
ABSTRACT
Developmental exposure to bisphenol A (BPA) has been shown to induce changes in DNA methylation in both mouse and human genic regions; however, the response in repetitive elements and transposons has not been explored. Here we present novel methodology to combine genomic DNA enrichment with RepeatMasker analysis on next-generation sequencing data to determine the effect of perinatal BPA exposure on repetitive DNA at the class, family, subfamily, and individual insertion level in both mouse and human samples. Mice were treated during gestation and lactation to BPA in chow at 0, 50, or 50,000 ng/g levels and total BPA was measured in stratified human fetal liver tissue samples as low (non-detect to 0.83 ng/g), medium (3.5 to 5.79 ng/g), or high (35.44 to 96.76 ng/g). Transposon methylation changes were evident in human classes, families, and subfamilies, with the medium group exhibiting hypomethylation compared to both high and low BPA groups. Mouse repeat classes, families, and subfamilies did not respond to BPA with significantly detectable differential DNA methylation. In human samples, 1251 individual transposon loci were detected as differentially methylated by BPA exposure, but only 19 were detected in mice. Of note, this approach recapitulated the discovery of a previously known mouse environmentally labile metastable epiallele, CabpIAP. Thus, by querying repetitive DNA in both mouse and humans, we report the first known transposons in humans that respond to perinatal BPA exposure.
Abbreviations
| Avy | = | agouti variable yellow |
| BPA | = | bisphenol A |
| IAP | = | intracisternal A particle |
| SINE | = | short interspersed element |
| LINE | = | long interspersed element |
| LTR | = | Long terminal repeat |
| ERV | = | endogenous retrovirus |
| FDR | = | false discovery rate. |
Introduction
DNA methylation as an epigenetic mark has been at the forefront of the integration of environmental exposures, gene expression patterns, and subsequent health effects. A growing number of studies report environmental shifts in genomic DNA methylation in multiple species following exposure to agents such as air pollution,Citation1,2 chemical exposures including bisphenol A (BPA)Citation3 and phthalates, radiation,Citation4 and nutritional deprivation.Citation5 Because the epigenome of mammals is reprogrammed in pre-implantation zygotes during early embryogenesis and in primordial germ cells (PGC) of the developing fetus,Citation6,7 perinatal exposures at even relatively low concentrations can have life course effects on the epigenome.
Research by our group and others has consistently shown genome-wide and locus specific effects following developmental exposures. Locus specific effects occur at the Agouti viable yellow (Avy) locus and a similar locus, CabpIAP, both controlled by the insertion of intracisternal A particle (IAP) transposons.Citation8-10 Interrogations of the methylome are now commonly performed in fields ranging from epidemiology to animal science, and mammalian studies often focus on DNA methylation of LINE1 elements as a proxy for global DNA methylation.Citation11,12 Conversely, studies using next-generation bisulfite sequencing often discard transposons due to the low mappability of certain repeat elements.Citation13 Despite the wide use of methylation detecting technologies, few researchers examine the repetitive portion of the genome in detail, even though it is known that repetitive elements are regulated with a high degree of specificityCitation14 and that developmental exposures can alter epigenetic programming of repetitive elements including the transposons such as Avy and CabpIAP.Citation9,15,16
Here we focus primarily on transposon repetitive elements, sequences that are capable of inserting themselves in new locations. Transposons are identified as part of a class, family, and subfamily, and are annotated at each individual insertion with their location in the genome. The main classes are long interspersed elements (LINEs), short interspersed elements (SINEs), long-terminal repeats (LTRs), and DNA elements. Unlike the transcriptome, the genomic complement of repetitive elements is highly species specific. For example, although both the mouse and human genomes consist of approximately 45% repetitive DNA, the human genome has over 1.1 million Alu elements, a primate specific short interspersed element of the class SINE, and no active endogenous retroviruses (class ERV), while the most active element in the mouse genome is an ERV of the family IAP and contains no Alu elements (although there is a similar element of the family B1). Approximately 20% of the mouse and human genomes are derived from long interspersed nuclear elements (class LINE). The remaining most common classes of repetitive DNA in both genomes are satellites, long terminal repeats (LTRs), DNA elements, and simple repeats. Following classes, families of transposons are identified based on homology and are named according to their parent class (e.g., LINE1, LINE2, ERVL, ERVL-MaLR, etc.). Subfamilies are the smallest groupings following similar naming patterns (e.g., AluYa5, AluYa8, etc.). Finally, individual repeat insertions are identified by their genomic location (e.g., AluYa5_chr15:52525124-52525577 in hg19).
Bisphenol A (BPA) is a high production volume chemical with endocrine disrupting properties. Recent investigations by us and others have found strong support for BPA as an epigenetic modifier in both perinatal and life course exposures in both mouse and humans.Citation17-19 Thus, to develop and test a novel methodology for identifying environmentally labile repetitive elements, we examined developmental BPA exposure in mouse, where BPA was added to maternal chow, and in human fetal liver stratified by natural BPA exposure, to examine the effect on transposon methylation.
Here we report a novel adaptation of next-generation sequencing data analysis to determine BPA's effect on classes, families, and subfamilies of transposon methylation, as well as its effects at the level of individual transposon methylation across exposure groups. While RNA-seq has been used in mouse to find transposons that are differentially methylated,Citation14 our method captures differential methylation independent of transcription and we examined both mouse and human samples. Our experimental design involved the digestion of DNA with methylation sensitive enzymes to enrich for GC rich regions and highly methylated CpG sites. The differential digestion followed by next-generation sequencing allowed read-counts to be used as a proxy for genomic methylation. Our pipeline of alignment to the genome followed by differential read-count analysis adapted from RNA-seq methods revealed DNA methylation differences in transposons (). By classifying the usually discarded multi-mapped reads using RepeatMasker, we identified the class, family, and subfamily of these reads and determined overall differences between exposure groups. Additionally, by selecting reads that span unique mappable regions and repetitive transposon regions, we determined methylation differences at unique transposon insertion loci. We report the first known group of over 1250 transposons responding to BPA exposure by differential DNA methylation in humans. We also recapitulate the discovery of the mouse CabpIAP metastable epiallele associated with a differentially methylated IAP transposon and report 19 transposons that respond to BPA exposure by modulating DNA methylation.
Results
MethylPlex efficiency
In order to detect differential DNA methylation in an unbiased manner using next-generation sequencing, we first enzymatically enriched genomic DNA for GC rich regions and methylated CpG sites. After sequencing, the enrichment efficiency was determined by comparison to reference genome CpG content. Sequenced reads identified as derived from repetitive DNA were compared across treatment groups as proxies for differential DNA methylation in classes, families, and subfamilies of repetitive DNA.
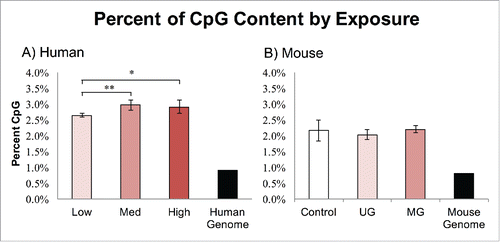
In mouse, 406,559,087 reads were sequenced covering 32 Gb across 12 samples. In human, 1,836,190,593 reads were sequenced covering 165 Gb across 18 samples. MethylPlex treatment results in increased methyl-CpG and high GC content; therefore, greater read counts correspond to higher methylation. MethylPlex enrichment increased CpG dinucleotide frequency from 0.80% in the reference mouse genome to an average of 2.17% in the mouse sequencing reads and from reference human genomic count of 0.91% CpG dinucleotide frequency to 2.84% in the human reads (). Only the human low BPA group showed a statistically significant reduction in efficiency, and the magnitude was small (P = 0.01 for low 2.65% vs. medium 2.97%, and P = 0.05 for low 2.65% vs. high 2.92%), while still being over 2.5x more enriched than the human genome. Individual lane enrichment efficiency is shown in Fig. S1. Overall, repetitive content in the sequencing reads averaged 48.74% in human samples, and 17.87% in mouse samples and were not significantly different across exposure groups (Fig. S2, Supplementary Files 4 and 5).
Figure 1. Flowchart for Experimental and Analysis Pipeline. Both humans and mice underwent MethylPlex enrichment prior to sequencing. Reads were Repeat Masked and used to identify group level differences, and aligned to reference genomes to identify individual differences in repeat DNA methylation. Asterisks denote significance, *P < 0.05, **P < 0.01.
Figure 2. Percent CpG Enrichment by Exposure. Both human and mouse samples exhibited increased CpG content after MethylPlex enrichment. (A) The human low BPA group was slightly but significantly lower than the medium group (P = 0.01), and the high group (P < 0.05). (B) The mouse samples exhibited no significant differences in CpG enrichment by exposure group.
Repeat class by exposure
Counts of individual reads consisting of >20 bp of annotated repetitive sequence allowed identification of class, family, and, usually, subfamily depending on length, and were used as a proxy to identify methylation differences by exposure group. P-values for differences between exposure levels for each class were calculated by Tukey's test. Identified repeats in humans were predominantly made up of SINEs (>40%), which displayed no significant differences across exposure groups. Human classes that were significantly differentially methylated by exposure group included LINEs (adjusted P = 0.014 low vs. medium), LTR elements (adjusted P = 0.008 medium vs. low), DNA elements (adjusted P < 0.001 medium vs. low, and P = 0.022 medium vs. high), and satellites (adjusted P = 0.043 low vs. high, and P = 0.006 low vs. medium) (). Importantly, all responding classes exhibited a non-monotonic response with the medium exposure level as hypomethylated compared to the low and high exposure levels.
Figure 3. Percent Reads by Repeat Class and Exposure. (A) A subset of classes in human was hypomethylated in the medium BPA group. (B) No classes were differentially methylated by exposure group in mice. Asterisks denote significance, *P < 0.05, **P < 0.01.
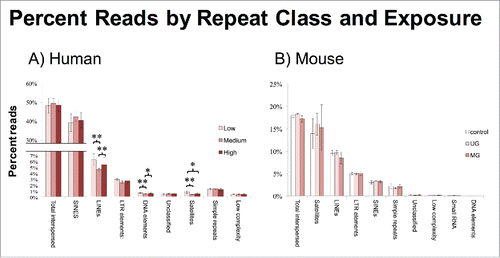
In mice, no classes of repeats were differentially methylated between exposure groups. Despite the lack of exposure differences, the MethylPlex treated mouse reads did exhibit a shift in enrichment by class type from the reference genome, an outcome explained by incomplete annotation of some repeat loci within the reference genome.
Disparity between genomic repeat coverage and enriched sequenced repeats
A large enrichment in reads annotated as satellites was observed, averaging 15.07% of reads per mouse, as compared to 0.06% annotated by RepeatMasker in the mm10 reference genome. In contrast, all other classes were depleted as compared to the mouse reference genome. LINEs were depleted, with an average of 9.29% of the reads, compared to 19.49% of the genome. SINEs were depleted, averaging 3.02% of the reads, compared to 7.35% of the genome. LTRs were depleted from 11.17% of the genome to 5.07% of the reads, and DNA transposons were depleted from 1.08% of the genome to 0.01% of the reads (Supplementary File 1). In humans, the total percentage of interspersed elements in the hg19 reference genome is 46% and the average across the 3 BPA groups is 48.74%. SINES were highly enriched, as expected, given their high CpG content, increasing from 13% in the genome to an average of 40.38% across all BPA groups. Conversely, LINEs were reduced from 21% in the genome to an average of just 4.82% of reads in the samples. Of the remaining classes, both LTR and DNA elements were reduced from a genomic 8.7% to average 2.39%, and a genomic 3% to average 0.49%, respectively. In stark contrast with the mouse, satellites were nearly unchanged, making up 0.45% of the human genome and 0.48% of the reads.
Repeat family by exposure
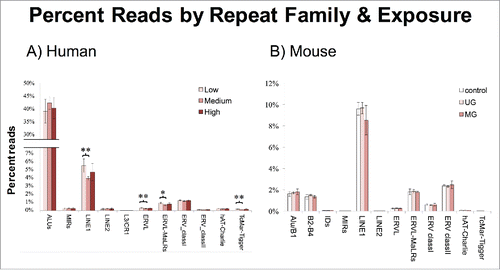
In humans, BPA exposure correlated with differential DNA methylation differences in the following repeat families: LINE1s (adjusted P = 0.008 low vs. medium), ERVLs (adjusted P = 0.005 low vs. medium), ERVL-MaLRs (adjusted P = 0.018 low vs. medium) and TcMar-Tigger (adjusted P = 0.004 low vs. medium) (). Similar to the results for classes in humans, all families exhibiting significantly different DNA methylation across exposure groups were between the low and medium BPA groups; the increased mean methylation in the medium vs. high comparison did not reach significance in any of the families. The results in mouse also mirrored the mouse class response, with no families exhibiting differences in DNA methylation across exposure groups.
Figure 4. Percent Reads by Repeat Family and Exposure. (A) A subset of repeat families was hypomethylated in the medium BPA group. (B) No families were differentially methylated by exposure group in mice. Asterisks denote significance, *P < 0.05, **P < 0.01.
Repeat subfamily by exposure
There are over 7000 subfamilies of human repeats described in the Repbase database. After filtering to remove simple repeats and subfamilies with <10 assignments in any group, the remaining 955 human subfamilies were analyzed. Only 15 subfamilies were significantly differentially methylated across BPA exposure groups. All 15 were in the Alu family and were relatively young at <44 million years old (). Ages were assigned according to previous reports (Citation20, Citation21). In contrast to the class and family results, subfamilies did not act in a consistent non-monotonic fashion. Element subfamilies that were significantly differentially methylated across all 3 levels varied in pattern. For example, AluSq increased in methylation from low to medium BPA, and decreased from medium to high with no significant difference between low and high. In the opposite pattern, AluSx decreased from low to medium and increased from medium to high, and was significantly increased between low and high as well. More information on directionality of change across exposure groups is available in . Interestingly, none of the subfamilies responded in the same direction. In mouse, 755 subfamilies were analyzed after filtering. Only one subfamily responded to BPA exposure, GSAT_MM, which was hypermethylated in the both the UG and MG groups compared to the control group ().
Table 1. Repeat subfamilies differentially methylated by exposure.
Individual transposon insertion DNA methylation by exposure
In the previous analyses, transposons and other repetitive DNA were examined in terms of their evolutionary relatedness, independently of their location within the genome, and aggregating the response of thousands to hundreds of thousands of elements to BPA exposure. In order to identify individual transposon insertions that may be especially sensitive to BPA exposure, we used a subset of reads that consisted of partial transposon sequence to measure the reads derived from specific locations in the genome.
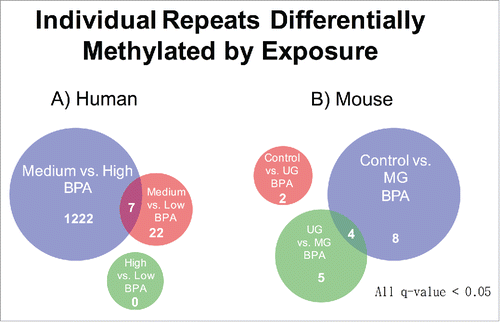
Counts of individual reads consisting of >20 bp of annotated repetitive sequence and >20 bp of uniquely mapped sequence were used as a proxy for locus specific methylation differences (). An average of 3.80% (1.8 billion) of reads passed these criteria in human, and 1.65% (101 million) in mice. The lower percentage and total number of passing reads in mice is due to the shorter read length (68 bp in mice vs. 90 bp in humans) and lower read depth for the mouse samples (GAIIx in mice vs. Hi-seq in humans). The identified reads were aligned to one of 5,298,092 unique repeat-associated loci across the human genome or 5,147,712 loci across the mouse genome. In humans, between the medium and high BPA groups, 1229 loci containing a transposon with significantly different DNA methylation were identified after correcting for multiple testing via FDR (q-value < 0.05) (, Supplementary File 6). For the remaining comparisons, 29 (medium vs. low) and 0 (high vs. low) transposons with differential DNA methylation were identified. In mice 12 (control vs. MG), 9 (UG vs. MG), and 2 (control vs. UG) differentially methylated transposons were observed (, Supplementary File 7). Two of the top hits in control vs. MG were intronic to the Cdk5rap1 gene, directly proximal to the previously known metastable transposon also intronic to this gene, called CabpIAP (Fig. S3). These loci are of the type B1_Mus2, and RMER12B, respectively.
Figure 5. Individual Transposon Detection Pipeline. Both mouse and human reads were filtered for reads containing partial unique and partial repetitive sequence. The unique sequence was mapped to the reference genome and read count differences corresponding to DNA methylation differences were tested for significance with EdgeR.
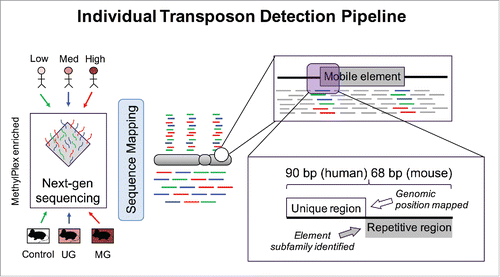
Figure 6. Individual Repeats Differentially Methylated by Exposure. (A) Number of human transposon insertions exhibiting significantly different read counts across BPA exposure groups. (B) Mouse transposon insertions exhibiting differential read counts across BPA treatment groups.
With respect to the genomic distribution of these elements, counts by genomic location are displayed in . Approximately 36–46% of these elements were located within introns in the human genome and 35-50% in mouse. Statistical deviation from expected genomic repeat distributions were only calculated for the human medium vs. high group as the other comparison counts were too low to draw significant inferences (Fig. S4). Within this comparison, an interesting pattern emerged relative to the general repetitive element abundance in the human genome. In the human medium vs. high group, more elements were found within promoters, 1 to 5 kb proximal to genes, or within introns, while intergenic regions were underrepresented in elements from this group.
Table 2. Genomic distribution counts of differentially methylated transposons.
Discussion
Here we have identified repetitive elements in both mouse and human that respond to perinatal BPA exposure using a novel approach with methylation-enriched next-generation sequencing data. The changes induced are not uniform, but rather impact specific classes, families, and subfamilies of repetitive elements. By mapping reads with both unique and repetitive content, a number of individual transposon insertions were identified that also exhibit significant exposure-dependent shifts in DNA methylation, including the first known transposons in humans that respond to perinatal BPA exposure.
The human and mouse exposure groups analyzed in this study span 4 orders of magnitude and overlap in the human high group (35.44 to 96.76 ng/g), and the mouse UG group (50 ng/g). In both species, the controls were an order of magnitude lower, human (non-detect to 0.83 ng/g) and mouse 0 ng/g. In mice, the high exposure group (MG) was 3 orders of magnitude higher (50,000 ng/g) than the highest measured human sample (96.76 ng/g). While the human results consistently found hypomethylation in some repeat classes, families, and subfamilies between the low and medium groups, the mouse results did not exhibit this pattern, despite the much higher top exposure dose. We did not analyze a mouse group equivalent to the human medium exposure group and therefore cannot extrapolate whether the mouse would respond similarly at the 35 to 97 ng/g range.
All repeat classes responding significantly to BPA exposure in human were hypomethylated at the medium exposure level, yet it was not a universal response and did not correlate with element frequency in the genome. For example, neither SINEs, a highly represented class, nor simple repeats, a lowly represented class, were differentially methylated. In contrast, all other classes representing transposons (LINEs, LTRs, DNA elements) as well as satellite repeats were hypomethylated in the medium group. Given the diversity of elements within these classes, we believe the hypomethylation seen at this BPA level is a global genomic response. As seen in our previous study focusing on mouse genic regions and BPA exposureCitation22,23, as well as in a number of studies reviewed by Vandenberg et al., non-monotonic dose responses are a frequent finding in endocrine disrupting compound studies.
The efficiency of MethylPlex GC and mCpG enrichment was measured by increased percentage CpG content compared to the reference genome. In humans, the low exposure group was slightly but significantly reduced in efficiency, yet was still 2.5-fold increased over the reference. The mouse groups showed no significant variation in CpG enrichment. More interestingly, while the total interspersed repeats found across all samples in human (48.74%) were similar in percentage to the reference genome (46%), mouse samples were made up of less than half the total interspersed repeat content in the reference genome (∼18% vs. ∼40%) (Fig S2). We believe this disparity is related to the differences in species-specific repeat complement, and may account for the lack of response to BPA at the class, family, and subfamily levels in mouse. Alternatively, the lower read depth and shorter read length used for the mouse groups may not have provided sufficient power to detect small magnitude responses over large numbers of elements as combined in larger groups (i.e., small numbers of highly responsive elements may not have exerted a large influence on the mean response of a class, family, or subfamily). MethylPlex treatment resulted in a non-uniform enrichment or depletion of certain classes of elements as expected by their sequence features. For example, human SINEs were greatly enriched compared to the reference genome, reflecting their increased CpG content. Unsurprisingly LINEs were reduced from 20% to 5% reflecting their AT richness and greater frequency outside of gene-rich regions, with a similar reduction also seen in the mouse. Conversely, the mouse SINEs were reduced in comparison to the genome, likely a consequence of their lost activity, and the less prominent role they play in Rodentia genomes, leading to their lower CpG density. While this approach allowed us to accurately test differential methylation between BPA exposure groups at single sites (or a group of sites), it did not allow for unbiased comparison between sites. Comparisons between sites (or families or classes) may be biased, because our coverage level is affected by both CpG density and mappability, both of which differ among sites and by class/family.
In mice, we previously found an increase in global DNA methylation with BPA exposure using the luminometric methylation assay (LUMA),Citation18 whereas here, in LINEs, often used as a proxy for global methylation, we see no difference in DNA methylation. This conflict arises from the fact that LUMA measures all CCGG sites within the genome. Similar disparities have been shown in mice fed high fat diets, where global DNA methylation was reduced in placentae as measured by LUMA, while neither LINE1 nor B1 elements were differentially methylated compared to control.Citation24 Therefore, we advise caution when extrapolating LINE1 methylation values as a proxy for global methylation. And while the progenitor LINE is an ancient paralog between mice and humans, its subsequent divergence and duplication within species clades and the characteristics of the host animal likely account for different responses we see here in mice and humans. Similarly, while LINE1 has been proven a useful tool in human epidemiology,Citation25 it is important to remember its limitations in assaying CpG methylation within the local genomic context.Citation26 We also note that the seeming extreme overrepresentation of mouse satellite reads in our sample data as compared to the reference mouse genome is an artifact of the incomplete assembly of highly repetitive centromeric regions. Mouse satellites annotated as GSAT_MM in RepeatMasker (γ-satellite) have been verified experimentally to exist in high copy number in these centromeres (but not in humans), and to contain highly conserved and highly methylated CpG sites.Citation27
Family level responses to BPA largely mirrored class differences, with no response in the mouse, and several human families exhibiting hypomethylation in the medium dose. These findings are unsurprising since most classes are made up predominantly of a single family. We note that LINE1 responded in human, while LINE2 elements were unaffected; this may be a consequence of their ancient heritage, ceasing transposition prior to the radiation of mammals. Interestingly, the family containing the most recently active endogenous retrovirally derived subfamily (HERV-K) is in the ERVL family, in which methylation was influenced by BPA exposure. The remaining LTR elements in the human reacted differently to BPA, with ERVL-MaLRs (mammalian apparent LTRs) becoming differentially methylated, while all other ERVs (Class I and II), were unaffected. Given the diverse origin and evolutionary history of ERVs in humans,Citation28 further investigation is warranted to determine why only a single family is environmentally responsive. Finally, the TcMar-Tigger family (an ancient and diverse group of short repeats) made up only a small fraction of reads (0.16%), and showed DNA methylation hypomethylation in the medium group. These ancient elements move via cut-and-paste and are phylogenetically and functionally distant from other transposons; however, they do contain transposase coding regions.Citation29 Given these results, we conclude that environmental response is not predicated on phylogenetic similarity, family age or activity, or method of replication.
Repbase classifies over 7000 subfamilies as the smallest grouping of related repeats.Citation30 The human subfamily story is intriguing in that only 15 subfamilies were detected as interacting with BPA, and all were Alu elements less than 44 million years old. Young elements have long been suspected, and more recently have been shown, to be more sensitive to stochastically differential methylation and more likely to lose their silencing than older elements.Citation31,32 Indeed, in a previous study we found that element age predicts DNA methylation level in mouse IAP elements.Citation10 While Alu elements are the most numerous transposon in the human genome, frequency cannot explain why element age (as indicated by subfamily) corresponds to BPA induced DNA methylation changes. We interpret these findings as a consequence of the relatively high CpG content in the medium to young subfamilies of Alu elements; thus, these elements were more likely to harbor responding CpGs, and note the known reduction in CpG content and decay rates in the older elements.Citation33 Furthermore, given the higher density of Alu elements found in gene rich regions,Citation34 any environmentally induced change in DNA methylation risks altering gene expression on a global scale. However, our results underscore that global DNA methylation analyses will be subject to subfamily specific effects if assays are not designed to pool a broad array of target elements, as also noted by other researchers.Citation35,36 In mice, unsurprisingly, the single subfamily detected with a significant BPA response is a member of the most frequently appearing class, a mouse major satellite. We believe that increased depth of sequencing would reveal additional subfamilies concordant with increased power. These results support the idea that BPA influences the transposome in a unique, species-specific, and wide-ranging way. We and others have shown that BPA influences the methylation of genic regions; however, these regions are conserved to a high degree across species. We are cognizant of the fact that transposon complement differs tremendously between mice and humans,Citation37 and, therefore, expect species-specific responses.
Until now, only a small number of transposons were known as epigenetically modifiable by environmental exposure in mice, and none were known in humans. All known instances of individual mouse transposons acting as epialleles are the result of IAP insertions, a type of ERV not present in the human genome.Citation3,9,38,39 Without a candidate class or subfamily to target in human, we chose to expand our search globally for differentially methylated transposons. In order to probe individual repetitive element insertions whose methylation status varied with BPA exposure, we extracted reads that spanned both unique, mappable, and repetitive transposon identified regions. In human samples, we were able to detect over 1000 individual elements with significant differential methylation between the medium and high groups, and no elements with differences between the low and high groups. This mirrors the group level analyses found in the classes and families, where the low and high groups were not significantly different (with the exception of human satellites). Fully half of the transposons different between the medium and high groups were Alu while the rest were spread across various families with no discernible pattern. Likewise, no pattern was found in the identity of elements different between low and medium exposure groups. Nevertheless, these elements represent the first candidate transposon loci that respond to environmental exposure by modulating DNA methylation. Our results in mice proved to be a good validation of our approach, as 2 of the 19 significantly different loci were found 3 kb proximal to a known epiallele, the IAP at the Cabp locus.
The genomic distribution of loci in one inter-group comparison (medium vs. high BPA) was compared to the overall genomic distribution of repeat elements in the human genome. While only this group provided sufficient regions for accurate statistics, the results were remarkable. We found a major deviation in the distribution of the 1229 elements from the differentially methylated regions between medium vs. high BPA. Repeat elements were preferentially found near genes, specifically within introns, promoters, and the region 1 to 5 kb proximal to a TSS and depleted in intergenic regions. Our observation of preferential variable repeat elements within control regions near genes is suggestive of a functional response in gene expression following BPA exposure. If transposons in these regions are particularly labile to the environment, their altered DNA methylation may be more likely to cause gene expression changes in their host genes and suggests that BPA's effects on transposon chromatin structure preferentially affects elements nearby genes. Using our novel approach, we have leveraged next-generation sequencing data to identify the first known transposons in humans that respond to BPA exposure. Though transposons are not usually thought of in terms of mediators between environmental insults and gene expression, our findings reveal that transposons, as a class and individually, do respond and may affect the expression of nearby genes. This method can be applied to discover other environmentally labile loci or transposons. In the future, these elements may serve as biomarkers or signatures of current and historical exposure. Finally, while other studies have estimated repeat enrichment, correlated to histone modifications, for example;Citation40 to our knowledge, this is the first genome-wide study examining the effects of an ubiquitous environmental agent (BPA) on the DNA methylation of repeats in a group context and individually.
Materials and methods
Mouse and human liver samples
Mice were obtained from a colony carrying the agouti viable yellow (Avy) epigenetic allele and the non-agouti (a) allele in a forced heterozygous state.Citation41 This colony consists of a/a and Avy/a congenic mice with 93% sequence identity to the C57Bl/6J strain.Citation42 Developmental BPA exposure and post-natal-day (PND) 22 liver extraction were previously described.Citation18 Briefly, virgin a/a dams were randomly assigned to one of 3 diets (base AIN-93G diet 95092 with 7% corn oil substituted for 7% soybean oil; Harlan Teklad, Madison, WI) supplemented with 0 BPA (control), 50 ng BPA/g (UG group), or 50,000 ng BPA/g (MG group). Dams were provided respective diets 2 weeks prior to mating with Avy/a males, and continued on diet through weaning. For this study, livers from PND 22 a/a “wild type” animals (n = 12) were collected for DNA extraction with a standard phenol-chloroform isolation protocol. The Avy/a offspring were excluded from this study because their known health disparities confound analysis. Mice from each treatment group were composed as follows: Control (2 male, 2 female), 50 ng BPA/g diet (2 male, 2 female), and 50,000 ng BPA/g diet (1 male, 3 female). Previously, we reported that perinatal exposure to these diets resulted in post-natal-day 22 liver total BPA concentrations (free plus conjugated) of 2.02 ng for the 50 ng/g BPA diet group and 441 ng/g for the 50,000 ng/g BPA diet.Citation18 Animals used in this study were maintained in accordance with the Guidelines for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and were treated humanely and with regard for alleviation of suffering. The study protocol was approved by the University of Michigan Committee on Use and Care of Animals.
As previously described,Citation18,22,43 human fetal liver samples (n = 18) were obtained from the NIH-funded University of Washington Birth Defects Research Laboratory fetal biobank (2R24 HD000836-47) and were flash frozen and stored in polycarbonate-free tubes at −80°C prior to DNA extraction and BPA quantitation. Total BPA concentrations (free plus conjugated) were measured by the Kannan Laboratory at the Wadsworth Center (New York State Department of Health) using 0.5 g of human fetal liver tissue with a high performance liquid chromatographer (HPLC) coupled with an API 2000 electrospray triple-quadruple mass spectrometer (ESI-MS/MS). To control for contamination in the sample preparation process, a negative blank of BPA-free water was processed identically as the human samples and resulted in free and conjugated BPA levels below the limit of quantification. For calculation of mean and median BPA concentrations, liver BPA levels below the limit of quantification were assigned a value of 0.071, which was estimated by dividing the LOQ (0.1 ng/g) by the square root of 2. Samples were stratified by BPA exposure into 3 groups of 6 samples each: low (total BPA ranging from non-detect to 0.83 ng/g), medium (3.5 to 5.79 ng/g), and high (35.44 to 96.76 ng/g) exposure groups. Samples met the criteria for IRB exception for human subjects research (UM IRB Exemption: HUM00024929) as no identifying clinical data was available on subjects except for gestational age and occasionally sex and race.
M-NGS library generation
Mouse and human DNA samples were enriched in methylated CpG sites using the MethylPlex kit from Rubicon Genomics Inc., Ann Arbor, MI (Patent Number US 2007/0031858 A1) that enables enrichment by methyl-sensitive restriction enzymes followed by depletion of low GC regions by enzymatic treatment. Specifically, MethylPlex relies upon thermo-selective denaturation of low GC regions in combination with primer ligation to double stranded, high GC regions and enzymatic degradation of single-stranded DNA. Previously MethylPlex combined with next-generation sequencing (M-NGS) has successfully identified regions of altered methylation in prostate cell lines and tissues, and genic regions in human fetal liverCitation22 and mouse liver DNA exposed to BPA.Citation17,44 As with RRBS, another enzyme-based method, we expect small fragment size <300 bp to include most CpG rich regions.Citation45 Briefly, for each sample, 50 nanograms of DNA were digested with methyl-sensitive enzymes and the resulting fragments ligated to synthetic adaptor sequences. Fragments were then amplified with PCR using universal primers. The resulting long fragments enriched in methylated DNA were subjected to secondary enzyme treatment to digest non-GC-rich DNA and underwent a second round of re-amplification using universal primers. After purification, DNA was prepared for sequencing by enzymatic digest to remove adaptor sequences and directly incorporated into the Illumina genomic DNA sequencing sample preparation kit procedure (Illumina Inc., San Diego, CA) at the end repair step, skipping the nebulization process. The samples were ligated to Illumina adaptors and run on 2% agarose gel and fragments in the 400 bp range were excised, gel-purified, and quality-checked on a Bioanalyzer (Agilent Technologies, San Diego, CA). Library material (10 nmol) was used to prepare flow-cells with approximately 30,000 clusters per lane, with the sequencing performed by the University of Michigan DNA Sequencing Core. For mouse, 80 cycles of single-end reads were run on an Illumina GAIIx and analyzed by the Illumina analysis pipeline. Mouse yielded an average 34 million reads per sample for 12 samples analyzed (32 Gb sequenced). For human, 100 cycles of single-end reads were run on an Illumina Hi-Seq and analyzed by the Illumina pipeline. Human samples yielded an average of 104 million reads each across 18 samples (165 Gb sequenced). Sequences were trimmed to 68 bp for mouse and 90 bp for human.
Global repeat analysis
A flowchart of the analysis pipeline is presented in . Sequencing reads from all samples were processed in the following pipeline with commands available in Supplementary File 1. Fastq files were converted to fasta format and split into 100 million line chunks for processing. RepeatMasker version open-4.0.5 was run against the Repbase database version (20090604) with the options “RepeatMasker -qq -xsmall -species (human or mouse as appropriate) <file>.” Nucleotide frequency analysis for CpG content was performed with galculator (http://www.bioinf.uni-leipzig.de/Software/galculator/). Repeat class and family frequencies were calculated with a series of awk scripts. Repeat subfamily analysis was preceded by filtering all simple repeats, and repeats with N/A counts or < 10 in any treatment group. ANOVA followed by Tukey's adjustment differentiated subfamilies with statistically significant differences (P < 0.05) by BPA exposure.
Individual repeat analysis
We adapted an RNA-seq pipeline as most appropriate for read count analysis of MethylPlex processed DNA for detecting differentially methylated repeats. Each read lane, corresponding to one individual, was processed to identify reads consisting of at least 20 bp of unique sequence (non-repetitive as flagged by RepeatMasker in uppercase), and 20 bp of repetitive sequence (repetitive as flagged by RepeatMasker in lowercase) by a custom perl script (Supplementary File 2). Reads were unified with their corresponding Repeat ID by merging sequencer masked file with the RepeatMasker. out file on the read identifier using “sharktopus.pl.” The combined output files were stripped of lowercase sequence to improve alignment of the unique regions to the genome. STAR aligner was used to align the unique regions of each read to the reference genome (Human hg19 or Mouse mm10) with the following parameters, “STAR –genomeLoad LoadAndKeep –outFilterMultimapNmax 1 –genomDir <genomeDir> –runThreadN <n-1 max threads> –readFilesIn <inputfile> –outFileNamePrefix <inputfile>.Star”.Citation46 The resulting sam files were sorted and converted and indexed into bam files using samtools.
Individual repeat analysis read count
Read count analysis allowed for detection of statistically significant counts at each locus as a proxy for genome methylation status. The RepeatMasker track was downloaded from UCSC Table Browser in GTF formatCitation47 and prepared for alignment by expanding the flanking region of each feature by ± 71 bp using awk (Supplementary File 1). Counts were generated per locus using featureCounts as part of the Subread packageCitation48 with the following parameters, “featureCounts -O -a <ucsc-repeat-track-expanded.gtf> -t -exon -g repeat_id -o <output.count.file>”.
Individual repeat analysis of read counts with edgeR
EdgeR was used to generate lists of statistically significant repeats based on read counts at each locus throughout the genome (Supplementary File 3).Citation49 Filtering steps were included to remove loci with low counts per million in an unbiased fashion. For human data which contained an average of 4 million reads per sample and 18 samples overall, we filtered out loci containing less than 5 reads in at least any 4 samples. For mouse data which contained an average of 0.5 million reads per sample and 12 samples overall, we filtered out loci containing less than 10 reads in at least any 3 samples. Generalized linear models were fit to account for the multi-level exposure analysis in edgeR using the glmFit function with significance calculated using the false discovery rate (q-value). Resulting top hit loci were visualized in SeqMonk (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/).
Individual repeat genomic distribution
Loci identified as significantly differentially methylated by edgeR were analyzed for their distribution throughout the hg19 and mm10 genomes for each species. The R software package ‘annotatr’ was used to match by overlap and summarize the individual transposon insertions by chromosomal location with the genomic features listed in (https://github.com/rcavalcante/annotatr).Citation50 Totals in are higher than each pairwise comparison's sum due to some repeat loci overlapping more than one genomic feature. Comparisons to the repeat distribution in the human genome were performed by extracting the hg19 rmask table from UCSC Genome Browser, i.e., a list of all known repeats with associated chromosomal locations. This list was annotated in the same manner as experimental results. P-values were calculated using the chi-square test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the University of Michigan DNA Sequencing Core for assistance with sample processing, Rubicon Genomics (Ann Arbor, MI) for access to MethylPlex technology, Dr. Kurunthachalam 710 Kannan at the Wadsworth Center, New York State Department of Health for liver tissue bisphenol A measurement, and the University of Washington Laboratory for the Study of Human Embryology (2R24 HD000836-47) for human tissue samples. We also thank Raymond Cavalcante for assistance with the “annotatr” software package.
Author's contributions
510 CF conceived the study. CF, MAS, and DCD designed the study. OSA performed mouse exposures. MSN led human BPA exposure analysis. CF, TRJ, and JK performed the DNA extraction and sequence preparation. CF and MAS designed and CF performed the bioinformatics pipeline. CF, MAS, and DCD 515 analyzed the data. CF drafted the manuscript and created the figures with editorial guidance from all authors.
KEPI_A_1183856_supplementary_data.zip
Download Zip (2.4 MB)Funding
This work was supported by NIH-NIEHS grant R01 ES017524 and the Michigan Lifestage Environmental Exposures and Disease (M-LEEaD) 700 NIEHS Center of Excellence P30 ES017885, Support for CF and MSN was provided by Institutional Training Grant T32 ES007062. Support for CF was also provided by NIH Grant K99 ES022221/R00 ES022221 (https://10 C. FAULK ET AL.www.niehs.nih.gov). The funders had no role in study design, data collec-705 tion and analysis, decision to publish, or preparation of the manuscript.
Related Research Data
References
- Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua A a., Suh HH, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med 2009; 179:572-8; PMID:19136372; http://dx.doi.org/10.1164/rccm.200807-1097OC
- Yauk C, Polyzos A, Rowan-Carroll A, Somers CM, Godschalk RW, Van Schooten FJ, Berndt ML, Pogribny IP, Koturbash I, Williams A, et al. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc Natl Acad Sci U S A 2008; 105:605-10; PMID:18195365; http://dx.doi.org/10.1073/pnas.0705896105
- Dolinoy DC, Das R, Weidman JR, Jirtle RL. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr Res 2007; 61:30-7; PMID:17413847; http://dx.doi.org/10.1203/pdr.0b013e31804575f7
- Bernal AJ, Dolinoy DC, Huang D, Skaar DA, Weinhouse C, Jirtle RL. Adaptive radiation-induced epigenetic alterations mitigated by antioxidants. FASEB J 2013; 27:665-71; PMID:23118028; http://dx.doi.org/10.1096/fj.12-220350
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A [Internet] 2008; 105:17046-9; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2579375&tool=pmcentrez&rendertype=abstract; PMID:18955703; http://dx.doi.org/10.1073/pnas.0806560105
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev [Internet] 2002; 117:15-23; Available from: http://www.sciencedirect.com/science/article/pii/S0925477302001818; PMID:12204247; http://dx.doi.org/10.1016/S0925-4773(02)00181-8
- Faulk C, Dolinoy DC. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics 2011; 6:791-7; PMID:21636976; http://dx.doi.org/10.4161/epi.6.7.16209
- Dolinoy DC. The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. NIH Public Access 2008; 66:1-8
- Druker R, Bruxner TJ, Lehrbach NJ, Whitelaw E. Complex patterns of transcription at the insertion site of a retrotransposon in the mouse. Nucleic Acids Res [Internet] 2004; 32:5800-8; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=528799&tool=pmcentrez&rendertype=abstract; PMID:15520464; http://dx.doi.org/10.1093/nar/gkh914
- Faulk C, Barks A, Dolinoy DC. Phylogenetic and DNA methylation analysis reveal novel regions of variable methylation in the mouse IAP class of transposons. BMC Genomics [Internet] 2013; 14:48; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3556122&tool=pmcentrez&rendertype=abstract; PMID:23343009; http://dx.doi.org/10.1186/1471-2164-14-48
- Andreotti G, Karami S, Pfeiffer RM, Hurwitz L, Liao LM, Weinstein SJ, Albanes D, Virtamo J, Silverman DT, Rothman N, et al. LINE1 methylation levels associated with increased bladder cancer risk in pre-diagnostic blood DNA among US (PLCO) and European (ATBC) cohort study participants. Epigenetics 2013; 9:404-15; PMID:24316677; http://dx.doi.org/10.4161/epi.27386
- Castro-Diaz N, Ecco G, Coluccio A, Kapopoulou A, Yazdanpanah B, Friedli M, Duc J, Jang SM, Turelli P, Trono D. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev [Internet] 2014; 28:1397-409; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24939876; PMID:24939876; http://dx.doi.org/10.1101/gad.241661.114
- Casa V, Gabellini D. A repetitive elements perspective in Polycomb epigenetics. Front Genet 2012; 3:1-16; PMID:22303408; http://dx.doi.org/10.3389/fgene.2012.00199
- Dunican DS, Cruickshanks HA, Suzuki M, Semple C a, Davey T, Arceci RJ, Greally J, Adams IR, Meehan RR. Lsh regulates LTR retrotransposon repression independently of Dnmt3b function. Genome Biol [Internet] 2013; 14:R146; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24367978/nhttp://www.ncbi.nlm.nih.gov/pmc/articles/PMC4054100/pdf/gb-2013-14-12-r146.pdf; PMID:24367978; http://dx.doi.org/10.1186/gb-2013-14-12-r146
- Dolinoy DC, Weidman JR, Waterland R a., Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect [Internet] 2006; 114:567-72; Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1440782/\nhttp://www.ehponline.org/ambra-doi-resolver/10.1289/ehp.8700; PMID:16581547; http://dx.doi.org/10.1289/ehp.8700
- Faulk C, Liu K, Barks A, Goodrich JM, Dolinoy DC. Longitudinal epigenetic drift in mice perinatally exposed to lead. Epigenetics [Internet] 2014; 9:934-41; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24786859; PMID:24786859; http://dx.doi.org/10.4161/epi.29024
- Kim JH, Sartor MA, Rozek LS, Faulk C, Anderson OS, Jones TR, Nahar MS, Dolinoy DC. Perinatal bisphenol A exposure promotes dose-dependent alterations of the mouse methylome. BMC Genomics [Internet] 2014; 15:30; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3902427&tool=pmcentrez&rendertype=abstract; PMID:24433282; http://dx.doi.org/10.1186/1471-2164-15-30
- Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, Weinhouse C, Rozek LS, Dolinoy DC. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen [Internet] 2012; 53:334-42; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3570056&tool=pmcentrez&rendertype=abstract; PMID:22467340; http://dx.doi.org/10.1002/em.21692
- Kim JH, Rozek LS, Soliman AS, Sartor MA, Hablas A, Seifeldin IA, Colacino JA, Weinhouse C, Nahar MS, Dolinoy DC. Bisphenol A-associated epigenomic changes in prepubescent girls: a cross-sectional study in Gharbiah, Egypt. Environ Health [Internet] 2013; 12:33; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3655072&tool=pmcentrez&rendertype=abstract; PMID:23590724; http://dx.doi.org/10.1186/1476-069X-12-33
- Kapitonov V, Jurka J. The age of Alu subfamilies. J Mol Evol [Internet] 1996; 42:59-65; Available from: http://www.ncbi.nlm.nih.gov/pubmed/8576965; PMID:8576965; http://dx.doi.org/10.1007/BF00163212
- Price AL, Eskin E, Pevzner PA. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res [Internet] 2004; 14:2245-52; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=525682&tool=pmcentrez&rendertype=abstract; PMID:15520288; http://dx.doi.org/10.1101/gr.2693004
- Faulk C, Kim JH, Jones TR, McEachin RC, Nahar MS, Dolinoy DC, Sartor MA. Bisphenol A-associated alterations in genome-wide DNA methylation and gene expression patterns reveal sequence-dependent and non-monotonic effects in human fetal liver. Environ Epigenetics [Internet] 2015; 1; dvv006; Available from: http://eep.oxfordjournals.org/content/1/1/dvv006.abstract; http://dx.doi.org/10.1093/eep/dvv006
- Vandenberg LN. Non-monotonic dose responses in studies of endocrine disrupting chemicals: bisphenol a as a case study. Dose Response [Internet] 2014; 12:259-76; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4036398&tool=pmcentrez&rendertype=abstract; PMID:24910584; http://dx.doi.org/10.2203/dose-response.13-020.Vandenberg
- Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, Lesage J, Boudadi E, Gross M-S, Taurelle J, Vigé A, et al. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS One [Internet] 2010; 5:e14398; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3006175&tool=pmcentrez&rendertype=abstract; PMID:21200436; http://dx.doi.org/10.1371/journal.pone.0014398
- Xiao-Jie L, Hui-Ying X, Qi X, Jiang X, Shi-Jie M. LINE-1 in cancer: multifaceted functions and potential clinical implications. Genet Med [Internet] 2015 [cited 2015 Nov 17]; 18:431-9; Available from: http://www.ncbi.nlm.nih.gov/pubmed/26334179; PMID:26334179
- Ruiz-Hernandez A, Kuo C-C, Rentero-Garrido P, Tang W-Y, Redon J, Ordovas JM, Navas-Acien A, Tellez-Plaza M. Environmental chemicals and DNA methylation in adults: a systematic review of the epidemiologic evidence. Clin Epigenetics [Internet] 2015; 7:55; Available from: http://www.clinicalepigeneticsjournal.com/content/7/1/55\nhttp://www.ncbi.nlm.nih.gov/pubmed/25984247; PMID:25984247; http://dx.doi.org/10.1186/s13148-015-0055-7
- Abdurashitov M a, Chernukhin V a, Gonchar D a, Degtyarev SK. GlaI digestion of mouse gamma-satellite DNA: study of primary structure and ACGT sites methylation. BMC Genomics 2009; 10:322; PMID:19607729; http://dx.doi.org/10.1186/1471-2164-10-322
- Crichton JH, Dunican DS, MacLennan M, Meehan RR, Adams IR. Defending the genome from the enemy within: Mechanisms of retrotransposon suppression in the mouse germline. Cell Mol Life Sci 2014; 71:1581-605; PMID:24045705; http://dx.doi.org/10.1007/s00018-013-1468-0
- Smit a F, Riggs a D. Tiggers and DNA transposon fossils in the human genome. Proc Natl Acad Sci U S A 1996; 93:1443-8; PMID:8643651; http://dx.doi.org/10.1073/pnas.93.4.1443
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462-7; PMID:16093699; http://dx.doi.org/10.1159/000084979
- Reiss D, Zhang Y, Rouhi A, Reuter M, Mager DL. Variable DNA methylation of transposable elements: The case study of mouse early transposons. Epigenetics 2010; 5:68-79; PMID:20083901; http://dx.doi.org/10.4161/epi.5.1.10631
- Reiss D, Mager DL. Stochastic epigenetic silencing of retrotransposons: Does stability come with age?. Gene 2007; 390:130-5; PMID:16987613; http://dx.doi.org/10.1016/j.gene.2006.07.032
- Xing J, Hedges DJ, Han K, Wang H, Cordaux R, Batzer M a. Alu element mutation spectra: molecular clocks and the effect of DNA methylation. J Mol Biol [Internet] 2004; 344:675-82; Available from: http://www.ncbi.nlm.nih.gov/pubmed/15533437; PMID:15533437; http://dx.doi.org/10.1016/j.jmb.2004.09.058
- Deininger P. Alu elements: know the SINEs. Genome Biol [Internet] 2011; 12:236; Available from: http://www.ncbi.nlm.nih.gov/pubmed/22204421; PMID:22204421; http://dx.doi.org/10.1186/gb-2011-12-12-236
- Hoad G, Harrison K. The Design and Optimization of DNA Methylation Pyrosequencing Assays Targeting Region-Specific Repeat Elements. Methods Mol Biol [Internet] 2015; Available from: http://www.ncbi.nlm.nih.gov/pubmed/26246355; PMID:26246355
- Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat Genet [Internet] 2013; 45:836-41; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3695047&tool=pmcentrez&rendertype=abstract; PMID:23708189; http://dx.doi.org/10.1038/ng.2649
- Huang CRL, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet [Internet] 2012; 46:651-75; Available from: http://www.annualreviews.org/doi/abs/10.1146/annurev-genet-110711-155616; PMID:23145912; http://dx.doi.org/10.1146/annurev-genet-110711-155616
- Juriloff DM, Harris MJ, Mager DL, Gagnier L. Epigenetic mechanism causes Wnt9b deficiency and nonsyndromic cleft lip and palate in the A/WySn mouse strain. Birth Defects Res A Clin Mol Teratol [Internet] 2014; 100:772-88; Available from: http://www.scopus.com/inward/record.url?eid=2-s2.0-84916219777&partnerID=tZOtx3y1; PMID:25257647; http://dx.doi.org/10.1002/bdra.23320
- Ekram MB, Kang K, Kim H, Kim J. Retrotransposons as a major source of epigenetic variations in the mammalian genome. Epigenetics 2012; 7:370-82; PMID:22415164; http://dx.doi.org/10.4161/epi.19462
- Day DS, Luquette LJ, Park PJ, Kharchenko P V. Estimating enrichment of repetitive elements from high-throughput sequence data. Genome Biol 2010; 11:R69; PMID:20584328; http://dx.doi.org/10.1186/gb-2010-11-6-r69
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol [Internet] 2003; 23:5293-300; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=165709&tool=pmcentrez&rendertype=abstract; PMID:12861015; http://dx.doi.org/10.1128/MCB.23.15.5293-5300.2003
- Weinhouse C, Anderson OS, Bergin IL, Vandenbergh DJ, Gyekis JP, Dingman MA, Yang J, Dolinoy DC. Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ Health Perspect 2014; 122:485-91; PMID:24487385
- Nahar MS, Liao C, Kannan K, Dolinoy DC. Fetal liver bisphenol A concentrations and biotransformation gene expression reveal variable exposure and altered capacity for metabolism in humans. J Biochem Mol Toxicol [Internet] 2013; 27:116-23; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3578057&tool=pmcentrez&rendertype=abstract; PMID:23208979; http://dx.doi.org/10.1002/jbt.21459
- Kim JH, Dhanasekaran SM, Prensner JR, Cao X, Robinson D, Kalyana-Sundaram S, Huang C, Shankar S, Jing X, Iyer M, et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res [Internet] 2011; 21:1028-41; Available from: http://genome.cshlp.org/cgi/doi/10.1101/gr.119347.110\npapers3://publication/doi/10.1101/gr.119347.110; PMID:21724842; http://dx.doi.org/10.1101/gr.119347.110
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc [Internet] 2011; 6:468-81; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21412275; PMID:21412275; http://dx.doi.org/10.1038/nprot.2010.190
- Dobin A, Davis C a, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics [Internet] 2013; 29:15-21; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3530905&tool=pmcentrez&rendertype=abstract; PMID:23104886; http://dx.doi.org/10.1093/bioinformatics/bts635
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res [Internet] 2004; 32:D493-6; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=308837&tool=pmcentrez&rendertype=abstract; PMID:14681465; http://dx.doi.org/10.1093/nar/gkh103
- Liao Y, Smyth GK, Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30:923-30; PMID:24227677; http://dx.doi.org/10.1093/bioinformatics/btt656
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139-40; PMID:19910308; http://dx.doi.org/10.1093/bioinformatics/btp616
- Cavalcante RG, Sartor MA. annotatr: Associating genomic regions with genomic annotations. bioRxiv [Internet] 2016; :1–9. Available from: http://biorxiv.org/lookup/doi/10.1101/039685