp and were designed close to the transcription start sites of the three AKAP12 isoforms (α, β, and γ) (Table S2). The MassARRAY EpiTYPER assay (Sequenom, San Diego, CA) was carried out as described before.Citation38,39 Briefly, the PCR amplified regions were in vitro transcribed, RNaseA cleaved, desalted, and subjected to matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS).
Reverse transcription and quantitative real-time PCR
Total RNA (1µg) was reverse transcribed using SuperScript II and oligo-dT primer (Invitrogen, Karlsruhe, Germany). Quantitative PCR was performed on a LightCycler 480 (Roche Diagnostics, Mannheim, Germany) using QuantiTect SYBR Green mix (Sigma, St. Louis, MO). Conditions for qRT-PCR and primer sequences are summarized in Table S3. Measurements were carried out in triplicates and relative expression levels were calculated using the ΔΔCT method. The reference genes HPRT, TBP, and ACTB were used for normalization.
Statistical analyses
To determine the significance of promoter CpG methylation levels between JMML and healthy controls, an unpaired t-test with Welch's correction was performed. P-values below 0.05 were assumed to be statistically significant. Expression values (mRNA) were compared using the non-parametric Kruskal-Wallis test with adjacent post-hoc Mann-Whitney U test. For the mutational analysis we first used the Kruskal-Wallis test and then the Mann-Whitney U test (post-hoc). All statistics was performed using GraphPad Prism software version 5.04 for Windows (GraphPad Software, La Jolla California USA, http:www.graphpad.com).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1145327_s02.docx
Download MS Word (24.9 KB)Acknowledgments
We thank Christopher C. Oakes, Céline Dutruel, Odilia Popanda and Peter Schmezer for helpful scientific discussions, Christiane Olk-Batz for sample preparation, Michael Rehli for providing the pCpGL-basic reporter construct, and all clinicians who provided patient samples and clinical data. DBL, CP, CF, and CMN were supported by the DFG Priority Program SPP1463 “Epigenetic regulation of normal hematopoiesis and its dysregulation in myeloid neoplasia.” CF was further supported by the DFG Collaborative Research Center “Medical Epigenetics” (CRC992-C05).
Related Research Data
References
- Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood 2015; 125:1083-90; PMID:25564399; http://dx.doi.org/10.1182/blood-2014-08-550483
- Locatelli F, Nollke P, Zecca M, Korthof E, Lanino E, Peters C, Pession A, Kabisch H, Uderzo C, Bonfim CS, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 2005; 105:410-9; PMID:15353481; http://dx.doi.org/10.1182/blood-2004-05-1944
- Boultwood J, Wainscoat JS. Gene silencing by DNA methylation in haematological malignancies. Br J Haematol 2007; 138:3-11; PMID:17489980; http://dx.doi.org/10.1111/j.1365-2141.2007.06604.x
- Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, Fernandez H, Tallman MS, Greally JM, Carraway H, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009; 114:3448-58; PMID:19652201; http://dx.doi.org/10.1182/blood-2009-01-200519
- Hasegawa D, Manabe A, Kubota T, Kawasaki H, Hirose I, Ohtsuka Y, Tsuruta T, Ebihara Y, Goto Y, Zhao XY, et al. Methylation status of the p15 and p16 genes in paediatric myelodysplastic syndrome and juvenile myelomonocytic leukaemia. Br J Haematol 2005; 128:805-12; PMID:15755284; http://dx.doi.org/10.1111/j.1365-2141.2005.05392.x
- Johan MF, Bowen DT, Frew ME, Goodeve AC, Reilly JT. Aberrant methylation of the negative regulators RASSFIA, SHP-1 and SOCS-1 in myelodysplastic syndromes and acute myeloid leukaemia. Br J Haematol 2005; 129:60-5; PMID:15801956; http://dx.doi.org/10.1111/j.1365-2141.2005.05412.x
- Olk-Batz C, Poetsch AR, Nollke P, Claus R, Zucknick M, Sandrock I, Witte T, Strahm B, Hasle H, Zecca M, et al. Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 2011; 117:4871-80; PMID:21406719; http://dx.doi.org/10.1182/blood-2010-08-298968
- Poetsch AR, Lipka DB, Witte T, Claus R, Nollke P, Zucknick M, Olk-Batz C, Fluhr S, Dworzak M, De Moerloose B, et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics 2014; 9:1252-60; PMID:25147919; http://dx.doi.org/10.4161/epi.29941
- Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature 2007; 449:1073-7; PMID:17960246; http://dx.doi.org/10.1038/nature06251
- Ordway JM, Williams K, Curran T. Transcription repression in oncogenic transformation: common targets of epigenetic repression in cells transformed by Fos, Ras or Dnmt1. Oncogene 2004; 23:3737-48; PMID:14990994; http://dx.doi.org/10.1038/sj.onc.1207483
- Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nat Biotechnol 2010; 28:1033-8; PMID:20944593; http://dx.doi.org/10.1038/nbt1010-1033
- Freedman MH, Cohen A, Grunberger T, Bunin N, Luddy RE, Saunders EF, Shahidi N, Lau A, Estrov Z. Central role of tumour necrosis factor, GM-CSF, and interleukin 1 in the pathogenesis of juvenile chronic myelogenous leukaemia. Br J Haematol 1992; 80:40-8; PMID:1311195; http://dx.doi.org/10.1111/j.1365-2141.1992.tb06398.x
- Kalaitzidis D, Gilliland DG. Going with the flow: JAK-STAT signaling in JMML. Cancer Cell 2008; 14:279-80; PMID:18835028; http://dx.doi.org/10.1016/j.ccr.2008.09.006
- Flotho C, Kratz C, Niemeyer CM. Targeting RAS signaling pathways in juvenile myelomonocytic leukemia. Curr Drug Targets 2007; 8:715-25; PMID:17584027; http://dx.doi.org/10.2174/138945007780830773
- Flotho C, Valcamonica S, Mach-Pascual S, Schmahl G, Corral L, Ritterbach J, Hasle H, Arico M, Biondi A, Niemeyer CM. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia 1999; 13:32-7; PMID:10049057; http://dx.doi.org/10.1038/sj.leu.2401240
- Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 2003; 34:148-50; PMID:12717436; http://dx.doi.org/10.1038/ng1156
- Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 2010; 42:794-800; PMID:20694012; http://dx.doi.org/10.1038/ng.641
- Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009; 114:1859-63; PMID:19571318; http://dx.doi.org/10.1182/blood-2009-01-198416
- Su B, Bu Y, Engelberg D, Gelman IH. SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a protein kinase C- Raf/MEK/ERK pathway. J Biol Chem 2010; 285:4578-86; PMID:20018890; http://dx.doi.org/10.1074/jbc.M109.073494
- Flotho C, Paulun A, Batz C, Niemeyer CM. AKAP12, a gene with tumour suppressor properties, is a target of promoter DNA methylation in childhood myeloid malignancies. Br J Haematol 2007; 138:644-50; PMID:17686059; http://dx.doi.org/10.1111/j.1365-2141.2007.06709.x
- Gelman IH. Emerging Roles for SSeCKS/Gravin/AKAP12 in the control of cell proliferation, cancer malignancy, and barriergenesis. Genes Cancer 2010; 1:1147-56; PMID:21779438; http://dx.doi.org/10.1177/1947601910392984
- Lin X, Nelson P, Gelman IH. SSeCKS, a major protein kinase C substrate with tumor suppressor activity, regulates G(1)–>S progression by controlling the expression and cellular compartmentalization of cyclin D. Mol Cell Biol 2000; 20:7259-72; PMID:10982843; http://dx.doi.org/10.1128/MCB.20.19.7259-7272.2000
- Nelson PJ, Gelman IH. Cell-cycle regulated expression and serine phosphorylation of the myristylated protein kinase C substrate, SSeCKS: correlation with culture confluency, cell cycle phase and serum response. Mol Cell Biochem 1997; 175:233-41; PMID:9350056; http://dx.doi.org/10.1023/A:1006836003758
- Coats SR, Covington JW, Su M, Pabon-Pena LM, Eren M, Hao Q, Vaughan DE. SSeCKS gene expression in vascular smooth muscle cells: regulation by angiotensin II and a potential role in the regulation of PAI-1 gene expression. J Mol Cell Cardiol 2000; 32:2207-19; PMID:11112996; http://dx.doi.org/10.1006/jmcc.2000.1246
- Streb JW, Kitchen CM, Gelman IH, Miano JM. Multiple promoters direct expression of three AKAP12 isoforms with distinct subcellular and tissue distribution profiles. J Biol Chem 2004; 279:56014-23; PMID:15496411; http://dx.doi.org/10.1074/jbc.M408828200
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA Methylome analysis. Cell Stem Cell 2014; 15:507-22; PMID:25158935; http://dx.doi.org/10.1016/j.stem.2014.07.005
- Lipka DB, Wang Q, Cabezas-Wallscheid N, Klimmeck D, Weichenhan D, Herrmann C, Lier A, Brocks D, von Paleske L, Renders S, et al. Identification of DNA methylation changes at cis-regulatory elements during early steps of HSC differentiation using tagmentation-based whole genome bisulfite sequencing. Cell Cycle 2014; 13:3476-87; PMID:25483069; http://dx.doi.org/10.4161/15384101.2014.973334
- Goeppert B, Schmezer P, Dutruel C, Oakes C, Renner M, Breinig M, Warth A, Vogel MN, Mittelbronn M, Mehrabi A, et al. Down-regulation of tumor suppressor A kinase anchor protein 12 in human hepatocarcinogenesis by epigenetic mechanisms. Hepatology 2010; 52:2023-33; PMID:20979053; http://dx.doi.org/10.1002/hep.23939
- Choi MC, Jong HS, Kim TY, Song SH, Lee DS, Lee JW, Kim NK, Bang YJ. AKAP12/Gravin is inactivated by epigenetic mechanism in human gastric carcinoma and shows growth suppressor activity. Oncogene 2004; 23:7095-103; PMID:15258566; http://dx.doi.org/10.1038/sj.onc.1207932
- Mori Y, Cai K, Cheng Y, Wang S, Paun B, Hamilton JP, Jin Z, Sato F, Berki AT, Kan T, et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterol 2006; 131:797-808; PMID:16952549; http://dx.doi.org/10.1053/j.gastro.2006.06.006
- Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013; 500:477-81; PMID:23925113; http://dx.doi.org/10.1038/nature12433
- Camus A, Mesbah K, Rallu M, Babinet C, Barra J. Gene trap insertion reveals two open reading frames in the mouse SSeCKS gene: the form predominantly detected in the nervous system is suppressed by the insertion while the other, specific of the testis, remains expressed. Mech Dev 2001; 105:79-91; PMID:11429284; http://dx.doi.org/10.1016/S0925-4773(01)00384-7
- Morgan MA, Dolp O, Reuter CW. Cell-cycle-dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood 2001; 97:1823-34; PMID:11238126; http://dx.doi.org/10.1182/blood.V97.6.1823
- Turtoi A, Mottet D, Matheus N, Dumont B, Peixoto P, Hennequiere V, Deroanne C, Colige A, De Pauw E, Bellahcene A, et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis 2012; 15:543-54; PMID:22584896; http://dx.doi.org/10.1007/s10456-012-9279-8
- Furlan I, Batz C, Flotho C, Mohr B, Lubbert M, Suttorp M, Niemeyer CM. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood 2009; 113:2867-8; PMID:19299654; http://dx.doi.org/10.1182/blood-2008-12-195693
- Niemeyer CM, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, Creutzig U, Haas O, Harbott J, Hasle H, Kerndrup G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 1997; 89:3534-43; PMID:9160658
- Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 2006; 1:127-30; PMID:17965610; http://dx.doi.org/10.4161/epi.1.3.3327
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A 2005; 102:15785-90; PMID:16243968; http://dx.doi.org/10.1073/pnas.0507816102
- Claus R, Lucas DM, Stilgenbauer S, Ruppert AS, Yu L, Zucknick M, Mertens D, Bühler A, Oakes CC, Larson RA, et al. Quantitative DNA Methylation Analysis Identifies a Single CpG Dinucleotide Important for ZAP-70 Expression and Predictive of Prognosis in Chronic Lymphocytic Leukemia. J Clin Oncol 2012; 30:2483-91; PMID:22564988; http://dx.doi.org/10.1200/JCO.2011.39.3090