
ABSTRACT
Several recent reports have described associations between gestational diabetes (GDM) and changes to the epigenomic landscape where the DNA samples were derived from either cord or placental sources. We employed genome-wide 450K array analysis to determine changes to the epigenome in a unique cohort of maternal blood DNA from 11 pregnant women prior to GDM development relative to matched controls. Hierarchical clustering segregated the samples into 2 distinct clusters comprising GDM and healthy pregnancies. Screening identified 100 CpGs with a mean β-value difference of ≥0.2 between cases and controls. Using stringent criteria, 5 CpGs (within COPS8, PIK3R5, HAAO, CCDC124, and C5orf34 genes) demonstrated potentials to be clinical biomarkers as revealed by differential methylation in 8 of 11 women who developed GDM relative to matched controls. We identified, for the first time, maternal methylation changes prior to the onset of GDM that may prove useful as biomarkers for early therapeutic intervention.
Introduction
Gestational diabetes (GDM) is a pregnancy-specific endocrine disorder with a prevalence of 3.5–14%.Citation1 Due to the worldwide obesity epidemic and recently modified diagnostic criteria, GDM is increasingly prevalent.Citation2 It occurs because of a mismatch between insulin production and requirement, leading to maternal hyperglycemia. Since glucose is able to cross the placenta, whereas insulin is not, the fetus is also exposed to hyperglycemic conditions. Women with GDM are at increased risk of caesarean section and stillbirth compared with healthy women.Citation3, 4 They are also more likely to develop type 2 diabetes (T2DM), dyslipidemia, and cardiovascular disease in later life,Citation5-7 while their offspring have an increased long-term risk of obesity and diabetes.Citation2
Epigenetic modifications, which may be causal of or associated with changes in gene expression, offer significant promise for understanding the underlying mechanisms of GDM. Indeed, and as an example, epigenetic changes in T2DM have been reported in genes involved in metabolism.Citation8-13 Since maternal epigenetic modifications are known to contribute to fetal programming,Citation14 recent studies have investigated the role of epigenetic alterations in offspring exposed to maternal hyperglycemia and found positive associations.Citation15-19 Furthermore, previous studies suggest that epigenetic modifications may play a role in the pathogenesis of GDM.Citation20,21
Epigenetic research in GDM has largely used targeted (candidate gene) approaches.Citation15,16,18,19 To date, only 2 studies have utilized genome-wide methodologyCitation17,22 and in these cases investigators examined placenta and cord blood samples from GDM pregnancies. Differentially methylated genes were identified between GDM and healthy pregnancies,Citation17,22 which provide evidence for the involvement of these genes and/or their differential methylation in GDM. However, there have been no genome-wide studies examining methylation differences between maternal tissue samples from GDM and healthy pregnancies. We decided to focus on maternal epigenetic profiles, as they would facilitate the assessment of the in utero environment and allow identification of predictive biomarkers that would enable targeted intervention to high risk groups.
On the basis of the current literature, we hypothesized the presence of pre-existing epigenetic markers in women who subsequently go on to develop GDM. In this study, and for the first time in this disease, we interrogated genome-wide DNA methylation in peripheral blood samples collected from women prior to the development of GDM and relative to matched healthy controls that did not develop GDM. Using this discovery cohort, our aim was to identify candidate genes with future promise as potential biomarkers for the prediction of GDM in early pregnancy.
Results
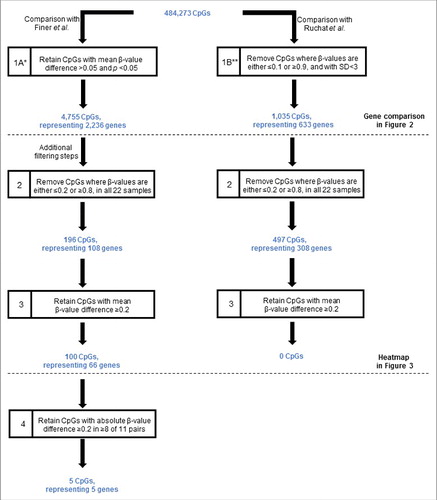
Our initial data analyses focused on comparison of our data in antenatal samples with the 2 recent genome-wide studies that investigated cord blood and placental tissue samples.Citation17,22 We compared our data with those of Finer et al.Citation22 and Ruchat et al.Citation17 separately due to the different approaches used for data processing by each study (). Using the filtering criteria shown in step 1A of , comparison of our data with those of Finer et al.Citation22 identified 4,755 differentially methylated CpGs (representing 2,236 genes) where the mean β-value difference between the GDM and healthy groups was >0.05 and statistically significant (P < 0.05). In contrast, comparison with the data of Ruchat et al.Citation17 (step 1B of ) identified 1,035 CpGs (representing 633 candidate genes). We also performed the same comparison after applying multiple testing adjustment using the false discovery rates, which showed no overlap of our data with these 2 studies.
Figure 1. Filtering criteria for the identification of CpGs differentially methylated between GDM and normal pregnancies. The starting number of CpGs (484,273) was derived through the removal of CpGs with high detection values (P > 0.05) and those with missing β-values in any one of the 22 samples, as described in the Materials and Methods. Horizontal line denotes additional filtering steps. *According to Finer et al. criteria.Citation22 **According to Ruchat et al. criteria.Citation17 GDM, gestational diabetes. SD, standard deviation.
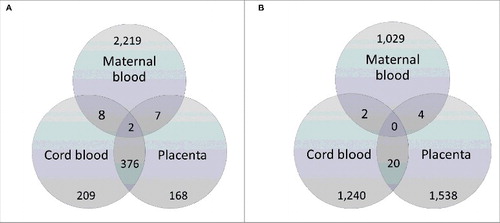
As shown in , by comparing the 2,236 genes identified as differentially methylated in our study with those reported by Finer et al.,Citation22 2 genes were common between maternal blood, umbilical cord, and placenta: Hook Microtubule-Tethering Protein 2 (HOOK2) and Retinol Dehydrogenase 12 (RDH12). Conversely, and as summarized by the Venn diagram in , there were no genes common to all 3 tissue types when we compared our data with that of Ruchat et al.Citation17
Figure 2. Venn diagrams illustrating comparison of genes differentially methylated in GDM using maternal blood with those identified in cord blood and placenta of GDM affected pregnancies from the cohorts of (A) Finer et al.Citation22 and (B) Ruchat et al.Citation17, respectively. The genes from our dataset that were common with the other study are shown in dark gray shading. Genes identified as differentially methylated in Finer et al.Citation22 were obtained from Supplementary file 2 of the published article, while the list of differentially methylated genes identified by Ruchat et al.Citation17 was kindly provided through personal communication with the corresponding author of Ruchat et al.Citation17
The 4,755 CpGs initially identified as differentially methylated were then subjected to further filtering (steps 2 and 3, ). Using this approach, we identified 100 unique CpGs (comprising 66 genes) that were differentially methylated between GDM and healthy pregnancies (the full annotated list is shown in Table S1). None of these CpGs have an annotated single-nucleotide polymorphism (SNP) in the probe. Closer examination of the 100 CpGs revealed that the majority (53%) were hypomethylated in GDM relative to healthy pregnancies. The observed differences in mean β-value showed a maximum difference of 0.38. The frequency and DNA methylation of these differentially methylated CpG sites in relation to their genomic location and CpG islands are shown in Figure S1. Of the differentially methylated CpGs, 45% were associated with a CpG island, shelf, or shore (Figure S1C).
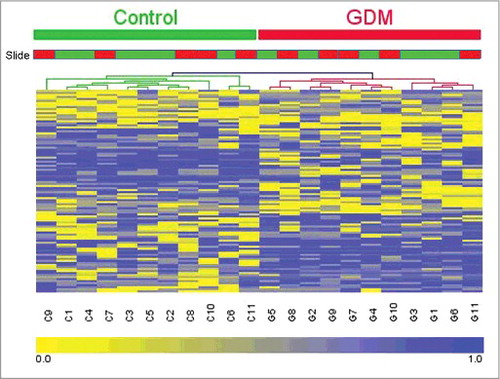
Hierarchical clustering was performed to determine whether the methylation patterns in these 100 CpGs can be used to distinguish between GDM and healthy pregnancies. The heatmap in illustrates that there are distinctive methylation patterns between GDM and healthy pregnancies, which segregate samples into 2 distinct groups comprising those from GDM and healthy populations. The slide type did not cause the clustering; therefore, our results were not due to batch effects. Calculation of the genomic inflation factor before and after normalization steps showed that removal of SNP containing probes and subset-quantile within array normalization (SWAN) by the minfi package reduced the genomic inflation.Citation23-25 Pre-normalization λ was estimated to be 1.189 (standard error of the estimation = 9.461 × 10−5); after normalization, the estimated λ was reduced to 1.132 (standard error of the estimation = 7.461 × 10−5). The remaining genomic inflation suggests that mild confounding stratification factors remain unaccounted for in the data.
Figure 3. Heatmap and dendrograms showing clusteringCitation45 for the 100 CpGs identified as differentially methylated (mean difference in β-values >0.2) between GDM and healthy pregnancies. DNA methylation across the 100 sites in each of the samples was analyzed by hierarchical clustering using the Euclidean distance and average linkage criteria. Each row represents an individual CpG site and each column a different sample. Healthy controls and GDM samples are shown by the green and red bars, respectively. Slide type is also shown with slide 1 in green and slide 2 in red. Color gradation from yellow to blue represents low to high DNA methylation respectively, with β-values ranging from 0 (no methylation; yellow) to 1 (complete methylation; blue). GDM, gestational diabetes.
Enrichment of gene ontology terms and biological pathways within the 66 genes associated with differentially methylated CpGs were assessed using DAVID online softwareCitation26 and identified 11 overrepresented pathways, with the top 3 (ranked by P-value) involved in cell adhesion molecules, type 1 diabetes mellitus, and keratin pathways. However, enrichment of these pathways was not statistically significant following adjustment for false discovery rates (Table S2).
Finally, we examined the absolute β-value differences across all 11 matched pairs. Using this stringent criteria, in 5 of the 100 CpGs identified, at least 8 of the 11 GDM pregnancies showed β-value differences of >0 .2 relative to matched controls. The 5 CpGs comprised of 5 genes (COPS8, PIK3R5, HAAO, C5orf34, and CCDC124) and their functions are shown in .
Table 1. Annotation for the 5 genes differentially methylated in 8 of 11 matched pairs, as determined by genome-wide DNA methylation analysis. *The official gene symbol, gene name and stated function were retrieved from the NCBI Gene database (accessed September 2015). **The absolute β-value difference range is the minimum to the maximum value of the individual absolute β-value differences for each differentially methylated CpG.
Discussion
We describe for the first time, genome-wide DNA methylation changes in maternal blood prior to the diagnosis of GDM. We identified 2 differentially methylated genes that shared identity with genes previously described in studies that interrogated placenta and umbilical cord blood samples and, in these cases, using the same array platforms.Citation17,22 Furthermore, using stringent filtering criteria, we identified 100 unique CpGs that segregated GDM and healthy pregnancies into distinct groups upon hierarchical clustering.
The strength of our study, in contrast to previous studies, is that we carefully matched each GDM pregnancy to a healthy one to ensure the samples were comparable.Citation17,22 Furthermore, as all samples were taken prior to development of pregnancy complications, there was limited sampling bias.
We were able to compare our data to those from 2 recent genome-wide studies in GDM using cord blood and placenta tissue.Citation17,22 Comparative analysis with Finer et al.Citation22 showed that HOOK2 and RDH12 were common to maternal blood, placenta and cord blood. HOOK2 codes for a linker protein that mediates binding to organelles and is responsible for morphogenesis of cilia and endocytosis.Citation27,28,29 RDH12 encodes a retinal reductase, which also plays a role in the metabolism of short-chain aldehydes.Citation27,30 In terms of KEGG orthology, it is involved in metabolic pathways as well as retinal metabolism.Citation31 These 2 genes, therefore, may represent important candidates for further study.
The disparity of candidate genes when comparisons are made to the previous studies might reflect the different data filtering criteria used by Ruchat et al.Citation17 and Finer et al.Citation22 Using the Finer et al. criteria, many of the differentially methylated CpGs are likely to have β-value differences <0.2, which could be difficult to reproduce either by alternative methodologies, such as pyrosequencing or in replication studies using independent patient cohorts. Moreover, we used a distinct patient population to the other 2 studies. We used samples from women prior to the development of their GDM, while both Ruchat et al.Citation17 and Finer et al.Citation22 used samples from women with established GDM. Furthermore, we used maternal blood samples, rather than placenta and cord blood samples. These disparities may have contributed to the differences in the absolute numbers of CpGs/genes identified.
Further analysis of our cohort identified 100 independent CpGs (comprising 66 genes), which were found to cluster GDM and healthy pregnancies separately. Reassuringly, these CpGs have no annotated SNPs in the probe. Enrichment of gene ontology terms and biological pathways of these 66 genes showed enrichment for genes involved in cell adhesion, type 1 diabetes mellitus, and keratin pathways.Citation26,32 Although the enrichment was not statistically significant following adjustment for false discovery rates, these are promising candidates, which are worth examining to elucidate the biological mechanisms behind GDM. In future work, it will be important to verify, in larger independent cohorts, the candidates identified herein and to determine the impact of differential methylation. This may in the future improve the understanding of GDM pathogenesis and aid in the development of therapy.
The design of this pilot study was to generate a list of genes of interest using a relatively small number of samples. In order to avoid type II errors (false negatives), we used uncorrected P-values to identify potential candidates in the preliminary screening. We then applied more stringent methodology (steps 2–4 of ) to identify candidate genes. A potential limitation of our study is the possibility of genomic inflation. Mild confounding stratification factors, such as changes in composition of blood during the pregnancy, the time of blood sampling, and parity, may have inflated the data. Therefore, we further validated the array data using an independent method with pyrosequencing in order to confirm our findings.
On closer inspection, 8 of 11 women who subsequently developed GDM showed differential methylation at 5 CpGs (consisting of COPS8, PIK3R5, HAAO, CCDC124, and C5orf34 genes) relative to matched controls. COPS8 encodes a regulator of multiple signaling pathways.Citation27,33 It is involved in protein binding and negative regulation of cell proliferation.Citation33,34 The PIK3R5 protein has important roles in cell growth, proliferation, motility, differentiation, survival, and intracellular trafficking.Citation27,35–37 The HAAO protein catalyzes the synthesis of quinolinic acid (QUIN). Increased cerebral levels of QUIN may participate in the pathogenesis of neurologic and inflammatory disorders, which may be mediated by HAAO.Citation27,38 This unique epigenetic signature may form the basis of future biomarker studies using a larger validation cohort. The CCDC124 protein is involved in cell cycle and division.Citation39 C5orf34 encodes for a protein that is highly conserved across species; however, its function remains uncharacterised.Citation27
In summary, for the first time, using a genome-wide approach in maternal blood, we have identified maternal methylation changes prior to the diagnosis of GDM. As a discovery-based study, our findings may prove useful toward developing simple biomarkers for predicting GDM, thus facilitating intervention strategies in the early antenatal period to improve the health of the mother and baby, both during pregnancy and in the long-term.
Materials and methods
Patients
Peripheral blood samples were obtained from women prospectively recruited at the University Hospital of North Midlands, UK, between 12–16 weeks gestation, prior to the diagnosis of any pregnancy complications as part of the EFFECT-M study.Citation40 At the end of pregnancy, we identified 11 women who had GDM and individually matched each one with a healthy woman who had a normal pregnancy. They were matched in terms of age, body mass index, ethnicity, smoking status, medications and folate supplementation (Table S3). The study was approved by the West Midlands (Black Country) Research Ethics Committee (REC reference no. 08/H1204/121).
Genome-wide DNA methylation profiling
We performed genome-wide analysis of DNA methylation using the Illumina HumanMethylation450 BeadChip (450K) array, which examines over 480,000 individual CpG sites. We first extracted genomic DNA from blood samples collected into potassium EDTA using standard phenol/chloroform procedures. Next, samples were sodium bisulfite convertedCitation41 and hybridized to arrays according to Illumina recommended protocols that we have previously described.Citation42 Methylation at individual CpGs is reported as a methylation β-value, which is a quantitative measure of methylation for each CpG site with range between 0 (no methylation) to 1 (completely methylated).
Validation by sodium bisulfite pyrosequencing
A technical validation between array β-values and methylation levels was determined by sodium bisulfite pyrosequencing in all 22 samples. To increase template quantity for pyrosequencing assays, whole genome amplification of bisulfite-converted DNA followed by touchdown PCR were performed as previously described.Citation42 A PyroMark Q24 instrument was used to run pyrosequencing assays according to the manufacturer's instructions (Qiagen). Analyses of Pyrograms were conducted on the PyroMark Q24 software (v 2.0.6., build 20; Qiagen). Seven CpGs representing 5 genes were chosen to provide a range of β-values. These demonstrated a strong positive correlation between β-values and percentage methylation by bisulphite sequencing (Spearman's r = 0.92, Fig. S2).
Data analysis
Each array passed quality control assessment based on the performance of internal array controls. Initial processing, probe type correction and assessment of array data was conducted using the minfi package and SWAN.Citation23,24 Probes with known SNPs were removed. All CpGs for which one or more of the 22 samples displayed detection P-values > 0.05 (indicating an unreliable site) or presented with missing β-values were excluded. The genomic inflation factor (λ, the ratio of the median of the observed distribution of the test statistic to the expected median) was calculated using the estlambda function of GenABEL.Citation25
We filtered the data using criteria shown in to identify differentially methylated sites between GDM and healthy pregnancies. In step the first analysis, we elected to use a minimum β-value difference of 0.05, in part to permit comparisons with a recent report describing DNA methylation in placenta and umbilical cord blood from GDM pregnancies also using the 450K array platform (step 1, ).Citation22 The genes identified as differentially methylated were obtained from the supplementary data of this particular publication. We also compared our data with a separate cohort of placenta and umbilical cord blood samples from GDM pregnancies.Citation17 We obtained their list of differentially methylated genes through personal communication with the corresponding author of the publication. Further filtering steps were applied to facilitate a more stringent analysis. To reduce the number of non-variable sites to improve the statistical power of subsequent analyses, we removed all sites with β-values ≥0 .8 and ≤0 .2 in all 22 samples (step 2, ). This is an approach that has been used by our group as well as by others.Citation41-44 As described previously by our group, we consider it a more robust methodology to remove from the data set CpGs that failed in any one of the samples, instead of eliminating specific failed CpGs from specific samples.Citation42 We retained only those CpGs that had a mean β-value difference of ≥0 .2 (step 3, ). Finally we examined the absolute β-values in each matched pairs. We used a cut-off of ≥0 .2 mean β-values difference to identify CpGs with considerable methylation differences.
Hierarchical clustering was performed utilizing Genesis software (v1.7.6) using Euclidian distance and average linkage criteria.Citation45 Enrichment of gene ontology terms and biological pathways within the genes associated with differentially methylated CpGs were assessed using DAVID online software.Citation26,32
Abbreviations
| GDM | = | Gestational diabetes |
| KEGG | = | Kyoto encyclopedia of genes and genomes |
| T2DM | = | Type 2 diabetes mellitus |
| QUIN | = | Quinolinic acid |
| SNP | = | Single-nucleotide polymorphism |
| SWAN | = | Subset-quantile within array normalization |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1166321_s02_tables.docx
Download MS Word (34.8 KB)KEPI_A_1166321_s02_legends.docx
Download MS Word (14.2 KB)KEPI_A_1166321_s02.zip
Download Zip (81.1 KB)Acknowledgment
The authors are grateful to Khaled Ismail, Angela Rooney, and Emma Hubball who obtained the EFFECT-M samples. This work was supported by funding provided by the University Hospital of North Midlands Charity.
Additional information
Funding
Related Research Data
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011; 34:S62-S9; PMID:21193628; https://doi.org/10.2337/dc11-S062
- National Collaborating Centre for Women's and Children's health. Diabetes in pregnancy. Management of diabetes and its complications from preconception to the postnatal period. NICE 2015; 3.
- Gorgal R, Gonçalves E, Barros M, Namora G, Magalhães Â, Rodrigues T, Montenegro N. Gestational diabetes mellitus: A risk factor for non-elective cesarean section. J Obstet Gynaecol Res 2012; 38:154-9; PMID:21995455; https://doi.org/10.1111/j.1447-0756.2011.01659.x
- Schmidt MI, Duncan BB, Reichelt AJ, Branchtein L. Gestational diabetes mellitus diagnosed with a 2-h 75-g oral glucose tolerance test and adverse pregnancy outcomes. Diabetes Care 2001; 24:1151-5; PMID:11423494; https://doi.org/10.2337/diacare.24.7.1151
- Bellamy L, Casas JP, Hingorani AD, Williams D. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet 2009; 373:1773-9; https://doi.org/10.1016/S0140-6736(09)60731-5
- Akinci B, Celtik A, Genc S, Yener S, Demir T, Secil M, Kebapcilar L, Yesil S. Evaluation of postpartum carbohydrate intolerance and cardiovascular risk factors in women with gestational diabetes. Gynecol Endocrinol 2011; 27:361-7; PMID:20540676; https://doi.org/10.3109/09513590.2010.492885
- Rivero K, Portal VL, Vieira M, Behle I. Prevalence of the impaired glucose metabolism and its association with risk factors for coronary artery disease in women with gestational diabetes. Diabetes Res Clin Pract 2008; 79:433-7; PMID:18045723; https://doi.org/10.1016/j.diabres.2007.10.015
- Ling C, Del Guerra S, Lupi R, Rönn T, Granhall C, Luthman H, Masiello P, Marchetti P, Groop L, Del Prato S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008; 51:615-22; PMID:18270681; https://doi.org/10.1007/s00125-007-0916-5
- Kulkarni SS, Salehzadeh F, Fritz T, Zierath JR, Krook A, Osler ME. Mitochondrial regulators of fatty acid metabolism reflect metabolic dysfunction in type 2 diabetes mellitus. Metabolism 2012; 61:175-85; PMID:21816445; https://doi.org/10.1016/j.metabol.2011.06.014
- Yang BT, Dayeh TA, Volkov PA, Kirkpatrick CL, Malmgren S, Jing X, Renström E, Wollheim CB, Nitert MD, Ling C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol 2012; 26:1203-12; PMID:22570331; https://doi.org/10.1210/me.2012-1004
- Yang BT, Dayeh TA, Kirkpatrick CL, Taneera J, Kumar R, Groop L, Wollheim CB, Nitert MD, Ling C. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA1c levels in human pancreatic islets. Diabetologia 2011; 54:360-7; PMID:21104225; https://doi.org/10.1007/s00125-010-1967-6
- Hall E, Dayeh T, Kirkpatrick CL, Wollheim CB, Dekker Nitert M, Ling C. DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med Genet 2013; 14:76; PMID:23879380; https://doi.org/10.1186/1471-2350-14-76
- Ribel-Madsen R, Fraga MF, Jacobsen S, Bork-Jensen J, Lara E, Calvanese V, Fernandez AF, Friedrichsen M, Vind BF, Højlund K, et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PloS one 2012; 7:e51302; PMID:23251491; https://doi.org/10.1371/journal.pone.0051302
- Gluckman PD, Hanson MA, Buklijas T, Low FM. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 2009; 5:401-8; PMID:19488075; https://doi.org/10.1038/nrendo.2009.102
- Bouchard L, Thibault S, Guay SP, Santure M, Monpetit A, St-Pierre J, Perron P, Brisson D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010; 33:2436-41; PMID:20724651; https://doi.org/10.2337/dc10-1024
- Bouchard L, Hivert MF, Guay SP, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers' blood glucose concentration. Diabetes 2012; 61:1272-80; PMID:22396200; https://doi.org/10.2337/db11-1160
- Ruchat SM, Houde AA, Voisin G, St-Pierre J, Perron P, Baillargeon JP, Gaudet D, Hivert MF, Brisson D, Bouchard L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 2013; 8:935-43; PMID:23975224; https://doi.org/10.4161/epi.25578
- Houde AA, Guay SP, Desgagné V, Hivert MF, Baillargeon JP, St-Pierre J, Perron P, Gaudet D, Brisson D, Bouchard L. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics 2013; 8:1289-302; PMID:24113149; https://doi.org/10.4161/epi.26554
- El Hajj N, Pliushch G, Schneider E, Dittrich M, Müller T, Korenkov M, Aretz M, Zechner U, Lehnen H, Haaf T. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes 2013; 62:1320-8; PMID:23209187; https://doi.org/10.2337/db12-0289
- Lehnen H, Zechner U, Haaf T. Epigenetics of gestational diabetes mellitus and offspring health: the time for action is in early stages of life. Mol Hum Reprod 2013; 19:415-22; PMID:23515667; https://doi.org/10.1093/molehr/gat020
- Ma RC, Tutino GE, Lillycrop KA, Hanson MA, Tam WH. Maternal diabetes, gestational diabetes and the role of epigenetics in their long term effects on offspring. Prog Biophys Mol Biol 2015; 118:55-68; PMID:25792090; https://doi.org/10.1016/j.pbiomolbio.2015.02.010
- Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, Sinha A, Williams D, Rakyan VK, Hitman GA. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet 2015; 24:3021-9; PMID:25634562; https://doi.org/10.1093/hmg/ddv013
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30:1363-9; PMID:24478339; https://doi.org/10.1093/bioinformatics/btu049
- Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalisation for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol 2012; 13:R44; PMID:22703947; https://doi.org/10.1186/gb-2012-13-6-r44
- Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007; 23:1294-6; PMID:17384015; https://doi.org/10.1093/bioinformatics/btm108
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008; 4:44-57; https://doi.org/10.1038/nprot.2008.211
- Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res 2005; 33:D54-8; PMID:15608257; https://doi.org/10.1093/nar/gki031
- Baron Gaillard CL, Pallesi-Pocachard E, Massey-Harroche D, Richard F, Arsanto JP, Chauvin JP, et al. Hook2 is involved in the morphogenesis of the primary cilium. Mol Biol Cell 2011; 22:4549-62; PMID:21998199; https://doi.org/10.1091/mbc.E11-05-0405
- Krämer H, Phistry M. Genetic analysis of hook, a gene required for endocytic trafficking in drosophila. Genetics 1999; 151:675-84.
- Haeseleer F, Jang GF, Imanishi Y, Driessen CAGG, Matsumura M, Nelson PS, Palczewski K. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem 2002; 277:45537-46; PMID:12226107; https://doi.org/10.1074/jbc.M208882200
- Kanehisa M, Susumu G. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28:27-30; PMID:10592173; https://doi.org/10.1093/nar/28.1.27
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; https://doi.org/10.1093/nar/gkn923
- Liu C, Guo LQ, Menon S, Jin D, Pick E, Wang X, Deng XW, Wei N. COP9 signalosome subunit Csn8 is involved in maintaining proper duration of the G1 phase. J Biol Chem 2013; 288:20443-52; PMID:23689509; https://doi.org/10.1074/jbc.M113.468959
- Rolland T, Taşan M, Charloteaux B, Pevzner Samuel J, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. A proteome-scale map of the human interactome network. Cell 2014; 159:1212-26; PMID:25416956; https://doi.org/10.1016/j.cell.2014.10.050
- Johnson C, Marriott SJ, Levy LS. Overexpression of p101 activates PI3Kγ signaling in T cells and contributes to cell survival. Oncogene 2007; 26:7049-57; PMID:17486067; https://doi.org/10.1038/sj.onc.1210504
- Shymanets A, Prajwal Vadas O, Czupalla C, LoPiccolo J, Brenowitz M, Ghigo A, Hirsch E, Krause E, Wetzker R, et al. Different inhibition of Gβγ-stimulated class IB phosphoinositide 3-kinase (PI3K) variants by a monoclonal antibody. Specific function of p101 as a Gβγ-dependent regulator of PI3Kγ enzymatic activity. Biochem J 2015; 469:59-69; PMID:26173259; https://doi.org/10.1042/BJ20150099
- Brock C, Schaefer M, Reusch HP, Czupalla C, Michalke M, Spicher K, Schultz G, Nürnberg B. Roles of Gβγ in membrane recruitment and activation of p110γ/p101 phosphoinositide 3-kinase γ. J Cell Biol 2003; 160:89-99; PMID:12507995; https://doi.org/10.1083/jcb.200210115
- Kohler C, Eriksson LG, Flood PR, Hardie JA, Okuno E, Schwarcz R. Quinolinic acid metabolism in the rat brain. Immunohistochemical identification of 3-hydroxyanthranilic acid oxygenase and quinolinic acid phosphoribosyltransferase in the hippocampal region. J Neurosci 1988; 8:975-87; PMID:3346732
- Telkoparan P, Erkek S, Yaman E, Alotaibi H, Bayik D, Tazebay UH. Coiled-coil domain containing protein 124 is a novel centrosome and midbody protein that interacts with the Ras-guanine nucleotide exchange factor 1B and is involved in cytokinesis. PloS One 2013; 8:e69289; PMID:23894443; https://doi.org/10.1371/journal.pone.0069289
- Fryer AA, Nafee TM, Ismail KM, Carroll WD, Emes RD, Farrell WE. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics 2009; 4:394-8; PMID:19755846; https://doi.org/10.4161/epi.4.6.9766
- Fryer AA, Emes RD, Ismail KM, Haworth KE, Mein C, Carroll WD, Farrell WE. Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics 2011; 6:86-94; PMID:20864804; https://doi.org/10.4161/epi.6.1.13392
- Glossop JR, Nixon NB, Emes RD, Haworth KE, Packham JC, Dawes PT, Fryer AA, Mattey DL, Farrell WE. Epigenome-wide profiling identifies significant differences in DNA methylation between matched-pairs of T- and B-lymphocytes from healthy individuals. Epigenetics 2013; 8:1188-97; PMID:24005183; https://doi.org/10.4161/epi.26265
- Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, Yang AS. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18:4808-17; PMID:19776032; https://doi.org/10.1093/hmg/ddp445
- Glossop JR, Emes RD, Nixon NB, Packham JC, Fryer AA, Mattey DL, Farrell WE. Genome-wide profiling in treatment-naive early rheumatoid arthritis reveals DNA methylome changes in T- and B-lymphocytes. Epigenomics 2015 Nov 10; 8(2):209-24. [Epub ahead of print]
- Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics 2002; 18:207-8; PMID:11836235; https://doi.org/10.1093/bioinformatics/18.1.207