
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
DNA methylation is a widely studied epigenetic mechanism and alterations in methylation patterns may be involved in the development of common diseases. Unlike inherited changes in genetic sequence, variation in site-specific methylation varies by tissue, developmental stage, and disease status, and may be impacted by aging and exposure to environmental factors, such as diet or smoking. These non-genetic factors are typically included in epigenome-wide association studies (EWAS) because they may be confounding factors to the association between methylation and disease. However, missing values in these variables can lead to reduced sample size and decrease the statistical power of EWAS. We propose a site selection and multiple imputation (MI) method to impute missing covariate values and to perform association tests in EWAS. Then, we compare this method to an alternative projection-based method. Through simulations, we show that the MI-based method is slightly conservative, but provides consistent estimates for effect size. We also illustrate these methods with data from the Atherosclerosis Risk in Communities (ARIC) study to carry out an EWAS between methylation levels and smoking status, in which missing cell type compositions and white blood cell counts are imputed.
Introduction
DNA methylation of cytosine residues at CpG dinucleotides has particular interest since it has a central role in normal human development and disease.Citation1 Epigenome-wide association studies (EWAS), analogous to genome-wide association studies (GWAS), are becoming increasingly common and popular to interrogate methylation change associated with disease status or related traits. Facing many of the same challenges as epidemiology studies, DNA methylation value can be highly related to the tissue, developmental stage, and disease status, and may be impacted by aging and exposure to environmental factors, such as diet or smoking, which may lead to potential confounding in association tests.Citation2 For example, in some DNA methylation data set, cases and controls typically differ in their cell type composition, which can result in spurious associations—those that merely tag the cell type rather than reveal more fundamental biological signals of interest.Citation3,4
To adjust for confounders in analysis of DNA methylation data, one approach is based on principal component analysis (PCA) and infers confounders using surrogate variables.Citation5-8 When the confounders are measured, they can be directly adjusted in a regression framework. However, incomplete data is a common problem in applied research. Restricting the analysis to complete cases will reduce the size of data and the power of association tests, which are critical for EWAS with stringent genome-wide significance level. Using the correlation between methylation values and the covariates, we can impute the missing covariate values based on a set of pre-selected CpG sites. For example, when a reference data set is available, Houseman et al.Citation9 proposed to infer the underlying composition of cell populations with distinct DNA methylation profiles via a projection-based approach.
In this paper, we explore statistical methods to impute missing covariate values in analysis of DNA methylation data, which is based on the multiple imputation (MI) approach.Citation10 Specifically, we apply variable selection to select a set of CpG sites from genome-wide methylation data and use the MI approach to impute missing covariates and perform association tests. We also extend the projection-based method by Houseman et al.,Citation9 treating the complete cases as the reference data. Through computer simulations, we show that the MI-based methods produce asymptotically unbiased estimates for the effect size, with slightly conservative power compared to the projection-based method. On the other hand, the projection-based method has the correct type 1 error rate, but may produce biased estimates under the alternative hypothesis. We further apply these methods to the DNA methylation data in the Atherosclerosis Risk in Communities (ARIC) study. We compare the CpG sites identified for association with smoking status, using different imputation approaches for missing cell type compositions and whole blood cell counts (WBC) in the analysis.
Results
Simulation results
Simulated DNA methylation values under the different simulation models are shown in supplemental . All the 3 imputation methods show non-inflated type 1 errors at different missing rates (). At the significance level of 0.05, the 2 MI-based methods are slightly conservative (type 1 error <5%) and have wider coverage (>95%) than expected (). The projection-based method has type 1 error rate close to the expected 5%. In contrast, the type 1 errors are inflated when the covariates Z are ignored in the association analysis and Z are correlated with both the variable of interest (X) and the methylation measure (Y) (supplemental Tables 1 and 2).
Table 1. Methods comparison for simulation model 1, missing rate is 30%, imputing time is 10 for MI method.
When the missing rate is relatively low (30%), the projection-based method has higher statistical power than the 2 MI-based methods (); when the missing rate is high (90%), the projection-based method and MI-PMM have similar power, which are higher than that of MI-Norm (). However, the projection-based method has downwards bias in estimated effect size and narrower coverage less than 95% (), especially when the missing rate is high (90%). When the effect size is large and missing rate is high, the coverage of estimated effect size from the projection-based method can be < 80%, compared to the expected 95% (). In MI-based methods, the estimated effect size is very close to the true value and the coverage is slightly wider than those of the full data analysis (96% vs. 95%). In addition, we compare the distribution of estimated effect size for different methods (). The two MI-based methods yield consistent estimates for the true parameter values, but slightly large variation compared to full data analysis, especially when the missing rate is high (90%). The projection-based method consistently underestimates the effect size under the alternative hypothesis, but the variation of estimates is similar to that from full data analysis.
Figure 1. Power under different effect size for simulation model 1. Comparisons between resulting power using the projection-based method, MI-Norm, MI-PMM, full data (assuming that we have all data without any missing), and complete-case analysis (excluding subjects with missing values). (A) Missing rate = 0.3. (B) Missing rate = 0.9.
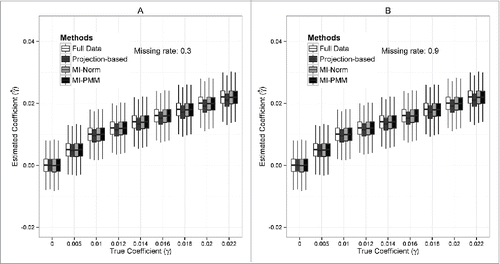
Figure 2. Boxplots for estimated effect size for simulation model 1. Comparisons between resulting estimated effect size using the projection-based method, MI-Norm, MI-PMM, full data (assuming that we have all data without any missing), and complete-case analysis (excluding subjects with missing values). (A) Missing rate = 0.3. (B) Missing rate = 0.9.
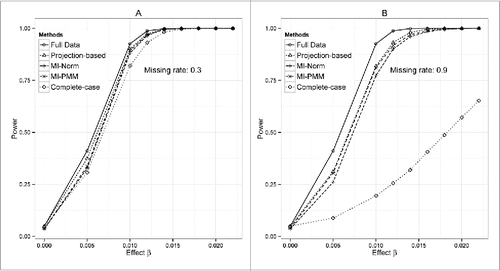
Table 2. Methods comparison for simulation model 1, missing rate is 90%, imputing time is 30 for MI method.
Note that simply excluding the subjects with missing covariates will lead to loss of power. also shows the statistical power of complete-case analysis, in which subjects with missing covariates are excluded. Comparing to the full data analysis or imputed data analysis (MI-Norm, MI-PMM, projection based method), complete-case analysis suffers from a loss of power, especially when the missing rate is high ().
We also evaluate the performance of various methods under different sample sizes (n = 500 or 1,000). The results are consistent (supplemental Tables 3, 4 and supplemental Fig. 2). When the missing covariate is binary, we applied the multiple-imputation based method (MI-Logreg), which shows similar performance as for continuous missing covariates (supplemental Tables 5, 6, 7 and supplemental Fig. 3, 4).
For the 2 MI-based methods, increasing the number of imputations from 10 to 30 does not change the results significantly when the missing rate is 30% (supplemental Table 8). When the missing rate is 90%, MI-PMM with 10 imputations shows small upwards bias in estimated effect size. The bias is minimized with 30 imputations (supplemental Table 9). The coverage of estimates using MI-Norm is also improved by using more imputations, 96% vs. 97% for 30 vs. 10 imputations, respectively. Supplemental Fig. 5 shows similar statistical power when 10 or 30 imputations are used.
When the covariates are only correlated with methylation levels but not the variable of interest, i.e., Z are independent predictors instead of confounders (simulation model 3), both the projection-based method and MI-based methods perform similarly to the full data analysis. All approaches have a controlled type 1 error, correct 95% coverage (supplemental Table 10), and comparable statistical power (supplement Fig. 6).
Data application: Association with smoking status
We investigate the association between DNA methylation and smoking status (never vs. current smoker), while adjusting for possible confounders, including cell type compositions, age, field center, gender, and white blood cell count. To adjust for batch effects, we run a linear mixed-effects model with chip and chip position as random effects. The majority of missingness in this data comes from cell type composition (∼91.1% missing rate), and total white blood cell (WBC) counts (∼14.2% missing rate).
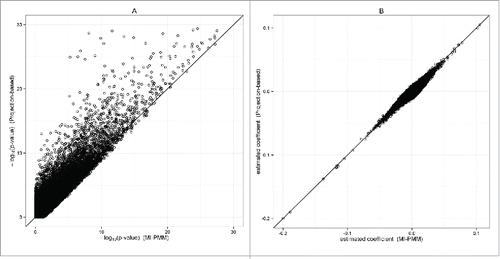
In the first analysis, we impute cell type composition using multiple imputation (30 imputations) and projection-based method, but only analyze samples with measured WBC. After imputation, we have 1,640 African American participants that are available for analysis. Using the projection-based method, we identify 2,552 CpG sites associated with smoking status with genome-wide significance (P-value < 10−7). As a comparison, MI-Norm finds 1,362 significant CpG sites and MI-PMM finds 1,747 sites. shows a scatter plot of the transformed association P-values for the projection-based method and MI-PMM, suggesting smaller P-values for the projection-based method than MI-PMM in general. shows a comparison of the estimated coefficients at each CpG site. MI-PMM and the projection-based method show comparable estimates, especially at CpG sites with strong significance. Results from MI-Norm are shown in supplemental , which are similar to those from MI-PMM. Most of the CpG sites identified by MI-PMM or MI-Norm in the first analysis remain genome-wide significant in at least one other imputation method (supplemental Fig. 9A).
Figure 3. EWAS of smoking status: MI-PMM vs. the projection-based method. The figure is based on 1,640 participants after imputing the missing cell type composite using MI-PMM or projection-based method. (A) Comparison of (P-value) from MI-PMM and from the projection-based method (P-values truncated at
). (B) Comparison of coefficient estimates from MI-PMM and from the projection-based method.
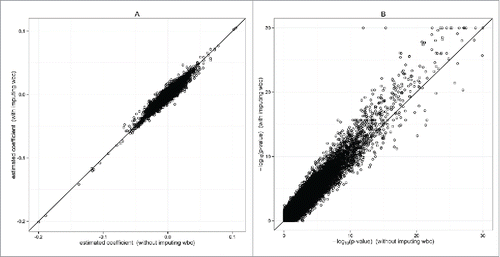
Our second analysis also imputes missing WBC values. The sample size after imputation increases to 1,932, which provides additional power for association tests. shows comparison of estimated coefficients and association P-values with and without imputed WBC. While the estimated coefficients are similar for the top signals, the association P-values are generally smaller in the second analysis. As shown in supplemental Fig. 8, MI-Norm has similar performance as MI-PMM. We identify 2,149 CpG sites genome-wide significant with MI-PMM. Among these, 1,590 sites overlap with the first analysis (supplemental Fig. 9B).
Figure 4. EWAS of smoking status with imputed WBC. Analysis is-based on MI-PMM. The data excluding observations with missing WBC contains 1,640 participants, while imputing missing WBC leads to 1,932 participants. (A) Comparison of coefficient estimate with and without imputing missing WBC values. (B) Comparison of –log10(P-value) with and without imputing missing WBC values (P-values truncated at 10−30).
Discussion
Missing data is a common problem in association studies, which can reduce the sample size and lead to decreased statistical power. Using genome-wide methylation data, we have proposed multiple imputation-based methods to impute missing covariates, which can be either continuous or categorical, using a set of selected CpG sites. We also extended an existing projection-based method for missing covariates imputation. Through computer simulations, we evaluated performance of the 2 types of methods. Our results suggest that although the projection-based method has the correct type 1 error rate and greater power than the MI-based methods, it may yield biased estimate and coverage for the effect size. In contrast, the MI-based methods are slightly conservative, but can provide consistent estimates for the effect size.
One key assumption of the projection-based method is “biological orthogonality”,Citation9 where the CpG sites used for missing phenotype imputation should not contain the sites that are associated with the covariate of interest. In practice, this assumption may not always be satisfied. In our simulations, 50 of the 250 CpG sites that are used to impute the missing covariates are correlated with the trait of interest, which leads to significant bias in the estimates of effect size. On the other hand, this assumption is not required for MI-based methods. In fact, the imputation model is recommended to include all variables that are in the analysis model to avoid estimation bias and maximize model certainty.Citation11-14
The MI-based methods take into account of imputation uncertainty by adding random noise to the imputed values and averaging over multiple imputed datasets. In the simulation study, the MI-based methods show slightly conservative results compared to the full data analysis. This conservatism has been shown previously by the theoretical results of Meng.Citation11 The projection-based method can be viewed as an imputation approach with a single imputed data set, and therefore ignores the uncertainty for imputed values. It can potentially underestimate the variance of estimated effect size and reduce the statistical power. However, our results show that the variance of estimates from the projection-based methods is similar to that from using the full data (), possibly because the imputation that is based on many CpG sites has high accuracy and little imputation uncertainty compared to biological variation among samples. In our simulations, the correlation between imputed and true values is ∼0.9 using the projection-based method.
For both the projection and MI-based methods, a pre-selected set of CpG sites is required which is associated with the covariates to be imputed. The projection-based method typically uses 100–500 CpG sites. In contrast, the MI-based method needs to limit the number of CpG sites to be much smaller than the sample size of full data, which can potentially reduce the imputation accuracy compared to the projection-based method. In our analysis, we limited the number of CpG sites to be no larger than 30 in both MI-Norm and MI-PMM.
The efficiency of MI-based methods depends on the proportion of missing data and number of imputations. Note that the variance of effect size is , where
is the average within-imputation variance and
is the between-imputations variance. The relative efficiency of infinitely many imputations compared to m imputation is
, where FMI is the fraction of missing information.Citation15 For a 5% loss of efficiency, we will need FMI/m < 0.05. For example, when FMI = 0.3, m needs to be 6 or higher. In our simulations, m = 10 is often sufficient, but m = 30 may improve the results when the missing rate is high. However, a large number of imputations will dramatically increase computation burden of EWASs by running the association tests multiple times genome-wide. Therefore, we suggest that an EWAS is first run with missing covariates being imputed using the projection-based method, and the CpG sites with genome-wide significance are re-evaluated using a MI-based method to obtain unbiased estimated effect size.
In addition to the imputation methods discussed in this paper, another class of statistical methods can infer unobserved confounders based on singular value decomposition (SVD) of the residual (and coefficient) matrix, for example, the surrogate variable methodsCitation5,6 and the “reference-free” method.Citation7 These methods are robust to model specification in association analysis; however, they do ignore the partial information on the covariates provided by complete cases in the missing data scenario, which can lead to loss of statistical power. In supplemental Figs. 10 and 11, we compared the power and parameter estimates from the “reference-free” method to those from imputation-based methods. The “reference-free” method showed relatively lower statistical power than both of the projection- and MI-based methods, even when the missing rate was high. The parameter estimates using the “reference-free” method are consistent with true values, but have larger variation than those from imputation-based methods.
In summary, we have proposed and evaluated imputation methods for missing values in covariates using high-dimensional DNA methylation data. The proposed methods will help to control for potential confounding and increase statistical power of epigenetic association studies. An R implementation of the methods is available at: https://github.com/ChongWu-Biostat/MethyImpute.
Methods
Our proposed method combines multiple imputation and variable selection in high-dimensional data. To deal with high-dimensional methylation data, we use a 2-step variable selection approach including a screen and a selection stage. Then standard multiple imputation approach is applied to impute the missing covariate values and account for the uncertainty of imputation.
Notation
We denote the DNA methylation measure Y. Here we assume all the measurements are known and on a “β value” scale, which can be interpreted as the proportion of methylated molecules at a given locus. But the proposed methods can be directly applied to alternative methylation measures, e.g., the M-value.Citation16 We have p covariates (C), including the covariate of interest (X), such as smoking status in our example below, and potential confounders (Z), such as the cell type composition and WBC. We assume for each covariate, say , the
is the observed part while
is the missing part. We use
to denote the set of non-missing covariates. In this paper, we consider 2 types of covariates with missing values, continuous and binary.
Variable selection
Given the large number of CpG sites surveyed in an epigenome-wide study, it is infeasible to include all CpG sites in a standard multiple imputation model. Buuren and Groothuis suggested that 15 variables are generally sufficient for imputation.Citation14 Hence, we use the following variable selection method, which includes a screen stage and a selection stage.
First, in the screen stage, for an incomplete covariate , we compute test statistics for association between
and DNA methylation level at each CpG site. We select the most differentially methylated CpGs with P-value <10−7, up to 100 sites. The P-value cutoff is based on the Bonferroni correction for genome-wide significance using the HM450 chip, but other cutoff value can also be considered.
Second, in the selection stage, we use a forward stepwise with BIC criterion to select the final predictor set from the selected CpG sites. In the forward selection, the CpG sites are added to the model (linear for continuous covariate and logistic for binary) one at a time,Citation17 with the site which produces the largest reduction in the BIC value. The selection will stop when 30 CpG sites are selected, if the BIC stopping rule is still not satisfied.
Note that the initial set of 100 CpG sites for variable selection and the 30 CpG sites for imputation are subjective. In practice, the actual numbers of sites can vary depending on the total number of CpG sites that are associated with the missing covariate, and the size of complete cases.
Multiple imputation
For each incomplete covariate, the missing data are replaced by m independent imputation sets of values. Here, for continuous missing covariates, we consider 2 methods: predictive mean matching (MI-PMM), a general semi-parametric imputation method,Citation18 and linear imputation method (MI-Norm).Citation10 In MI-PMM, imputation is restricted to the observed values and can preserve non-linear relations even if the structural part of the imputation model is misspecified.Citation14 For categorical missing covariate variables, we consider using logistic regression (MI-Logreg) to impute the missing values.
In MI-Norm, for a specific covariate C, we regress on the set of selected K CpG sites (Yi1, Yi2, …, YiK) and the covariates with complete information, say Yi = (Yi1, Yi2, …, YiK, X, Cc). Here, we assume the missing covariate
follows a normal distribution:
where
and
can be estimated from
. Considering the uncertainty caused by missing data, we draw the imputation parameters
,
from the joint posterior distribution.Citation19 Specifically,
is drawn as
, where
is a random draw from a Chi square distribution with
degrees of freedom, and p is the total number of predictors in the model. Then,
is drawn as
, where
is a row vector of p independent random draws form a standard normal distribution, and
the estimated covariance matrix of
. We impute the missing observations based on the normal distribution described above and the imputation parameters
,
.
In MI-PMM, we match each missing to the respondent with the closet predicted mean and then impute using the respondent's value directly.Citation18
In MI-Logreg, instead of fitting a linear model as in MI-Norm, we fit a logistic regression model. For each missing observation , let
, and an imputed value is drawn as 1 if
, otherwise as 0, where ui is a random number from uniform(0, 1).Citation10
We analyze each of the m imputed data sets separately to test for association between DNA methylation and trait of interest. We then use Rubin's ruleCitation19 to combine the m estimates into an overall estimate. The combined estimate is the average of the individual estimates:
The total variance of is calculated as follows:
where
is the within-imputation variance (, uncertainty about the results from one imputed data set) and is the between-imputation variance (
, uncertainty due to the missing information).
Projection-based method
We tailor the method proposed by Houseman et al.Citation9 to a more general situation. Briefly, the Houseman algorithm identifies 100-300 CpG sites that discriminated cellular composition in sorted normal human cell populations (consisting of B cells, granulocytes, monocytes, NK, and CD4+ and CD8+ T cells). The method fits a linear model at each of these CpG sites using a reference dataset to estimate the coefficient for each cellular component. It then uses a matrix projection approach to map these estimated coefficients to the relative proportions of each cellular component in the samples without cell type composition information. This method can be applied to other phenotypes besides cell type composition, if a reference data set is available with both DNA methylation data and phenotype of interest. In the case of missing data, we use the set of samples with complete information () as the reference dataset, and apply the projection approach to impute the missing values (
).
Simulation
We conducted a simulation study to evaluate the performance of proposed methods for the type 1 error, statistical power, bias and coverage of the estimates. For each subject, the variable of interest, X, was generated from a uniform distribution between 0.1 and 0.9. A set of 3 covariate, Z1, Z2, and Z3, were also simulated which may contain missing values.
For continuous confounders, we considered 3 simulation models (), which generated methylation data for a total of 400 CpG sites on 2,000 subjects. Specifically, we simulated data for 50 CpG sites under model 1, 200 CpG sites under model 2, and 150 CpG sites under model 3. These CpG sites were analyzed together as from a single data set. In our analyses, we used the first 250 CpG sites from model 1 and 2 to impute missing values in covariates Z for the projection-based method; for the 2 MI-based methods, up to 30 CpG sites among the 250 sites were selected to carry out multiple imputation. The association was then tested at each of the 400 CpG sites adjusting for imputed covariates. We further considered sample size of 500 and 1,000 subjects.
Figure 5. Simulation models 1-3. X: covariate of interest; Y: DNA methylation level; Z: Covariate(s) that contain missing values.
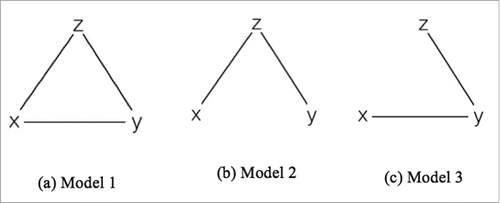
In simulation model 1 and 2, the covariates Z1, Z2, and Z3 were correlated with X, but they were independent of X in model 3. We masked a proportion of samples as missing in these 3 covariates, with equal missing rate at 0.3 or 0.9. In simulation model 1, we simulated methylation levels at 50 CpG sites as follows:where both β and γ are positive (). In model 2, we simulated methylation levels at 200 CpG sites using the same model above, with β > 0 but γ = 0 (). In these 2 models, the covariates Z confound the relationship between methylation level Y and variable X. We used the 50 CpG sites from model 1 to evaluate the statistical power of different imputation methods, and the 200 CpG sites from model 2 to evaluate the type 1 error in presence of confounding factors. In model 3, we simulated data at another 150 CpG sites where their methylation levels were correlated with covariates Z and X, but Z was independent of X (). Under this simulation model, we evaluated the statistical power of imputation methods when the missing covariates are independent risk factors.
To consider the effect of the categorical confounders, we further simulated binary covariates (simulation model 4), which was correlated with
. In this model, we simulated methylation levels at 50 CpG sites as follows:
where
and both β and γ are positive (similar to simulation model 1). In model 5, we simulated methylation levels at 200 CpG sites using the same model above, with β > 0 but γ = 0 (similar to simulation model 2). In these 2 models, the covariate Z4 confounds the relationship between methylation level Y and variable X, and is set as the missing covariate with 30% missing rate. Note that projection based method can still be applied in these 2 models by treating the binary covariate as continuous (a linear probability model).
We simulated 100 datasets and averaged the association test results of these 100 data sets and multiple CpG sites within each data set to calculate bias and uncertainty of estimation. For each of imputation method, the coverage of estimation was calculated as the proportion of simulations that the 95% confidence interval contained the true value of covariate estimates. The power was calculated as the proportion of simulations that the association P-value is less than 0.05 in the simulation model 1, 3 and 4, and the type 1 error rate was calculated using data in model 2 and 5. We compared the results to those from the full data analysis, in which we assumed that we have all data without any missing value and adjust the covariates (Z) through a linear regression. We further compared the results to those from the complete-case analysis, in which subjects with missing value were excluded.
ARIC data
The Atherosclerosis Risk in Communities (ARIC) study is a prospective cohort study of cardiovascular disease risk in 4 US communities.Citation20 Between 1987 and 1989, 7,082 men and 8,710 women aged 45–64 y were recruited from Forsyth County, North Carolina; Jackson, Mississippi (African Americans only); suburban Minneapolis, Minnesota; and Washington County, Maryland. Before conducting the study, the institutional review boards of each participating university approved the protocol. After written informed consent including that for genetic studies was obtained, participants underwent a baseline clinical examination (Visit 1) and 3 subsequent follow-up clinical exams at intervals of roughly 3 y (Visits 2-4). An additional follow-up exam (Visit 5) was conducted in 2011-13.
Followed manufacturer instructions, genomic DNA were extracted from peripheral blood leukocyte samples using the Gentra Puregene Blood Kit (Qiagen). Bisulphite conversion of 1 μg genomic DNA was performed using the EZ-96 DNA Methylation Kit (Deep Well Format) (Zymo Research) according to the manufacturers instructions (www.zymoresearch.com). Bisulphite conversion efficiency was determined by PCR amplification of the converted DNA before proceeding with methylation analyses on the Illumina platform using Zymo Research Universal Methylated Human DNA Standard and Control Primers.
Using the Illumina HM450 chip, bisulphite-converted DNA from 2,905 African American participants at Visit 2 (1990-92; n = 2,504) or Visit 3 (1993-95; n = 441) was measured for methylation status. Using Illumina GenomeStudio 2011.1, Methylation module 1.9.0 software, we determined the degree of methylation.
Individuals (n = 71) were excluded from analysis if a pass rate for the DNA sample for the study participant was less than 99% (probes with a detection P-value > 0.01/all probes on the array), or the sample failed gender or genotype consistency checking. Probes on the HM450 chip for which the pass rate was less than 99% (sample with a detection P-value > 0.01 at probe/all samples) were not analyzed (n = 5,170).
In an epigenome-wide analysis, we tested the association between DNA methylation (Y) and smoking status (never vs. current smoker) (X) at each CpG site using linear mixed effects model (LMM). The covariates (Z) included cell type composition, age, field center, gender, and white blood cell count. Technical variables such as chip and chip position were included as random effects. Among the covariates, cell type composition and total white blood cell (WBC) counts contained considerable missing values, with ∼91% and 14% missing respectively.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1145328_s02.docx
Download MS Word (3.4 MB)Acknowledgments
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). The authors thank the staff and participants of the ARIC study for their important contributions. Methylation profiling of the ARIC samples was also supported by the National Institutes of Health (NIH) American Recovery and Reinvestment Act of 2009 (ARRA) Building on GWAS for NHLBI-diseases: the US. CHARGE consortium (5RC2HL102419) (PI: E. Boerwinkle). This work was carried out in part using computing resources at the University of Minnesota Supercomputing Institute.
Related Research Data
References
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011; 11:726–34; PMID:21941284; http://dx.doi.org/10.1038/nrc3130
- Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet 2012; 13:705–19; PMID:22986265; http://dx.doi.org/10.1038/nrg3273
- Koestler DC, Christensen B, Karagas MR, Marsit CJ, Langevin SM, Kelsey KT, Wiencke JK, Houseman EA. Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics 2013; 8:816–26; PMID:23903776; http://dx.doi.org/10.4161/epi.25430
- Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013; 31:142–7; PMID:23334450; http://dx.doi.org/10.1038/nbt.2487
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 2007; 3:1724–35; PMID:17907809; http://dx.doi.org/10.1371/journal.pgen.0030161
- Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics 2011; 27:1496–505; PMID:21471010; http://dx.doi.org/10.1093/bioinformatics/btr171
- Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 2014; 30:1431–9; PMID:24451622; http://dx.doi.org/10.1093/bioinformatics/btu029
- Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J. Epigenome-wide association studies without the need for cell-type composition. Nat Methods 2014; 11:309–11; PMID:24464286; http://dx.doi.org/10.1038/nmeth.2815
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13:86; PMID:22568884; http://dx.doi.org/10.1186/1471-2105-13-86
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: Issues and guidance for practice. Stat Med 2011; 30:377–99; PMID:21225900; http://dx.doi.org/10.1002/sim.4067
- Meng X-L. Multiple-imputation inferences with uncongenial sources of input. Stat Sci 1994; 9:538–58; http://dx.doi.org/10.1214/ss/1177010269
- Schafer JL. Analysis of incomplete multivariate data. CRC Press 1997
- Collins LM, Schafer JL, Kam CM. A comparison of inclusive and restrictive strategies in modern missing data procedures. Psychol Methods 2001; 6:330–51; PMID:11778676; http://dx.doi.org/10.1037/1082-989X.6.4.330
- Buuren S, Groothuis-Oudshoorn K. mice: Multivariate imputation by chained equations in R. J Stat Soft 2011; 45; PMID:22289957; http://dx.doi.org/10.18637/jss.v045.i03
- Schafer JL. Analysis of incomplete multivariate data. London: Chapman & Hall; 1997.
- Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587; PMID:21118553; http://dx.doi.org/10.1186/1471-2105-11-587
- Derksen S, Keselman H. Backward, forward and stepwise automated subset selection algorithms: Frequency of obtaining authentic and noise variables. Br J Math Stat Psychol 1992; 45:265–82; http://dx.doi.org/10.1111/j.2044-8317.1992.tb00992.x
- Little RJ. Missing-data adjustments in large surveys. J Bus Eco Stat 1988; 6:287–96; http://dx.doi.org/10.1080/07350015.1988.10509663
- Rubin DB. Multiple imputation for nonresponse in surveys. John Wiley & Sons; 2004.
- The Atherosclerosis Risk in Communities (ARIC). Study: design and objectives. The ARIC investigators. Am J Epidemiol 1989; 129:687–702; PMID:2646917