
ABSTRACT
The WNT signaling pathway has an essential role in colorectal carcinogenesis and progression, which involves a cascade of genetic and epigenetic changes. We aimed to analyze DNA methylation affecting the WNT pathway genes in colorectal carcinogenesis in promoter and gene body regions using whole methylome analysis in 9 colorectal cancer, 15 adenoma, and 6 normal tumor adjacent tissue (NAT) samples by methyl capture sequencing. Functional methylation was confirmed on 5-aza-2′-deoxycytidine-treated colorectal cancer cell line datasets. In parallel with the DNA methylation analysis, mutations of WNT pathway genes (APC, β-catenin/CTNNB1) were analyzed by 454 sequencing on GS Junior platform. Most differentially methylated CpG sites were localized in gene body regions (95% of WNT pathway genes). In the promoter regions, 33 of the 160 analyzed WNT pathway genes were differentially methylated in colorectal cancer vs. normal, including hypermethylated AXIN2, CHP1, PRICKLE1, SFRP1, SFRP2, SOX17, and hypomethylated CACYBP, CTNNB1, MYC; 44 genes in adenoma vs. NAT; and 41 genes in colorectal cancer vs. adenoma comparisons. Hypermethylation of AXIN2, DKK1, VANGL1, and WNT5A gene promoters was higher, while those of SOX17, PRICKLE1, DAAM2, and MYC was lower in colon carcinoma compared to adenoma. Inverse correlation between expression and methylation was confirmed in 23 genes, including APC, CHP1, PRICKLE1, PSEN1, and SFRP1. Differential methylation affected both canonical and noncanonical WNT pathway genes in colorectal normal-adenoma-carcinoma sequence. Aberrant DNA methylation appears already in adenomas as an early event of colorectal carcinogenesis.
Introduction
Colorectal cancer (CRC) is one of the most frequent cancers world-wide with an annual incidence of approximately 1 300 000 newly diagnosed cases, and a very high global mortality of more than 690 000/year.Citation1 Although more and more CRC-associated molecular alterations are determined, epidemiological data emphasize the necessity of further research of this disease. CRC can develop through several distinct molecular pathways (adenoma-carcinoma sequence, serrated neoplastic pathway or on the basis of long-standing inflammatory bowel disease) involving a cascade of genetic, epigenetic events affecting gene expression changes.Citation2-4 The analysis of these pathways can improve our understanding of colorectal carcinogenesis and can reveal potential diagnostic, prognostic, and therapeutic predictive markers.
Abnormalities of epigenetic regulation such as DNA methylation alterations can contribute to malignant transition.Citation5-6 It is well known that global hypomethylation and promoter hypermethylation of several tumor suppressor genes are characteristic in cancers.Citation7 Furthermore, with the advent of whole methylome analysis, several studies allude to the important role of methylation alterations in the transcribed regions of genes (gene body region)Citation7 with a remarkable proportion of differentially methylated CpG sites located thereCitation5-6 and influencing the regulation of gene expression.Citation8-9
The WNT signaling pathway (, Supplementary Table 1) has an essential role in colorectal carcinogenesis and progression, which is characterized by accumulation of genetic and epigenetic changes. The most known WNT pathway alterations, such as APC and AXIN2 inactivation by mutations or loss of heterozygosity, affect the APC-GSK3-Axin complex leading to β-catenin stabilization, thereby resulting in the constitutive activation of canonical WNT signaling in CRC.Citation10 Promoter hypermethylation of negative regulators of canonical WNT pathway like secreted frizzled-related proteins (SFRPs),Citation11-18 dickkopf family proteins (DKKs),Citation11,17,19-21 and WIF1 WNT inhibitory factorCitation11,17,21 has also been described in malignancies, including CRC. Methylation patterns of CRC and normal adjacent tissue samples were compared in most of the previous global methylation studies focusing on WNT signaling pathway.Citation20,22 Colorectal adenoma samples were involved in methylation analyses for selected WNT pathway genesCitation11,15,17,23,24 focusing mainly on promoter methylation level alterations. Serrated adenoma samples—representing another neoplastic pathway of CRC development—were also profiled in several previous methylation projects.Citation23,25-27
Table 1. Analyzed WNT pathway genes.
Illumina BeadChip technology is a frequently used method in global methylation studiesCitation22,28,29 and allows the determination of the methylation status of >485,000 CpG sites at single-nucleotide resolution. Next generation sequencing of the methylated DNA regions, such as methyl capture sequencing (MethylCap-seq), is another option for genome-wide methylation analysis for revealing novel differentially methylated regions (DMRs).Citation30,31 MethylCap-seq provides a more extensive overview of whole-genome methylation than BeadChip methylation arrays; however, it has lower resolution and sensitivity.Citation31
In this study, we aimed to analyze the most frequent genetic and global DNA methylation alterations of WNT pathway genes in parallel during colorectal normal-adenoma-carcinoma sequence progression. We tested both promoter and gene body DNA methylation alterations using MethylCap-seq and studied the potential regulatory role of the identified DMRs on mRNA expression of WNT signaling pathway genes. Approximately 15-20% of CRCs belong to the distinct molecular subtype of cancers called CpG island methylator phenotype (CIMP) with high degree of DNA methylation of certain genes (CIMP-high).Citation7 However, the DNA methylation changes also affect CIMP-negative (CIMP-zero or CIMP-low) CRCs, which represent the majority (approximately 75%) of sporadic CRCs.Citation7,17 In this study, we focused on CIMP-negative CRCs and their precancerous lesions in order to find common DNA methylation alterations of WNT pathway genes typical in development and progression of sporadic colorectal cancer.
Results
Determination of microsatellite instability (MSI) and CpG island methylation phenotype (CIMP) status
According to the results of immunohistochemical staining for MMR genes (MLH1, MSH2, MSH6, PMS2), all samples were found to be microsatellite stable (MSS). Using dichotomized CIMP status determination [CIMP-negative (CIMP-zero or CIMP-low) or CIMP-positive (CIMP-high)], all tumor samples were proven to be CIMP-negative, according to both classicCitation7,32 and Weisenberger CIMP status panel.Citation33
DNA methylator analysis of WNT signaling pathway genes
Methylation status of 160 WNT pathway-related genes (Gene symbols shown in and detailed gene information shown in Supplementary Table 1) was analyzed at 100 bp resolution in 30 colorectal tissue samples [9 colorectal cancer (CRC), 15 adenoma (AD), 6 normal adjacent tissue (NAT)] by MethylCap-seq ( and ).
Table 2. Clinical data of colorectal tissue samples used in MethylCap-seq study.
Table 3. Samples used in the study.
Methylated CpG containing DNA fragments were effectively isolated using MethylCap protein (Supplementary Figure 1A). Recovery was 12.51 ± 10.14% in methyl-captured DNA samples, while the unmethylated fraction was slightly detected (0.41 ± 0.16%). Sequencing results of all samples fulfilled the quality criteria, according to the MEDIPS quality control protocol.Citation34 Pearson correlation coefficient of saturation analysis exceeded the 0.5 threshold for every sample (0.71 ± 0.07). Coverage of at least 5% of the methylation sites (11.43 ± 2.56%) was more than 5 times for each sample during the CpG coverage analysis. According to the CpG enrichment test, the efficacy of the methyl capture was proven to be appropriate as all samples showed value well above the 1.4 threshold (enrichment score relH: 2.87 ± 0.42) (Supplementary Figure 1B, C, D).
During the determination of DMRs both in promoter and in gene body regions, methylation changes were considered significant if P-values were under 0.05 and at least a 10% methylation difference could be measured (absolute value of Δβ was > 0.1).
The promoter regions of 33 out of the 160 analyzed WNT pathway genes were differentially methylated in CRC compared to NAT, including hypermethylated AXIN2, CHP1, PRICKLE1, SFRP1, SFRP2, SOX17, and hypomethylated CACYBP, CTNNB1, MYC (, ). In AD vs. NAT comparison, altered promoter methylation of 44 WNT signaling genes was detected, including hypermethylated APC, AXIN2, DAAM2, DKK4, PRICKLE1, SOX17, SFRP1, SFRP2 and SFRP4, and hypomethylated CACYBP, FZD3 (, ). Forty-one genes were identified showing different promoter methylation levels between CRC and AD samples. Hypermethylation of AXIN2, DKK1, VANGL1, and WNT5A gene promoters was increased in CRC compared to AD, while promoter methylation of SOX17, PRICKLE1, DAAM2, and MYC genes was higher in AD than in CRC (). The whole gene list, including genomic positions and P- and β-values for DMRs, is shown in Supplementary Table 2A (CRC vs. NAT), 2B (AD vs. NAT), and 2C (CRC vs. AD).
Figure 1. Differentially methylated regions in WNT pathway gene promoters. Methylation probabilities (β-values) of 100 bp long analyzed regions were calculated with respect to genome wide CpG density dependent Poisson distributions, and are represented on a 0–1 scale. Promoter DMRs are shown A. in colorectal cancer (CRC) vs. normal adjacent tissue (NAT) comparison B. in adenoma (AD) vs. NAT comparison and C. between CRC vs. AD samples. Methylation intensities are illustrated on a color scale: high methylation levels are marked with red, low methylation levels are represented by green. Gene symbols mentioned in the text are listed on the heat maps, the whole gene lists with genomic positions, P- and β-values of DMRs can be seen in Supplementary Table 2.
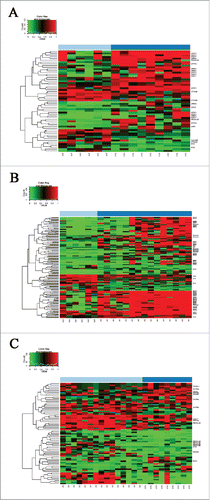
Promoter methylation alterations have been firstly mentioned, since their effect on gene expression is well described, but most of the detected differentially methylated CpG sites were localized in gene body regions. In CRC samples, 93.75% (150 out of 160) of the analyzed WNT pathway genes had DMRs in their gene body region (all together, 3788 DMRs). In the adenoma group, a similar proportion (96.88%) of WNT pathway genes showed aberrant gene body methylation, with approximately 1.5-fold higher number of DMRs (total of 5737 DMRs).
In silico DNA methylation analysis of WNT pathway genes
CpG sites belonging to the significant DMRs in our MethylCap-sey study were searched using the Illumina BeadChip 450K array data sets from Luo et al.Citation28 (GEO accession number: GSE48684; ) and P- and Δβ-values were determined in CRC vs. NAT, AD vs. NAT, and CRC vs. AD comparisons. Approximately one third of significant promoter DMRs detected by MethylCap-seq were represented in the Illumina BeadChip 450K data set by at least one cg ID (one CpG site; ). Within the represented regions, 58% of the promoter methylation changes could be validated between CRC and NAT samples, 83% between AD and NAT samples, and 30% in CRC vs. AD comparison. In the case of gene body alterations, only approximately 3% of significant DMRs obtained by MethylCap-seq were represented in the BeadChip 450K array, and 25–51% of the methylation alterations could be confirmed between the diagnostic groups ().
Table 4. Comparison of BeadChip 450KCitation28 data and MethylCap-seq results for WNT pathway genes.
Gene expression analysis of WNT signaling pathway genes
Gene expression data of colonic tissue samples from the Gene Expression Omnibus (GEO) database were involved in in silico mRNA expression analysis of 160 WNT pathway genes (GEO accession numbers: GSE37364,Citation35 GSE8671,Citation36 GSE18105,Citation37 GSE32323,Citation38 GSE22242,Citation39 GSE9348)Citation40 (). The log Fold Change (logFC) data of significantly differentially expressed WNT pathway genes, together with the methylation data, are shown in Supplementary Table 2 for AD vs. NAT, CRC vs. NAT, and CRC vs. AD comparisons. Compared to promoter methylation alterations, the expression of 10 WNT signaling genes, including CACYBP, CTNNB1, MYC, PRICKLE1, PSEN1, and SFRP1, changed oppositely in CRC vs. NAT comparison (). The expression of 14 WNT pathway genes, including APC, CHP1, DAAM2, PRICKLE1, RUVBL1, SFRP1, SFRP2, SFRP4, and TLE3, were inversely correlated with promoter methylation status in AD vs. NAT samples (). In CRC vs. AD tissues, the expression of 8 genes, including CTNNB1, CXXC4, PRICKLE1, VANGL1, and SFRP2, was opposed to promoter methylation differences (). Inverse relation between promoter methylation and gene expression was found in CACYBP, CHP1, CTNNB1, CXXC4, PRICKLE1, SFRP1, SFRP2, and TLE3 genes, at least in 2 of the 3 comparisons (CRC vs. NAT, AD vs. NAT, and CRC vs. AD).
Table 5. WNT pathway genes showing inverse relation between DNA methylation and mRNA expression (CRC vs. NAT).
Table 6. WNT pathway genes showing inverse relation between DNA methylation and mRNA expression (adenoma vs. NAT).
Table 7. WNT pathway genes showing inverse relation between DNA methylation and mRNA expression (CRC vs. adenoma).
Demethylation analysis on colon cancer cell lines
Gene expression data of 5 5-aza-2′-deoxycytidine-treated colon cancer cell lines were evaluated (GEO accession numbers: GSE29060,Citation41 GSE32323,Citation38 GSE14526,Citation42 and GSE41588Citation43)().
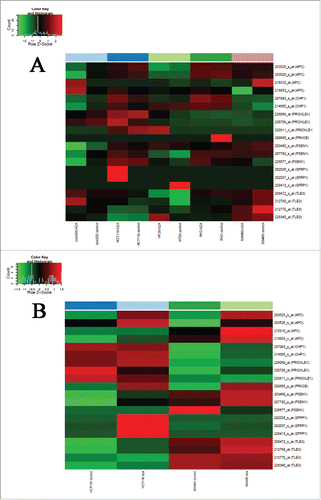
Upregulation of mRNA expression was observed—at least in one of the analyzed colon cancer cell lines after 5-aza-2′-deoxycytidine demethylation treatment—for 7 genes (APC, CHP1, PRICKLE1, PRKCB, PSEN1, SFRP1, and TLE3) showing promoter hypermethylation and downregulated mRNA expression in benign and/or malignant colonic tissue samples. In SW480 and HCT116 colorectal adenocarcinoma cell lines, 86–100% of the genes showed opposite expression changes caused by the demethylation treatment (). GSE29060 and GSE41588 data sets were also evaluated, but mRNA expression of the above-mentioned genes was not restored in HT29 cell line by the demethylating treatment.
Figure 2. Effect of 5-aza-2′-deoxycytidine demethylation treatment on colon cancer cell lines. Gene expression data of colon cancer cell lines under 5-aza-2′-deoxycytidine demethylation treatment analyzed on Affymetrix HGU133 Plus 2.0 microarrays are shown (GEO accession numbers of gene expression data sets: GSE32323,Citation38 GSE14526Citation42). Demethylation agent treatment reversed (upregulated) mRNA expression of 7 WNT pathway genes showing promoter hypermethylation and downregulated mRNA expression in benign and/or malignant colonic tissue samples, principally in SW480 and HCT116 colorectal adenocarcinoma cell lines in varying degrees. A. GSE32323 data set B. GSE14526 data set. Samples are shown in columns, selected transcripts are represented in rows. High mRNA expression intensities are marked in red, low expression levels are shown in green. AZA = 5-aza-2′-deoxycytidine.
APC and β-catenin mutation analysis
Mutation analysis of APC and β-catenin genes was performed on the same sample set that was used in the whole methylome profiling. APC mutations were detected in 0% of NAT, 27% of adenoma, and 29% of CRC samples, while none of the samples showed β-catenin mutation. Two truncated protein resulting in frameshift mutations (COSM13113 and COSM296347) and 2 nonsense mutations with G/T substitution (COSM18775 and COSM18759) were detected. A SNP with G/A substitution (SNPrs41115) was found in 53% of adenoma samples and in 71% of CRC samples at the targeted APC sequence region. The list and details of the detected mutations can be seen in .
Table 8. Detected mutations in analyzed colonic tissue samples.
Discussion
Similarly to other malignancies, colorectal cancer arises as a consequence of accumulating genetic and epigenetic alterations. Beside the conventional mutations, altered DNA methylation has also been revealed as a causative factor in carcinogenesis and tumor progression. Gene silencing mediated by aberrant promoter DNA methylation is one of the key features of carcinogenesis.Citation8 Gene body regions are also strongly methylated, which can positively correlate with the gene expression.Citation8,9 The importance of DNA methylation is further enhanced by the recently initiated trials using low-dose DNA demethylating agents as anticancer drugs.Citation9
We focused on DNA methylation analysis of WNT signaling genes/gene promoters in CIMP-negative CRCs and precancerous adenomas. Although since the establishment of CIMP conception the main focus has been on DNA methylation analysis of CIMP-positive proximal colon cancers, notable epigenetic alterations, including promoter DNA hypermethylation, can be detected also in CIMP-negative CRCs representing the majority of sporadic CRCs.Citation4,44 A reliable plasma methylation marker test with high specificity and sensitivity—such as Epi proColon 2.0 analyzing SEPT9 promoter methylationCitation45—should be based on systematic DNA methylation alterations identified and detectable both in CIMP-negative and CIMP-positive CRCs.
In this study, systematic analysis of DNA methylation of WNT signaling pathway genes in relation to CRC development and progression was performed using methyl capture sequencing (MethylCap-seq) and whole genomic expression microarrays. MethylCap-seq can test both promoter and gene body regions in the entire genome. For comparing previous and our DNA methylation results, the in silico analysis of Luo et al.Citation28 data, also including adenomas besides CRC and NAT samples, was performed. Similarly to the work of Luo et al.,Citation28 we compared a group of NAT samples to CRC and adenoma sample groups, instead of doing a pairwise comparison of CRC and NAT samples.
When compared to the methylation array, MethylCap-seq provides a more extensive overview of whole genome methylation, although its resolution and sensitivity is lower than that of BeadChip methylation arrays.Citation31 Concerning WNT signaling genes in our experiments, approximately one third of the significant DMRs was represented and could be analyzed on BeadChip array for gene promoters; only ∼3% of the identified DMRs for gene body regions was represented. The coexisting compatible regions in both test systems showed high DNA methylation similarity between the diagnostic groups, especially between adenoma and NAT samples (83%). Concordance between CRC and NAT groups was 58%, but only 30% between CRC and AD. This partial discordance was most probably related to the difference between our patient cohorts concerning CIMP and MSI or mutation status. Regarding resolution, the Illumina BeadChip 450K array detects the methylation status of a given CpG site, while MethylCap-seq measures the summation of methylated CpG sites located within the given 100 bp long analysis window.
In our results, promoter methylation changes affected both canonical (WNT/β-catenin) and noncanonical [planar cell polarity (PCP) and WNT/Ca2+] WNT signaling pathways. In CRC samples, significant DMRs were found in 20.63% of the WNT pathway gene promoters, which was even higher in adenoma samples (27.50%). Aberrant promoter methylation was mainly detected in the canonical WNT (22.3%) and WNT/Ca2+ pathway genes (22.9%) in CRC, but the PCP pathway was also affected (18.8%). In AD samples, promoter methylation changes were principally observed in WNT/Ca2+ pathway genes (40%), while high number of DMRs were also found in canonical and PCP WNT pathways genes (28.1%).
Similarly to our results, promoter hypermethylation of canonical WNT inhibitory genes, such as SFRP1,Citation11,12 SFRP2,Citation11,13-15 and SOX17,Citation11,24 was detected both in CRC and adenoma samples. However, in contrast to the findings of Silva et al.,Citation11 in our study these genes showed elevated promoter methylation in adenoma samples compared to CRC tissue. In accordance with our previous bisulfite sequencing findings,Citation18 SFRP1 and SFRP2 promoter methylation in CRC and AD tissue samples was also detected by MethylCap-seq at the same regions ().
Table 9. Methyl capture and bisulfite sequencingCitation18 data for SFRP1 and SFRP2 promoter regions (overlapping regions).
Correspondingly to the previous findings,Citation11,17, 19-20 we found that another known inhibitor of canonical WNT pathway, DKK2, was significantly hypermethylated in CRC vs. NAT and AD vs. NAT comparisons. However, our promoter estimation considering the sequence in the region between 2000 bp upstream and 1000 bp downstream from the transcription start site (if the positive DNA strand was the coding strand) identified ‘active promoter’ in at least one of the 9 analyzed cell lines, according to the ENCODE ChromHMM results.Citation46 The studied DMRs in DKK2 promoter were excluded because they were labeled as ‘poised promoters’ by ENCODE, despite their significance (P-value was between 0.00001 and 0.035 in CRC vs. NAT; P < 0.01 in AD vs. NAT) and high Δβ-values (0.41–0.72 in CRC vs. NAT and 0.49–0.60 in AD vs. NAT). The lack of significant changes in mRNA expression data between groups also supports the tested DKK2 region as an inactive promoter.
Our MethylCap-seq results indicate that promoter DMRs of APC, AXIN1, AXIN2, and CSNK1 genes may participate in the impairment/reduction of the APC-Axin-CSNK1-β-catenin complex leading to the constitutive activation of the canonical WNT pathway. APC hypermethylation has been already described in adenoma and CRC tissue and CRC plasma samples.Citation19,47,48 In accordance with findings of Judson et al.,Citation48 hypermethylation in the active promoter of the APC gene in adenoma samples was detected in our study as well (). Similar alteration appeared in CRC samples, but in a different region upstream to the APC gene, which was considered as a ‘weak promoter’ or ‘enhancer’. Reduced APC mRNA levels on microarrays indicate silencing by promoter hypermethylation, which impairs APC tumor suppressor function during colorectal carcinogenesis, besides the frequent APC mutations. DNA methylation differences in APC promoter and mutation hot spot regions could not been detected between samples with or without APC mutations. This supports that APC mutations and APC promoter hypermethylation are not exclusive factors, but rather 2 parallel mechanisms leading to the loss of APC function. We found APC mutations in approximately one third of the CRC samples, which is below the mutation rate described by others (75–80%Citation22). However, we only analyzed a limited region [BLAST: chr5:112,175,118–112,176,026 (Feb. 2009 GRCh37/hg19 Assembly)] with the most prevalent APC mutations. Still, our results were similar to those of Luo et al.Citation28 finding APC mutations in 29.7% of CRC and 27.8% of adenoma tissue samples using larger sample size and testing longer APC regions.
Increased promoter methylation of the AXIN2 gene is thought to be associated with carcinogenesis of MSI+ CRC.Citation49 We also detected AXIN2 promoter hypermethylation with MethylCap-seq both in CRC and adenoma tissues compared to NAT samples, but the prevalence of DMRs was higher in gene body regions (25 hypermethylated DMRs, 13 hypomethylated DMRs). In silico analysis also revealed a remarkable upregulation of AXIN2 mRNA in both disease groups, suggesting the tumor promoting function of AXIN2,Citation49 especially in MSS CRC cases.
We also found β-catenin hypomethylation in CRC compared to NAT tissues, which was also detected earlier by others using Illumina methylation array and in methylation-sensitive high resolution melting analysis.Citation20 The significantly hypomethylated CpG site (41240163, Illumina ID: cg09678212) in the previous analysisCitation20 is located within WNT_CTNNB1_41240101 100 bp long analysis window in our study. In silico evaluation of Luo et al.Citation28 Illumina methylation array data also resulted in significant (P<0.001) hypomethylation of this CpG site in CRC compared to NAT (Δβ = -0.20) and adenoma samples (Δβ = -0.12). Besides identifying the only β-catenin promoter hypomethylated CpG site found by the Illumina platform, our MethylCap-seq analysis revealed significant hypomethylation in the 300 bp region (WNT_CTNNB1_41239901; WNT_CTNNB1_41240001 and WNT_CTNNB1_41240101) of the CTNNB1 gene promoter. Hypomethylation of this region in CRC compared to NAT and adenoma samples suggests its contribution to aberrantly elevated intracellular β-catenin level in colorectal carcinogenesis. Supporting this, significant elevation of CTNNB1 mRNA levels were observed in CRC compared to normal samples with logFC: 0.38–0.94, along with increased nuclear translocation of β-catenin protein as a sign of the activation of the canonical WNT pathway during malignant transformation. Oncogenic function of canonical WNT pathway activation is known to occur at early stages of carcinogenesis, and can be promoted by DNA methylation alterations from the precancerous adenoma stage.
The PCP pathway, as a noncanonical WNT signaling, can play multiple roles in oncogenesis.Citation50 At an early stage, PCP pathway has a tumor suppressive effect as it can antagonize the canonical pathway.Citation50 However, during tumor progression, it can be tumorigenic by promoting tumor cell motility and invasion, angiogenesis induction, and metastatic spreading.Citation50 DNA methylation changes both in promoters and transcribed regions of PCP pathway genes were detected at the adenoma stage. DMRs were observed in FZD2, FZD7, PRICKLE1, and WNT5A gene promoters, both in adenoma and CRC samples, while promoter hypermethylation on DAAM2 and downregulation of its mRNA level were typical only in adenoma tissue compared to NAT samples. Increased FZD2 and WNT5A levels were measured in cancer cell lines, including CRC and advanced tumors, and were shown to promote epithelial-mesenchymal transition and metastasis.Citation50,51 FZD7 can enhance migration and invasion of CRC mediated by noncanonical WNT signaling.Citation50 PRICKLE1, a disheveled (Dvl) associated protein, may have a tumor suppressor function by antagonizing Dvl recruitment by Frizzled.Citation52 It can bind to Dvl3 and facilitate its ubiquitination-mediated degradation, suggesting that PRICKLE1 is a negative regulator of WNT/β-catenin pathway.Citation52,53 Promoter hypermethylation of PRICKLE1, found in adenoma and CRC samples in our methylation study, also support this idea. DAAM2 and PRICKLE1 seems to be DNA methylation regulated genes as inverse correlation was found between their promoter hypermethylation and reduced mRNA expression.
The noncanonical WNT/Ca2+ pathway, which can antagonize β-catenin-dependent signaling and stimulate cell migration,Citation53 was the most affected by promoter methylation changes in adenomas. This involved the WNT5A (ligand), FZD1, 2 and 7 (receptors), and downstream elements, such as PLCB1, PRKCB, and NFAT5 genes in AD samples, whereas promoter hypermethylation of FZD2, FZD7, PLCB1, and PRKCB genes was in silico-validated in adenoma samples.Citation28 Hypermethylation of FZD2, FZD7, PLCB1, and WNT5A gene promoters was also detected in CRC, compared to NAT samples. However, in many cases it was not accompanied by downregulation of mRNA expression. WNT5A has a dual function: it can act as tumor suppressor or as oncogene in a context-dependent manner. Depending on the type and level of co-receptors, it can activate noncanonical or β-catenin-dependent WNT signaling. Although its promoter was found to be hypermethylated both in adenoma and CRC tissues, with only moderate difference in the gene body region, it was probably not a DNA methylation-regulated gene. The remarkable increase on its mRNA expression (AD vs. NAT: logFC: 0.65–1.02; CRC vs. NAT logFC: 1.21–2.58) may be regulated by other mechanisms. FZD2 and FZD7 genes, which were upregulated in CRC (FZD7 is already in adenomas) may promote migration and metastatic spreading.Citation51 The connection between methylation and mRNA expression of these genes was controversial, since both increased mRNA expression and promoter hypermethylation were measured in CRC, which was further elevated in adenoma tissue samples.
An inverse relation was observed between gene expression and promoter DNA methylation in WNT-related 23 genes. Among others, APC, CHP1, PRICKLE1, PRKCB, PSEN1, and SFRP1 genes were identified as methylation-regulated WNT pathway genes during colorectal normal-adenoma-carcinoma sequence progression in view of 5-aza-2′-deoxycytidine (5-aza) demethylation treatment data of colon cancer cell lines, specifically of HCT116 and SW480. SW480 is a MSS, CIMP-negative colorectal adenocarcinoma cell lineCitation54 most similar to the clinical samples analyzed in our MethylCap-seq study. Demethylation treatment was capable of inducing re-expression of these genes in CRC cell lines, suggesting a casual relationship between promoter hypermethylation and expression silencing of these genes. Restoration of APC, PRKCB, and SFRP1 expression by 5-aza treatment was also observed earlier in different colon cancer cell lines.Citation55-57
Despite having detected most of the DMRs in gene body regions, we discuss these only shortly here because their contribution is not well known or controversial. Approximately 95% of the analyzed WNT pathway genes showed aberrant gene body methylation in adenoma and CRC compared to the NAT samples with ∼1.5-fold higher number of DMRs in adenomas. Gene body methylation may positively regulate gene expressionCitation8,9 and can be involved in splicing regulation.Citation8,58 However, it can also contribute to gene silencing, according to the whole genomic bisulfite and RNA sequencing data of Lou et al.Citation59 In the recent study of Wang et al. about the roles of genic methylation in prostate tumorigenesis, 12 groups of genes with collaborative differential methylation patterns, including hyper-methylated promoter/hypermethylated gene body and hypermethylated promoter/hypomethylated gene body, were identified, assuming a gene activating role for gene body methylation.Citation60 In line with these findings, we identified strongly hypermethylated gene body associated with upregulated mRNA expression in WNT pathway genes, such as AXIN2, CSNK1E, MYC, NKD1, which expression was downregulated after 5-aza-2′-deoxycytidine treatment in colon cancer cell lines. Also, in other genes, reduced expression was associated with intense gene body hypermethylation reversed by 5-aza demethylation agent (e.g., PPARD) in CRC cell lines. Our study resulted in gene body methylation alterations between CRC and NAT samples in LRP5, TCF7L1, WNT2, and WNT6 WNT signaling pathway genes, on the same, mainly intronic regions, as in a previous study by Farkas et al.Citation20
Further research focusing on disease-specific methylation alterations and gene expression regulatory mechanisms is required to clarify the selective functions of DNA methylation at the gene body and other non-promoter regions.Citation59
Conclusions
DNA methylation alterations affect both canonical and noncanonical WNT signaling pathway genes at their promoter and gene body regions. Using MethylCap-seq, which allows extensive discovery of DNA methylation in the entire genome, we identified new aberrant methylation patterns affecting genes including CTNNB1, DAAM2, and PRICKLE1, characterizing the colorectal normal-adenoma-dysplasia sequence progression. Previously described methylation changes in WNT pathway genes during colorectal carcinogenesis, such as SFRP1, SFRP2, SOX17, and APC, could also be successfully verified.
Our results suggest that DNA methylation changes of canonical and noncanonical WNT signaling pathways also contribute to the development and progression of CIMP-negative CRC, besides the frequent APC mutations. Aberrant DNA methylation already appears in adenomas, indicating DNA methylation as an early event of colorectal carcinogenesis. The genomic regions showing epigenetic changes identified in this study might provide the basis for further studies revealing potential early diagnostic and therapeutic targets.
Patients and methods
Clinical samples
A total of 30 colonic tissue samples [15 adenoma, 9 CRC, 6 normal adjacent tissue (NAT)] were included in the MethylCap-seq study (, ). All adenoma and part of CRC samples were collected during screening colonoscopy; further surgically removed CRC and NAT tissue samples were also analyzed.
After informed consent of untreated patients, colonic biopsy samples were taken during endoscopic intervention and stored in RNALater Stabilization Solution (Ambion, ThermoFisher Scientific, AM7024) at −80°C until use. Biopsy samples from the same site were immediately fixed in buffered formalin for histological evaluation. Histological diagnoses were established by experienced pathologists. Tissue samples from untreated CRC patients were also obtained from surgically removed colon or rectum tumors and from histologically NAT that originated from the area farthest available from the tumor. Tissue samples were snap frozen in liquid nitrogen directly after surgery and were stored at -80°C. Detailed patient specification is described in . The study was conducted according to the Helsinki declaration and approved by the local ethics committee and government authorities [Regional and Institutional Committee of Science and Research Ethics (TUKEB) Nr.: 69/2008, 202/2009 and 23970/2011 Semmelweis University, Budapest, Hungary].
MSI status was determined according to the results of immunohistochemical staining as described earlierCitation17 with monoclonal antibodies for MLH1 (1:80), MSH2 (1:80), MSH6 (1:80), PMS2 (1:150, all from Novocastra/Leica Biosystems NCL-L-MLH1, NCL-MSH2, NCL-L-MSH6, NCL-L-PMS2) markers. A tumor sample was scored MSS if all of the 4 above MMR proteins showed expression.
CIMP category was specified using both classic (CDKN2A, MLH1, MINT1, MINT2, and MINT31)Citation7,32 and Weisenberger (CACNA1G, IGF2, NEUROG1, RUNX3, SOCS1) marker panels,Citation33 according to the MethylCap-seq data overlapping with regions of CIMP panel methylation-specific PCRs.Citation7,32,33 A sample was considered as CIMP-positive (CIMP-high) when at least 2 of the 5 markers (in case of the classic panel) and at least 3 of the 5 markers (in case of the new panel) were found to be methylated.
DNA isolation
Tissue samples were homogenized in 2% sodium dodecyl sulfate, and digested with 4 mg/mL proteinase K for 16 hat 56°C. Genomic DNA was isolated using High Pure PCR Template Preparation Kit (Roche Applied Science, 11796828001) according to the manufacturer's instructions.Citation17 DNA was eluted in 2x with 100 μl RNase- and DNase-free water and stored at -20°C. Isolated DNA samples was quantified by Qubit fluorometer using Qubit dsDNA HS Assay (Invitrogen, ThermoFisher Scientific, Q32854).
Methyl capture sequencing
After fragmentation of 3 μg genomic DNA samples (100–400 bp) using Bioruptor Sonicator (Diagenode, UCD-300), the DNA fragments containing methylated CpGs were isolated in SX-8G IP-Star® Compact Automated System using Auto MethylCap kit (Diagenode, C02020011) using High Elution Buffer according to the manufacturer's instructions.Citation61
The methylated DNA fraction was purified on QIAquick PCR purification columns (Qiagen, 28104). SYBR Green quantitative RT-PCR was performed using Human meDNA primer pair (TSH2B) (Diagenode, pp-1041-500) for detection of methylated DNA and Human unDNA primer pair (GAPDH) (Diagenode, pp-1044-500) for showing unmethylated DNA. Library preparation was carried out using Illumina TruSeq ChIP Sample Preparation kit (Illumina, IP-202). Cluster generation was done on cBot instrument using TruSeq SR Cluster Kit v3-cBot-HS (Illumina, GD-401-3001) which provides reagents that bind samples to complementary adapter oligos for cluster amplification on single-read flow cells. Next generation sequencing of the methylated DNA fragments was performed on HiScanSQ instrument using TruSeq SBS v3-HS reagents (Illumina, FC-401-3001) according to the manufacturer's instructions. Bowtie2Citation62 with default settings was used to map the 100 bp paired and 50 bp unpaired reads to the hg19 human genome reference assembly.Citation63 The generated bam files were sorted and indexed by samtools.Citation64 Data were processed by the MEDIPSCitation65 bioconductor R package. After quality control, unpaired reads were extended to the length of the average fragment length of the paired samples (250 bp). PCR duplicates were removed, then the coverage data were binned with 100 bp window size. Methylation probabilities (β-values hereafter) were calculated with respect to genome wide CpG density dependent Poisson distributions.
In silico DNA methylation analysis of WNT pathway genes
Illumina BeadChip 450K DNA methylation array data sets from Luo et al.Citation28 were downloaded from NCBI Gene Expression Database and analyzed (GEO accession number: GSE48684). This data set contains the methylation results of 120 colorectal tissue samples including CRC, AD, and NAT samples (). CpG sites belonging to the significant DMRs in our MethylCap-sey study were searched on the Illumina BeadChip 450K array, and P- and Δβ-values were determined in CRC vs. NAT, AD vs. NAT and CRC vs. AD comparisons.
Gene expression analysis
Gene expression data of colonic tissue samples from the Gene Expression Omnibus (GEO) database were included in the in silico mRNA expression analysis of WNT pathway genes. WNT pathway genes were selected according to the KEGG pathway database. GSE37364 set contains Affymetrix HGU133 Plus2.0 whole transcriptome data of 94 colonic biopsy samples (38 healthy normal, 27 colorectal adenocarcinoma, and 29 tubulovillous/villous adenoma) previously hybridized by our research group.Citation35 Five GEO data sets made by others on the same platform were also analyzed (GSE8671,Citation36 GSE18105,Citation37 GSE32323,Citation38 GSE22242,Citation39 GSE9348Citation40). Gene expression data of CRC and healthy normal (N)/ normal adjacent tissue (NAT) samples are available under serial accession numbers GSE18105, GSE32323 and GSE9348. GSE8671 data set contains mRNA expression data of adenoma and healthy normal biopsy samples, GSE22242 dataset includes transcriptome analysis results of pooled normal, adenoma, and CRC biopsy samples. Differentially expressed genes were determined.
Demethylation analysis on colorectal cancer cell lines
Gene expression data of 5-aza-2′-deoxycytidine (5-aza)-treated colorectal cancer cell lines (HT29, COLO320, HCT116, RKO, SW480) were downloaded and in silico analyzed [GEO accession numbers: GSE29060 (previously performed by our research group),Citation41 GSE32323,Citation38 GSE14526,Citation42 and GSE41588Citation43]. GSE29060 and GSE41588 data sets contain the expression data of HT29 cells treated with 5-aza at higher concentration (5–10 μM) for 72 hours and 5 days respectively. In another 2 studies (GSE32323 and GSE14526) 5-aza was applied for 72 hours at lower concentrations (0.5 μM and 3 μM, respectively). Differentially expressed genes after 5-aza demethylation treatment were determined.
Mutation analysis
Mutation hot spot regions of APC and β-catenin genes were amplified using custom made PCR primers (shown in Supplementary Table 3) and amplicons were sequenced with a GS Junior instrument (Roche), as described earlier.Citation66 Briefly, Rapid Library Molecular Identifier (RL_MID) adaptors were ligated to the PCR products during library preparation providing a sample specific sequence motif. The quality checking of PCR libraries were made on Agilent Bioanalyzer instrument using High Sensitivity DNA Chip (Agilent, G2939AA, 5067-4626). Emulsion PCR amplification of the amplicon libraries was performed using the Lib-L emPCR Kit (Roche, 05996481001), with 2 DNA molecules per bead ratio following the manufacturer's instructions. Bead enrichment and sequencing were carried out using GS Junior Titanium Sequencing Kit (Roche, 05996554001) according to the method described in the Sequencing Method Manual, GS FLX Titanium Series. Amplicon Variant Analyzer software (Roche) was applied for identification of variants.
Statistical analysis
During statistical evaluation of gene expression and DNA methylation data of 160 WNT signaling pathway genes, false discovery rate (FDR) was applied for Student t-test with the criteria P < 0.05 in all paired comparisons. In case of gene expression for LogFC calculation, the differences between the averages of samples groups were considered (abs ≥ 1 criteria). Methylation alterations between diagnostic groups were characterized by Δβ-values (the differences of the average β-values of sample groups).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A__1190894_supplemetary_data.zip
Download Zip (225.4 KB)Funding
This study was supported by the National Research, Development and Innovation Office (KMR-12-1-2012-0216 grant) and the validation study by the Hungarian Scientific Research Fund (OTKA-K111743 grant).
Related Research Data
References
- GLOBOCAN 2012. Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. 2012. http://globocan.iarc.fr/Pages/online.aspx
- Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Patholog Res Int 2012; 2012:509348; PMID:22888469; http://dx.doi.org/10.1155/2012/509348
- Sipos F, Mũzes G, Patai AV, Fũri I, Péterfia B, Hollósi P, Molnár B, Tulassay Z. Genome-wide screening for understanding the role of DNA methylation in colorectal cancer. Epigenomics 2013; 5:569-81; PMID:24059802; http://dx.doi.org/10.2217/epi.13.52
- Patai AV, Molnár B, Kalmár A, Schöller A, Tóth K, Tulassay Z. Role of DNA methylation in colorectal carcinogenesis. Dig Dis 2012; 30:310-5; PMID:22722557; http://dx.doi.org/10.1159/000337004
- Kim MS, Lee J, Sidransky D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev 2010; 29:181-206; PMID:20135198; http://dx.doi.org/10.1007/s10555-010-9207-6
- Patai AV, Molnár B, Tulassay Z, Sipos F. Serrated pathway: alternative route to colorectal cancer. World J Gastroenterol 2013; 19:607-15; PMID:23431044; http://dx.doi.org/10.3748/wjg.v19.i5.607
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004; 4:988-93; PMID:15573120; http://dx.doi.org/10.1038/nrc1507
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; http://dx.doi.org/10.1038/nrg3230
- Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014; 26:577-90; PMID:25263941; http://dx.doi.org/10.1016/j.ccr.2014.07.028
- Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006; 25:7531-7; PMID:17143297; http://dx.doi.org/10.1038/sj.onc.1210059
- Silva AL, Dawson SN, Arends MJ, Guttula K, Hall N, Cameron EA, Huang TH, Brenton JD, Tavaré S, Bienz M, et al. Boosting Wnt activity during colorectal cancer progression through selective hypermethylation of Wnt signaling antagonists. BMC Cancer 2014; 14:891; PMID:25432628; http://dx.doi.org/10.1186/1471-2407-14-891
- Chen YZ, Liu D, Zhao YX, Wang HT, Gao Y, Chen Y. Aberrant promoter methylation of the SFRP1 gene may contribute to colorectal carcinogenesis: a meta-analysis. Tumour Biol 2014; 35:9201-10; PMID:24929326; http://dx.doi.org/10.1007/s13277-014-2180-x
- Vatandoost N, Ghanbari J, Mojaver M, Avan A, Ghayour-Mobarhan M, Nedaeinia R, Salehi R. Early detection of colorectal cancer: from conventional methods to novel biomarkers. J Cancer Res Clin Oncol 2016; 142:341-51; PMID:25687380; http://dx.doi.org/10.1007/s00432-015-1928-z
- Kalmár A, Péterfia B, Hollósi P, Wichmann B, Bodor A, Patai ÁV, Schöller A, Krenács T, Tulassay Z, Molnár B. Bisulfite-Based DNA methylation analysis from recent and archived formalin-fixed, paraffin embedded colorectal tissue samples. Pathol Oncol Res 2015; 21:1149-56; PMID:25991403; http://dx.doi.org/10.1007/s12253-015-9945-4
- Sui C, Wang G, Chen Q, Ma J. Variation risks of SFRP2 hypermethylation between precancerous disease and colorectal cancer. Tumour Biol 2014; 35:10457-65; PMID:25053594; http://dx.doi.org/10.1007/s13277-014-2313-2
- Samaei NM, Yazdani Y, Alizadeh-Navaei R, Azadeh H, Farazmandfar T. Promoter methylation analysis of WNT/β-catenin pathway regulators and its association with expression of DNMT1 enzyme in colorectal cancer. J Biomed Sci 2014; 21:73; PMID:25107489; http://dx.doi.org/10.1186/s12929-014-0073-3
- Patai ÁV, Valcz G, Hollósi P, Kalmár A, Péterfia B, Patai Á, Wichmann B, Spisák S, Barták BK, Leiszter K, et al. Comprehensive DNA methylation analysis reveals a common ten-gene methylation signature in colorectal adenomas and carcinomas. PLoS One 2015; 10:e0133836; PMID:26291085; http://dx.doi.org/10.1371/journal.pone.0133836
- Kalmár A, Péterfia B, Hollósi P, Galamb O, Spisák S, Wichmann B, Bodor A, Tóth K, Patai ÁV, Valcz G, et al. DNA hypermethylation and decreased mRNA expression of MAL, PRIMA1, PTGDR and SFRP1 in colorectal adenoma and cancer. BMC Cancer 2015; 15:736; PMID:26482433; http://dx.doi.org/10.1186/s12885-015-1687-x
- Silva TD, Vidigal VM, Felipe AV, DE Lima JM, Neto RA, Saad SS, Forones NM. DNA methylation as an epigenetic biomarker in colorectal cancer. Oncol Lett 2013; 6:1687-92; PMID:24260063; http://dx.doi.org/10.3892/ol.2013.1606
- Farkas SA, Vymetalkova V, Vodickova L, Vodicka P, Nilsson TK. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/β-catenin signaling pathway genes. Epigenomics 2014; 6:179-91; PMID:24811787; http://dx.doi.org/10.2217/epi.14.7
- Taniguchi H, Yamamoto H, Hirata T, Miyamoto N, Oki M, Nosho K, Adachi Y, Endo T, Imai K, Shinomura Y. Frequent epigenetic inactivation of Wnt inhibitory factor-1 in human gastrointestinal cancers. Oncogene 2005; 24:7946-52; PMID:16007117; http://dx.doi.org/10.1038/sj.onc.1208910
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID:22810696; http://dx.doi.org/10.1038/nature11252
- Murakami T, Mitomi H, Saito T, Takahashi M, Sakamoto N, Fukui N, Yao T, Watanabe S. Distinct WNT/β-catenin signaling activation in the serrated neoplasia pathway and the adenoma-carcinoma sequence of the colorectum. Mod Pathol 2015; 28:146-58; PMID:24925057; http://dx.doi.org/10.1038/modpathol.2014.41
- Voorham QJ, Janssen J, Tijssen M, Snellenberg S, Mongera S, van Grieken NC, Grabsch H, Kliment M, Rembacken BJ, Mulder CJ, et al. Promoter methylation of Wnt-antagonists in polypoid and nonpolypoid colorectal adenomas. BMC Cancer 2013; 13:603; PMID:24350795; http://dx.doi.org/10.1186/1471-2407-13-603
- Muto Y, Maeda T, Suzuki K, Kato T, Watanabe F, Kamiyama H, Saito M, Koizumi K, Miyaki Y, Konishi F, et al. DNA methylation alterations of AXIN2 in serrated adenomas and colon carcinomas with microsatellite instability. BMC Cancer 2014; 14:466; PMID:24964857; http://dx.doi.org/10.1186/1471-2407-14-466
- Fang Y, Wang L, Zhang Y, Ge C, Xu C. Wif-1 methylation and β-catenin expression in colorectal serrated lesions. Zhonghua Bing Li Xue Za Zhi 2014; 43:15-9; PMID:24713243; http://dx.doi.org/10.3760/cma.j.issn.0529-5807.2014.01.004
- Fu X, Li L, Peng Y. Wnt signalling pathway in the serrated neoplastic pathway of the colorectum: possible roles and epigenetic regulatory mechanisms. J Clin Pathol 2012; 65:675-9; PMID:22412046; http://dx.doi.org/10.1136/jclinpath-2011-200602
- Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S, Carter KT, Morris SM, Wang J, Willis JE, Makar KW, Ulrich CM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology 2014; 147:418-29; PMID:24793120; http://dx.doi.org/10.1053/j.gastro.2014.04.039
- Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, Alekseev MV, Shelygin YA, Govorun VM. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics 2013; 8:921-34; PMID:23867710; http://dx.doi.org/10.4161/epi.25577
- Zhao Y, Sun J, Zhang H, Guo S, Gu J, Wang W, Tang N, Zhou X, Yu J. High-frequency aberrantly methylated targets in pancreatic adenocarcinoma identified via global DNA methylation analysis using methylCap-seq. Clin Epigenetics 2014; 6:18; PMID:25276247; http://dx.doi.org/10.1186/1868-7083-6-18
- De Meyer T, Bady P, Trooskens G, Kurscheid S, Bloch J, Kros JM, Hainfellner JA, Stupp R, Delorenzi M, Hegi ME, et al. Genome-wide DNA methylation detection by MethylCap-seq and Infinium HumanMethylation450 BeadChips: an independent large-scale comparison. Sci Rep 2015; 5:15375; PMID:26482909; http://dx.doi.org/10.1038/srep15375
- Lee S, Cho NY, Yoo EJ, Kim JH, Kang GH. CpG island methylator phenotype in colorectal cancers: comparison of the new and classic CpG island methylator phenotype marker panels. Arch Pathol Lab Med 2008; 132:1657-65; PMID:18834226; http://dx.doi.org/10.1043/1543-2165(2008)132[1657:CIMPIC]2.0.CO;2
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38:787-93; PMID:16804544; http://dx.doi.org/10.1038/ng1834
- Rodriguez BA, Frankhouser D, Murphy M, Trimarchi M, Tam HH, Curfman J, Huang R, Chan MW, Lai HC, Parikh D, et al. Methods for high-throughput MethylCap-Seq data analysis. BMC Genomics 2012; 13Suppl 6:S14; PMID:23134780; http://dx.doi.org/10.1186/1471-2164-13-S6-S14
- Galamb O, Wichmann B, Sipos F, Spisák S, Krenács T, Tóth K, Leiszter K, Kalmár A, Tulassay Z, Molnár B. Dysplasia-carcinoma transition specific transcripts in colonic biopsy samples. PLoS One 2012; 7:e48547; PMID:23155391; http://dx.doi.org/10.1371/journal.pone.0048547
- Sabates-Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, Rehrauer H, Laczko E, Kurowski MA, Bujnicki JM, Menigatti M, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res 2007; 5:1263-75; PMID:18171984; http://dx.doi.org/10.1158/1541-7786.MCR-07-0267
- Matsuyama T, Ishikawa T, Mogushi K, Yoshida T, Iida S, Uetake H, Mizushima H, Tanaka H, Sugihara K. MUC12 mRNA expression is an independent marker of prognosis in stage II and stage III colorectal cancer. Int J Cancer 2010; 127:2292-9; PMID:20162577; http://dx.doi.org/10.1002/ijc.25256
- Khamas A, Ishikawa T, Shimokawa K, Mogushi K, Iida S, Ishiguro M, Mizushima H, Tanaka H, Uetake H, Sugihara K. Screening for epigenetically masked genes in colorectal cancer Using 5-Aza-2′-deoxycytidine, microarray and gene expression profile. Cancer Genomics Proteomics 2012; 9:67-75; PMID:22399497; http://dx.doi.org/1109-6535/2012 $2.00+.40
- Tang H, Guo Q, Zhang C, Zhu J, Yang H, Zou YL, Yan Y, Hong D, Sou T, Yan XM. Identification of an intermediate signature that marks the initial phases of the colorectal adenoma-carcinoma transition. Int J Mol Med 2010; 26:631-41; PMID:20878084; http://dx.doi.org/10.3892/ijmm_00000508
- Hong Y, Downey T, Eu KW, Koh PK, Cheah PY. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Exp Metastasis 2010; 27:83-90; PMID:20143136; http://dx.doi.org/10.1007/s10585-010-9305-4
- Spisák S, Kalmár A, Galamb O, Wichmann B, Sipos F, Péterfia B, Csabai I, Kovalszky I, Semsey S, Tulassay Z, et al. Genome-wide screening of genes regulated by DNA methylation in colon cancer development. PLoS One 2012; 7:e46215; PMID:23049694; http://dx.doi.org/10.1371/journal.pone.0046215
- Yagi K, Akagi K, Hayashi H, Nagae G, Tsuji S, Isagawa T, Midorikawa Y, Nishimura Y, Sakamoto H, Seto Y, et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res 2010; 16:21-33; PMID:20028768; http://dx.doi.org/10.1158/1078-0432.CCR-09-2006
- Xu X, Zhang Y, Williams J, Antoniou E, McCombie WR, Wu S, Zhu W, Davidson NO, Denoya P, Li E. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinformatics 2013; 14Suppl 9:S1; PMID:23902433; http://dx.doi.org/10.1186/1471-2105-14-S9-S1
- Exner R, Pulverer W, Diem M, Spaller L, Woltering L, Schreiber M, Wolf B, Sonntagbauer M, Schröder F, Stift J, Wrba F, Bergmann M, Weinhäusel A, Egger G. Potential of DNA methylation in rectal cancer as diagnostic and prognostic biomarkers. Br J Cancer 2015; 113:1035-45; PMID:26335606; http://dx.doi.org/10.1038/bjc.2015.303
- Tóth K, Sipos F, Kalmár A, Patai AV, Wichmann B, Stoehr R, Golcher H, Schellerer V, Tulassay Z, Molnár B. Detection of methylated SEPT9 in plasma is a reliable screening method for both left- and right-sided colon cancers. PLoS One 2012; 7:e46000; http://dx.doi.org/10.1371/journal.pone.0046000
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43-9; PMID:21441907; http://dx.doi.org/10.1038/nature09906
- Syed Sameer A, Shah ZA, Abdullah S, Chowdri NA, Siddiqi MA. Analysis of molecular aberrations of Wnt pathway gladiators in colorectal cancer in the Kashmiri population. Hum Genomics 2011; 5:441-52; PMID:21807601; http://dx.doi.org/10.1186/1479-7364-5-5-441
- Judson H, Stewart A, Leslie A, Pratt NR, Baty DU, Steele RJ, Carey FA. Relationship between point gene mutation, chromosomal abnormality, and tumour suppressor gene methylation status in colorectal adenomas. J Pathol 2006; 210:344-50; PMID:16902913; http://dx.doi.org/10.1002/path.2044
- Wu ZQ, Brabletz T, Fearon E, Willis AL, Hu CY, Li XY, Weiss SJ. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc Natl Acad Sci USA 2012; 109:11312-7; PMID:22745173; http://dx.doi.org/10.1073/pnas.1203015109
- Wang Y. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther 2009; 8:2103-9; PMID:19671746; http://dx.doi.org/10.1158/1535-7163.MCT-09-0282
- Gujral TS, Chan M, Peshkin L, Sorger PK, Kirschner MW, MacBeath G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014; 159:844-56; PMID:25417160; http://dx.doi.org/10.1016/j.cell.2014.10.032
- Chan DW, Chan CY, Yam JW, Ching YP, Ng IO. Prickle-1 negatively regulates Wnt/beta-catenin pathway by promoting Dishevelled ubiquitination/degradation in liver cancer. Gastroenterology 2006; 131:1218-27; PMID:17030191; http://dx.doi.org/10.1053/j.gastro.2006.07.020
- Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog 2011; 10:5; PMID:21483657; http://dx.doi.org/10.4103/1477-3163.78111
- Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013; 2:e71; PMID:24042735; http://dx.doi.org/10.1038/oncsis.2013.35
- Fang JY, Lu J, Chen YX, Yang L. Effects of DNA methylation on expression of tumor suppressor genes and proto-oncogene in human colon cancer cell lines. World J Gastroenterol 2003; 9:1976-80; PMID:12970888; http://dx.doi.org/10.3748/wjg.v9.i9.1976
- Hagiwara K, Ito H, Murate T, Miyata Y, Ohashi H, Nagai H. PROX1 overexpression inhibits protein kinase C beta II transcription through promoter DNA methylation. Genes Chromosomes Cancer 2012; 51:1024-36; PMID:22833470; http://dx.doi.org/10.1002/gcc.21985
- Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 2002; 31:141-9; PMID:11992124; http://dx.doi.org/10.1038/ng892
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res 2010; 20:320-31; PMID:20133333; http://dx.doi.org/10.1101/gr.101907.109
- Lou S, Lee HM, Qin H, Li JW, Gao Z, Liu X, Chan LL, Kl Lam V, So WY, Wang Y, et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol 2014; 15:408; PMID:25074712; http://dx.doi.org/10.1186/s13059-014-0408-0
- Wang Y, Jadhav RR, Liu J, Wilson D, Chen Y, Thompson IM, Troyer DA, Hernandez J, Shi H, Leach RJ, Huang TH, Jin VX. Roles of Distal and Genic Methylation in the Development of Prostate Tumorigenesis Revealed by Genome-wide DNA Methylation Analysis. Sci Rep 2016; 6:22051; PMID:26924343; http://dx.doi.org/10.1038/srep22051
- Diagenode. Innovating Epigenetic Solutions. http://www.diagenode.com/media/catalog/file/MethylCap_kit_manual.pdf
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357-9; PMID:22388286; http://dx.doi.org/10.1038/nmeth.1923
- National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078-9; PMID:19505943; http:dx.doi.org/10.1093/bioinformatics/btp352
- Chavez L, Lienhard M, Dietrich J. MEDIPS: (MeD)IP-seq data analysis. R package version 1.14.0. 2013
- Patai ÁV, Barták BK, Péterfia B, Micsik T, Horváth R, Sumánszki C, Péter Z, Patai Á, Valcz G, Kalmár A, et al. Comprehensive DNA methylation and mutation analyses reveal a methylation signature in colorectal sessile serrated adenomas. Pathol Oncol Res 2016 (in press)