
ABSTRACT
We evaluated the association between methylation of 9 genes, SCGB3A1, GSTP1, RARB, SYK, FHIT, CDKN2A, CCND2, BRCA1, and SFN in tumor samples from 720 breast cancer cases with clinicopathological features of the tumors and survival. Logistic regression was used to estimate odds ratios (OR) of methylation and Cox proportional hazards models to estimate hazard ratios (HR) between methylation and breast cancer related mortality. Estrogen receptor (ER) and progesterone receptor (PR) positivity were associated with increased SCGB3A1 methylation among pre- and post-menopausal cases. Among premenopausal women, compared with Stage 0 cases, cases of invasive cancer were more likely to have increased methylation of RARB (Stage I OR = 4.7, 95% CI: 1.1–19.0; Stage IIA/IIB OR = 9.7, 95% CI: 2.4–39.9; Stage III/IV OR = 5.6, 95% CI: 1.1–29.4) and lower methylation of FHIT (Stage I OR = 0.2, 95% CI: 0.1–0.9; Stage IIA/IIB OR = 0.2, 95% CI: 0.1–0.8; Stage III/IV OR = 0.6, 95% CI: 0.1–3.4). Among postmenopausal women, methylation of SYK was associated with increased tumor size (OR = 1.7, 95% CI: 1.0–2.7) and higher nuclear grade (OR = 2.0, 95% CI 1.2–3.6). Associations between methylation and breast cancer related mortality were observed among pre- but not post-menopausal women. Methylation of SCGB3A1 was associated with reduced risk of death from breast cancer (HR = 0.41, 95% CI: 0.17–0.99) as was BRCA1 (HR = 0.41, 95% CI: 0.16–0.97). CCND2 methylation was associated with increased risk of breast cancer mortality (HR = 3.4, 95% CI: 1.1–10.5). We observed differences in methylation associated with tumor characteristics; methylation of these genes was also associated with breast cancer survival among premenopausal cases. Understanding of the associations of DNA methylation with other clinicopathological features may have implications for prevention and treatment.
Abbreviations
| SCGB3A1 | = | secretoglobin, family 3A, member 1 |
| GSTP1 | = | glutathione S-transferase pi 1 |
| RARB | = | retinoic acid receptor, beta |
| SYK | = | spleen tyrosine kinase |
| FHIT | = | fragile histidine triad |
| CDKN2A | = | cyclin-dependent kinase inhibitor 2A |
| CCND2 | = | cyclin D2 |
| BRCA1 | = | breast cancer gene 1 |
| SFN | = | stratifin |
Introduction
Understanding how epigenetic changes are related to breast cancer clinicopathological features could lead to better understanding of carcinogenesis and possibly to improvements in prevention, diagnosis, and treatment.Citation1-3 CpG islands that are normally unmethylated may become methylated in cancer cells, which can result in altered expression, including silencing of tumor suppressor or DNA repair genes.Citation4
Over 100 genes have been identified as aberrantly methylated in breast tumors or breast cancer cell lines.Citation4 These genes include functionally important genes, such as tumor suppressor genes, DNA repair genes, genes related to the detoxification of xenobiotics, genes involved in hormone and receptor mediated signaling, and genes involved in cell-cycle control.Citation2,5-8 However, there is limited evidence regarding the extent to which alterations in methylation are associated with other, better studied clinicopathological features, such as hormone receptor status,Citation9-11 human epidermal growth factor receptor (HER2) expression,Citation1,12 stage,Citation13 metastasis,Citation11,13 histologic grade,Citation14 nuclear grade,Citation14 tumor size,Citation2 and overall survival.Citation9 Prior studies of DNA methylation in breast tumors that utilized pyrosequencing were limited by small sample size and included a relatively small number of genes; most were hospital-based studies.Citation9,Citation15-20,Citation21 Furthermore, inconsistent results have been reported, which may be due to varying populations and patient characteristics. Limitations in laboratory methods and differences between tumor and blood DNA are also potential sources of inconsistent findings. Citation1,9,11,22,23
In order to better understand the role of methylation in breast carcinogenesis, we evaluated the association between methylation in breast tumors of 9 genes of significance to regulation of cellular processes with clinicopathological features of tumors for breast cancer cases in a large population-based study. We also assessed the association between methylation in breast tumors and all-cause and breast cancer related mortality.
Results
Descriptive characteristics of the Western New York Exposures and Breast Cancer (WEB) study breast cancer cases included in these analyses compared with those excluded are displayed in . Characteristics were similar for most measures. Among premenopausal cases, those with methylation information available for any of the analyses compared with those not included tended to have tumors that were larger than 2 cm (37% vs. 22%), metastatic disease at presentation (37% vs. 26%), and of a higher stage (Stage III/IV: 10% vs. 7%). Among postmenopausal cases, those with data available were more likely to have tumors larger than 2 cm (22% vs. 16%), metastatic disease (22% vs. 16%), and to be Caucasian (6% vs. 10%).
Table 1. Selected characteristics of WEB study cases by inclusion in methylation analysesFootnotea.
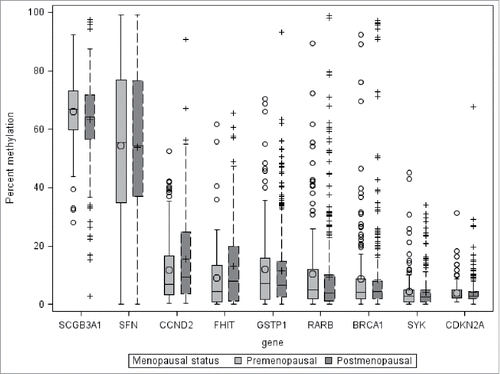
Methylation values for individual CpG sites were correlated to the average for each gene. Spearman's ρ values ranged from 0.38–0.95. The median correlation was 0.75. For the remainder of these analyses, we examined mean methylation for each gene, averaging the data for the several sites within one gene. Distributions of average methylation by gene are depicted in . Most genes were highly skewed toward no methylation, although there was considerable variability between genes.
Figure 1. Distribution of average percent DNA methylation in breast tumors, selected genes, WEB study. The distance between the 25th and 75th percentiles, the inter-quartile range, is represented by the length of the box. The vertical lines represent 1.5 times the 25th and 75th percentile. The symbols outside the vertical lines represent values outside 1.5 times the inter-quartile range. The mean is represented by the symbol within the box and the median by the horizontal line within the box.
Six of the 9 genes analyzed were associated (P < 0.05) with one or more clinicopathological features after adjusting for potential covariates and are presented in the final tables. Adjusted odds ratios (OR) for methylation greater than the median value by clinicopathological feature among premenopausal cases are presented in . Methylation of SCGB3A1 was strongly associated with increased likelihood of ER positive (OR = 5.0, 95% CI: 2.1–12.1) and PR positive (OR = 4.7, 95% CI: 2.1–10.5) tumors. Compared with Stage 0 cases, cases of invasive cancer were more likely to have tumors with percent methylation of RARB above the median value (Stage I vs. Stage 0 OR = 4.7, 95% CI: 1.1–19.0, Stage IIA/IIB vs. Stage 0 OR = 9.7, 95% CI: 2.4–39.9, Stage III/IV vs. Stage 0 OR = 5.6, 95% CI: 1.1–29.4). Cases of invasive breast cancer tended to have lower methylation of FHIT than those with Stage 0 breast cancer, although the confidence interval included the null value when comparing Stage III /IV to Stage 0 (OR = 0.6, 95% CI: 0.1–3.5).
Table 2. Association of methylation above the median for selected genes with selected clinicopathological factors for breast cancer, premenopausal women WEB study 1996–2001.
Adjusted ORs for methylation greater than the median value by clinicopathological feature among postmenopausal cases are presented in . Methylation of SCGB3A1 was associated ER positive (OR = 2.9, 95% CI: 1.5–5.6) and PR positive (OR = 2.3; 95% CI: 1.4–4.0) tumors and the absence of vascular/lymph invasion (OR = 0.6, 95% CI: 0.3–1.0). Tumors larger than 2 cm tended to have more methylation of SYK (OR = 1.7, 95% CI: 1.2–3.6). Additionally, tumors that had a nuclear grade of III were more likely to have high methylation of SYK than nuclear grade I tumors (OR = 2.0, 95% CI: 1.2–3.6). Postmenopausal cases with vascular/lymph invasion were more likely to have high methylation of CCND2 (OR = 2.2, 95% CI: 1.0–4.5).
Table 3. Association of methylation above the median for selected genes with selected clinicopathological factors for breast cancer, postmenopausal women WEB study 1996–2001.
We also assessed the association between clinicopathological features and breast cancer excluding Stage 0 cases. When Stage 0 cases were excluded most results were similar in direction and magnitude with some difference in statistical significance because of the smaller sample size. Methylation of RARB and FHIT were associated with higher stage in the primary analyses but when Stage 0 cases were excluded we did not observe evidence of an exposure-response gradient between stage and methylation.
Tumor DNA methylation information was obtained for 107 of the 170 breast cancer cases who died during follow-up. Sixty-two of these cases died from breast cancer. The mean follow-up for breast cancer cases included in this analysis was 89 months (standard deviation 21 months). Methylation status was not associated with all-cause mortality (results not shown). Results of methylation and breast cancer related mortality among premenopausal women with invasive breast cancer are presented in . Among premenopausal cases, methylation of CCND2 was associated with increased risk of death from breast cancer (HR = 3.41, 95% CI: 1.11–10.48). Methylation of SCGB3A1 and BRCA1 were associated with reduced risk of breast cancer related mortality (SCGB3A1: HR = 0.41, 95% CI: 0.17–0.99, BRCA1: HR = 0.40, 95% CI: 0.16–0.97). When analyses were further adjusted for TNM stage and ER status results were similar (CCDND2: HR = 2.27, 95% CI: 0.74–7.03, SCGB3A1: HR = 0.44, 95% CI: 0.17–1.14, BRCA1: HR = 0.37, 95% CI: 0.15–0.96). However, the associations between methylation of CCND2, SCGB3A1 and breast cancer mortality were attenuated and statistically non-significant. We did not observe any association between methylation status of the selected genes and breast cancer related mortality among postmenopausal cases of invasive breast cancer ().
Table 4. Hazard ratios (HR) and 95% confidence intervals (CI) for death from breast cancer by methylation of selected genes, WEB study premenopausal cases of breast cancer.
Table 5. Hazard ratios (HR) and 95% confidence intervals (CI) for death from breast cancer by methylation of selected genes, WEB study postmenopausal cases of invasive breast cancer.
Discussion
We evaluated associations between methylation of 9 selected genes and clinicopathological features of primary breast cancer cases. Functions of these genes are summarized in . We did not observe a clear pattern between overall methylation and clinicopathological features. Several associations were identified and were gene-specific, and in some instances depended on menopausal status. For instance, ER and/or PR positive tumors tended to have greater methylation of SCGB3A1 regardless of menopausal status, while associations between methylation of RARB, SYK, and FHIT and prognostic factors depended on menopausal status. Additionally, we found that methylation of the selected genes was only associated with breast-cancer related mortality among premenopausal cases. It is increasingly clear that aberrant methylation is a critical component to carcinogenesis and that factors affecting methylation of different genes may differ. Understanding of the factors related to these changes may provide important insight into both prevention and treatment. Our study provides information on a large, population-based sample of breast tumors, stratified by menopausal status, with a quantitative measure of methylation provided.
Table 6. Summary of functions of genes included in analysis.
In examination of associations with methylation of specific genes, we found increased methylation of SCGB3A1 was strongly associated with ER and PR positive tumors, which is consistent with previous studies.Citation9,19,23,24 Methylation of SCGB3A1 has been frequently reported in breast tumors, and absent in adjacent normal breast tissue and has been posited as an early event in breast carcinogenesis (reviewed in ref.Citation25). We also observed that methylation of SCGB3A1 was associated with reduced breast cancer related mortality among premenopausal women only.
Methylation of RARB was higher among invasive breast cancer tumors than Stage 0 tumors, among premenopausal women only. Methylation of RARB has previously been associated with poor prognostic factors, including high histological grade, high proliferation rate, increased tumor size, and lymph node metastasis; menopausal status was not addressed in the prior study.Citation13 However, we only observed an association between TNM stage and methylation of RARB among premenopausal cases.
Methylation of SYK was associated with larger tumor size and higher nuclear grade among postmenopausal cases. SYK is involved in cell growth signaling and is considered a tumor suppressor gene.Citation26 Loss of expression of SYK has previously been associated with increased breast cancer cell proliferation, invasivenessCitation27 and overall poor breast cancer prognosis.Citation28,29 Our finding that higher methylation of SYK was associated with increased tumor size suggests that methylation of this gene may contribute to tumor growth.
Methylation of FHIT has previously been found to be associated with several clinicopathological features among both pre- and postmenopausal women including poorly differentiated breast tumors,Citation30 higher TNM stage,Citation30-32 increased likelihood of lymph node involvement,Citation31 and negative ER and PR status.Citation31 In a previous study,Citation20 there was no association between methylation of FHIT and clinicopathological features. However, this study had only 60 participants and did not stratify on menopausal status. In our analyses there was some indication that methylation of FHIT was associated with TNM stage among premenopausal cases. The number of premenopausal cases with Stage III/IV tumors was small and a pattern of association was not readily apparent. Stage I and IIA/IIB tumors had lower methylation FHIT than Stage 0 (ductal carcinoma in situ) while Stage III/IV tumors had higher methylation of FHIT than Stage 0. Conversely, among postmenopausal women, those with vascular or lymph invasion at diagnosis tended to have higher methylation of FHIT.
BRCA1 methylation was associated with decreased risk of breast cancer related mortality among premenopausal women and larger tumor size among postmenopausal women. Reports of methylation of BRCA1 in breast tumors and clinicopathological features are inconsistent. Methylation of BRCA1 has been found to be associated with decreased expression,Citation33,34 but was not associated with clinicopathological features in a previous study of 49 breast tumors.Citation33 Furthermore, in a study of 139 incident breast cancer cases, triple negative status was the only clinicopathological feature associated with BRCA1 methylation; BRCA1 methylation has also been reported to be associated with poor overall and breast-cancer specific survival.Citation34 Our findings of an association between methylation of BRCA1 and a decreased risk of breast cancer related mortality among premenopausal women are not consistent with these reports and warrant further investigation.
CCND2 methylation was associated with increased risk of breast cancer related mortality among premenopausal women. Among postmenopausal women, tumors with vascular/lymph invasion had higher methylation of CCND2. CCND2 has previously been reported to be methylated in breast tumors.Citation35,36 To our knowledge, ours is the first study to assess methylation of CCND2 and breast cancer related mortality.
We did not observe any association between methylation of CDKN2A or SFN and clinicopathological features or survival. These genes have previously been reported to be methylated in breast tumors.Citation1,21,33,37-39 Results of prior studies of methylation of CDKN2A and breast cancer have been mixed. In one study, methylation of CDKN2A was associated with higher stage of breast cancer and HER2 positive breast cancer,Citation21 while in another study, there was no association between CDKN2A methylation and clinicopathological features.Citation40 A previous studyCitation39 found increased methylation of SFN to be associated with poor prognosis. Our results are not consistent with this finding as we did not observe any association between methylation status of SFN and clinicopathological features or survival.
In our study, methylation status of GSTP1 was not associated with any clinicopathological feature or breast cancer survival. Promoter methylation of GSTP1 leads to inhibition of GSTP1 expression and decreased capability to detoxify estrogens. Methylation of GSTP1 in breast tumors has also previously been associated with poor prognostic factors including lymph node metastasis,Citation1,41 larger tumor size,Citation41 HER2 positive statusCitation42 and higher stage.Citation21,41 In our sample we did not observe an association of those poor prognostic factors and methylation of GSTP1. Furthermore, our findings are different from those of previous studies reporting an association between methylation of GSTP1 in breast tumor DNA and poor prognosis.Citation43,44. Previous studies that reported an association with poor prognosis and methylation of GSTP1 used MethylightCitation43 or immunohistochemistry.Citation44 A recent analysis comparing the Methylight assay and pyrosequencing reported that the Methylight assay may not detect a substantial proportion of methylated GSTP1, which could explain inconsistent findings regarding methylation status of GSTP1 and breast cancer prognosis and clinicopathological features.Citation45 Although differences between assays have been less studied for other genes, overall several of our findings are not consistent with previous reports, suggesting that assay technologies may contribute a large amount of variability between studies and should be assessed in future studies.
Our study has limitations that should be considered in interpretation of these findings. We were unable to obtain methylation information for all cases in the WEB study because of participants whose tumor tissue was not available or the existing tissue was not useable for the laboratory assay. Those included tended to have larger tumors and poorer prognostic factors than those not included. Another limitation is that some participants did not have methylation data for all of the genes, limiting statistical power. Further, associations for premenopausal women need to be interpreted with caution because of the smaller sample size for that group.
Our study also has several important strengths. Our study sample was relatively big, larger than most prior reports of methylation and clinicopathological features of breast cancer. Rather than a hospital-based study, the samples came from a population-based sample, which included most of the hospitals of the study region. We used pyrosequencing to measure methylation, a continuous and highly sensitive measure.Citation46 Stratification by menopausal status allowed us to examine differences by that variable.
In conclusion, we found that methylation of particular genes was associated with clinicopathological features of breast tumors, particularly SCGB3A1, RARB, SYK, FHIT, CCND2, and BRCA1. We also observed limited evidence that methylation of SCGB3A1, BRCA1, and CCND2 were associated with breast cancer related mortality among premenopausal women only. Given the differences in findings for each gene studied, it appears that there are differences in factors leading to methylation of each gene as well as differences between factors related to methylation and to the other clinicopathological features. There may be potentially important modification of these associations by menopausal status that should be considered further. Future studies on methylation in a larger number of genes with a greater sample size are warranted. Several of our findings were inconsistent with previous studies, which could be the result of differences between samples, limitations in study design, or differences in assay technology and laboratory methods. Understanding of the processes leading to altered methylation and the associations of aberrant methylation with other clinicopathological features is of importance to better understand both carcinogenesis and prognosis for breast tumors.
Materials and methods
Detailed study methods of the Western New York Exposures and Breast Cancer (WEB) study have been published previously.Citation14,47 Briefly, the WEB study was a population-based case-control study of breast cancer, conducted from 1996 to 2001. The current analyses of tumor methylation were restricted to breast cancer cases. Cases were 1,170 women, residents of Erie and Niagara counties, with primary, histologically confirmed, incident breast cancer, aged 35–79 y at diagnosis, and interviewed within one year of diagnosis. Cases of Stage 0 breast cancer or ductal carcinoma in situ (DCIS) were included because aberrant methylation is expected to occur early in carcinogenesis.Citation48,49 All participants provided informed consent, and the study protocol was approved by the Institutional Review Boards of the University at Buffalo and all participating institutions.
Participants completed extensive in-person interviews and self-administered questionnaires that included queries on medical history, breast cancer risk factors and other potential confounding factors. Information on tumor size, histologic grade, and tumor-node-metastasis (TNM) stage was abstracted from medical records by trained research nurses using a standard protocol. Estrogen receptor (ER) and progesterone receptor (PR) status were determined by immunohistochemistry at Georgetown University as previously described.Citation14,50 Vital status for WEB study participants was obtained via linkage with the National Death Index through December 31, 2006. Cause of death was broadly categorized as breast cancer, other cancer, cardiovascular, or other cause.
Pyrosequencing
Candidate genes for the methylation analyses were selected because of their significance to cellular regulation and because they had been previously reported to be aberrantly methylated in breast cancer.Citation19-21,34,37,39,41,51 Tumor blocks were obtained from 922 of the women with breast cancer in the WEB study. Of these tumor blocks, some were depleted in previous analyses. Methylation data were available for 720 cases. Sample size varied for each gene primarily because individual samples did not pass quality control for some of the assays. The genes assessed in these analyses were as follows: secretoglobin, family 3A, member 1 (SCGB3A1); glutathione S-transferase pi 1 (GSTP1); retinoic acid receptor, β (RARB); spleen tyrosine kinase (SYK); fragile histidine triad (FHIT); cyclin-dependent kinase inhibitor 2A (CDKN2A); cyclin D2 (CCND2); breast cancer gene 1 (BRCA1); and stratifin (SFN).
Bisulfite pyrosequencing is a quantitative technique used to measure DNA methylation, which has been identified as having high reproducibility and sensitivity.Citation46 We used the PyroMark MD Pyrosequencing system (Qiagen, Valencia, CA) to detect methylated CpG sites in sequencing reactions.Citation52 Tumor samples were microdissected from fixed microscope slides. Genomic DNA (500 ng) was treated with sodium bisulfite using the EZ DNA Methylation kit (Zymo Research, Orange, CA). Bisulfite-treated DNA was amplified with specific primers for each gene of interest. The Pyro Mark Assay Design program and the Pyro Q-CpG software were used for primer designs and data analysis, respectively. Ten percent of total samples were assayed in duplicate for each gene for quality control purposes. Additionally, as a positive control, human methylated and non-methylated DNA (Zymo Research, Orange, CA) was used along with water blanks as a negative control. Primer sets, SYK (Cat. # PM00151816), SCGB3A1 (Cat. # PM00022687), GSTP1 (Cat. # PM00151809), CDKN2A (Cat. # 972012), and CCND2 (Cat. # PM00051674) were purchased from Qiagen.
The intraclass correlation coefficient between duplicate samples for methylation of estrogen receptor 1 (ESR1) and Ras association domain family member 1 (RASSF1) genes were both below 0.50 and so they were not analyzed further. The intraclass correlation coefficient for the remaining 9 genes included in these analyses ranged from 0.68 to 0.98.
Statistical analysis
Quality control was assessed by calculating an intraclass correlation coefficient for the 10% of samples that were analyzed in duplicate. Site specific methylation percentages were available for 4–10 CpG sites for each gene. Methylation of individual CpG sites was averaged for each gene for each participant. The association between methylation of individual CpG sites of a gene and the average methylation for each gene was measured with Spearman's correlation. Student's t-tests and Χ2 tests were used to assess differences in descriptive characteristics among WEB study cases with information on methylation and those not included in any of the analyses.
All analyses of the association between methylation of select genes and clinicopathological features were stratified by menopausal status because of previous reports of a difference in etiology.Citation53 Unconditional logistic regression was employed to estimate odds ratios (OR) and 95% confidence intervals (CI) for odds of increased methylation with regards to selected features. We initially considered methylation greater than 15% as the outcome of interest, as had been done previously.Citation54 However, these models were not feasible; there were too many categories with very few or no participants above 15%. We then considered methylation above the median methylation value for each gene. Covariates that were considered as potential confounders were age, age at menarche, race, smoking status, body mass index (BMI), and parity. Variables that changed the association between methylation of any gene and any clinicopathological feature by at least 10 percent were included in adjusted models. Final models were adjusted for age in years, age at menarche, race, smoking status, and BMI.
Hazard ratios (HR) and 95% confidence intervals for breast cancer and all-cause mortality were calculated using Cox proportional hazards models. Non-invasive (DCIS) cases were excluded from the breast cancer related mortality analyses. Covariates considered for inclusion in these models were, age, age at menarche, race, smoking status, BMI, and parity. Covariates that changed the association between breast cancer mortality and methylation of any of the genes by at least 10% were included in the final models. We also further adjusted for ER status and TNM stage to determine if the association was mediated through these factors.
All P-values reported are 2-sided and statistical significance was defined as P < 0.05. Statistical analyses were conducted using SPSS version 21.00 (IBM Corp. Released 2012. IBM SPSS Statistics for Windows, Armonk, NY: IBM Corp) and SAS Enterprise Guide version 4.3 (SAS Institute, Cary, NC).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Sunami E, Shinozaki M, Sim MS, Nguyen SL, Vu AT, Giuliano AE, Hoon DS. Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res 2008; 10:16; PMID:18485221; http://dx.doi.org/10.1186/bcr2098
- Dworkin AM, Huang TH, Toland AE. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol 2009; 19:165-71; PMID:19429480; http://dx.doi.org/10.1016/j.semcancer.2009.02.007
- Saxena A, Dhillon VS, Shahid M, Khalil HS, Rani M, Prasad DT, Hedau S, Hussain A, Naqvi RA, Deo SV, et al. GSTP1 methylation and polymorphism increase the risk of breast cancer and the effects of diet and lifestyle in breast cancer patients. Exp Ther Med 2012; 4:1097-103; PMID:23226781; http://dx.doi.org/10.3892/etm.2012.710
- Jovanovic J, Ronneberg JA, Tost J, Kristensen V. The epigenetics of breast cancer. Mol Oncol 2010; 4:242-54; PMID:20627830; http://dx.doi.org/10.1016/j.molonc.2010.04.002
- Chen KM, Stephen JK, Raju U, Worsham MJ. Delineating an epigenetic continuum for initiation, transformation and progression to breast cancer. Cancers 2011; 3:1580-92; PMID:21776373; http://dx.doi.org/10.3390/cancers3021580
- Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 2008; 659:40-8; PMID:18407786; http://dx.doi.org/10.1016/j.mrrev.2008.02.004
- Lodygin D, Hermeking H. The role of epigenetic inactivation of 14-3-3sigma in human cancer. Cell Res 2005; 15:237-46; PMID:15857578; http://dx.doi.org/10.1038/sj.cr.7290292
- McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res 2009; 15:3927-37; PMID:19509173; http://dx.doi.org/10.1158/1078-0432.CCR-08-2784
- Xu J, Shetty PB, Feng W, Chenault C, Bast RC, Jr., Issa JP, Hilsenbeck SG, Yu Y. Methylation of HIN-1, RASSF1A, RIL and CDH13 in breast cancer is associated with clinical characteristics, but only RASSF1A methylation is associated with outcome. BMC Cancer 2012; 12:1471-2407; PMID:22695491; http://dx.doi.org/10.1186/1471-2407-12-243
- Kajabova V, Smolkova B, Zmetakova I, Sebova K, Krivulcik T, Bella V, Kajo K, Machalekova K, Fridrichova I. RASSF1A Promoter Methylation Levels Positively Correlate with Estrogen Receptor Expression in Breast Cancer Patients. Transl Oncol 2013; 6:297-304; PMID:23730409; http://dx.doi.org/10.1593/tlo.13244
- Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, Maher ER, Latif F. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res 2011; 71:2988-99; PMID:21363912; http://dx.doi.org/10.1158/0008-5472.CAN-10-4026
- Fiegl H, Millinger S, Goebel G, Muller-Holzner E, Marth C, Laird PW, Widschwendter M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res 2006; 66:29-33; PMID:16397211; http://dx.doi.org/10.1158/0008-5472.CAN-05-2508
- Marzese DM, Hoon DSB, Chong KK, Gago FE, Orozco JI, Tello OM, Vargas-Roig LM, Roqué M. DNA methylation index and methylation profile of invasive ductal breast tumors. J Mol Diagn 2012; 14:613-22; PMID:22925694; http://dx.doi.org/10.1016/j.jmoldx.2012.07.001
- Tao MH, Shields PG, Nie J, Millen A, Ambrosone CB, Edge SB, Krishnan SS, Marian C, Xie B, Winston J, et al. DNA hypermethylation and clinicopathological features in breast cancer: the Western New York Exposures and Breast Cancer (WEB) Study. Breast Cancer Res Treat 2009; 114:559-68; PMID:18463976; http://dx.doi.org/10.1007/s10549-008-0028-z
- Halvorsen AR, Helland A, Fleischer T, Haug KM, Grenaker Alnaes GI, Nebdal D, Syljuasen RG, Touleimat N, Busato F, Tost J, et al. Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int J Cancer 2014; 135:2085-95; PMID:24658971; http://dx.doi.org/10.1002/ijc.28862
- Watanabe Y, Maeda I, Oikawa R, Wu W, Tsuchiya K, Miyoshi Y, Itoh F, Tsugawa K, Ohta T. Aberrant DNA methylation status of DNA repair genes in breast cancer treated with neoadjuvant chemotherapy. Genes Cells 2013; 18:1120-30; PMID:24581343; http://dx.doi.org/10.1111/gtc.12100
- Foedermayr M, Sebesta M, Rudas M, Berghoff AS, Promberger R, Preusser M, Dubsky P, Fitzal F, Gnant M, Steger GG, et al. BRCA-1 methylation and TP53 mutation in triple-negative breast cancer patients without pathological complete response to taxane-based neoadjuvant chemotherapy. Cancer Chemother Pharmacol 2014; 73:771-8; PMID:24526178; http://dx.doi.org/10.1007/s00280-014-2404-1
- Ghosh S, Gu F, Wang CM, Lin CL, Liu J, Wang H, Ravdin P, Hu Y, Huang TH, Li R. Genome-wide DNA methylation profiling reveals parity-associated hypermethylation of FOXA1. Breast Cancer Res Treat 2014; 147:653-9; PMID:25234841; http://dx.doi.org/10.1007/s10549-014-3132-2
- Feng W, Orlandi R, Zhao N, Carcangiu ML, Tagliabue E, Xu J, Bast RC, Jr., Yu Y. Tumor suppressor genes are frequently methylated in lymph node metastases of breast cancers. BMC Cancer 2010; 10:1471-2407; PMID:20642860; http://dx.doi.org/10.1186/1471-2407-10-378
- Jeong YJ, Jeong HY, Lee SM, Bong JG, Park SH, Oh HK. Promoter methylation status of the FHIT gene and Fhit expression: association with HER2/neu status in breast cancer patients. Oncol Rep 2013; 30:2270-8; PMID:23969757; http://dx.doi.org/10.1593/tlo.09148
- Klajic J, Fleischer T, Dejeux E, Edvardsen H, Warnberg F, Bukholm I, Lonning PE, Solvang H, Borresen-Dale AL, Tost J, et al. Quantitative DNA methylation analyses reveal stage dependent DNA methylation and association to clinico-pathological factors in breast tumors. BMC Cancer 2013; 13:1471-2407; PMID:24093668; http://dx.doi.org/10.1186/1471-2407-13-456
- Park SY, Kwon HJ, Lee HE, Ryu HS, Kim SW, Kim JH, Kim IA, Jung N, Cho NY, Kang GH. Promoter CpG island hypermethylation during breast cancer progression. Virchows Arch 2011; 458:73-84; PMID:21120523; http://dx.doi.org/10.1007/s00428-010-1013-6
- Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, Tse CK, Swift-Scanlan T, Geradts J, Troester MA, et al. DNA methylation profiling in the Carolina Breast Cancer Study defines cancer subclasses differing in clinicopathological features and survival. Breast Cancer Res 2014; 16:450; PMID:25287138; http://dx.doi.org/10.1186/s13058-014-0450-6
- Feng W, Shen L, Wen S, Rosen DG, Jelinek J, Hu X, Huan S, Huang M, Liu J, Sahin AA, et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res 2007; 9:R57; PMID:17764565; http://dx.doi.org/10.1186/bcr1762
- Dai D, Dong XH, Cheng ST, Zhu G, Guo XL. Aberrant promoter methylation of HIN-1 gene may contribute to the pathogenesis of breast cancer: a meta-analysis. Tumour Biol 2014; 35:8209-16; PMID:24850174; http://dx.doi.org/10.1007/s13277-014-2055-1
- Golouh R, Cufer T, Sadikov A, Nussdorfer P, Usher PA, Brunner N, Schmitt M, Lesche R, Maier S, Timmermans M, et al. The prognostic value of Stathmin-1, S100A2, and SYK proteins in ER-positive primary breast cancer patients treated with adjuvant tamoxifen monotherapy: an immunohistochemical study. Breast Cancer Res Treat 2008; 110:317-26; PMID:17874182; http://dx.doi.org/10.1007/s10549-007-9724-3
- Yuan Y, Liu H, Sahin A, Dai JL. Reactivation of SYK expression by inhibition of DNA methylation suppresses breast cancer cell invasiveness. Int J Cancer 2005; 113:654-9; PMID:15455373; http://dx.doi.org/10.1002/ijc.20628
- Blancato J, Graves A, Rashidi B, Moroni M, Tchobe L, Ozdemirli M, Kallakury B, Makambi KH, Marian C, Mueller SC. SYK allelic loss and the role of Syk-regulated genes in breast cancer survival. PLoS One 2014; 9:e87610; PMID:24523870; http://dx.doi.org/10.1371/journal.pone.0087610
- Martens JW, Nimmrich I, Koenig T, Look MP, Harbeck N, Model F, Kluth A, Bolt-de Vries J, Sieuwerts AM, Portengen H, et al. Association of DNA methylation of phosphoserine aminotransferase with response to endocrine therapy in patients with recurrent breast cancer. Cancer Res 2005; 65:4101-17; PMID:15899800; http://dx.doi.org/10.1158/0008-5472.CAN-05-0064
- Raish M, Dhillon VS, Ahmad A, Ansari MA, Mudassar S, Shahid M, Batra V, Gupta P, Das BC, Shukla N, et al. Promoter Hypermethylation in Tumor Suppressing Genes p16 and FHIT and Their Relationship with Estrogen Receptor and Progesterone Receptor Status in Breast Cancer Patients from Northern India. Transl Oncol 2009; 2:264-70; PMID:19956388; http://dx.doi.org/10.1593/tlo.09148
- Zaki SM, Abdel-Azeez HA, El Nagar MR, Metwally KA, MM SA. Analysis of FHIT Gene Methylation in Egyptian Breast Cancer Women: Association with Clinicopathological Features. Asian Pac J Cancer Prev 2015; 16:1235-9; PMID:25735361; http://dx.doi.org/10.7314/APJCP.2015.16.3.1235
- Syeed N, Husain SA, Sameer AS, Chowdhri NA, Siddiqi MA. Mutational and promoter hypermethylation status of FHIT gene in breast cancer patients of Kashmir. Mutat Res 2011; 707:1-8; PMID:21095196; http://dx.doi.org/10.1016/j.mrfmmm.2010.11.001
- Li Q, Wei W, Jiang YI, Yang H, Liu J. Promoter methylation and expression changes of in cancerous tissues of patients with sporadic breast cancer. Oncology letters 2015; 9:1807-13; PMID:25789047; http://dx.doi.org/10.3892/ol.2015.2908
- Hsu NC, Huang YF, Yokoyama KK, Chu PY, Chen FM, Hou MF. Methylation of BRCA1 promoter region is associated with unfavorable prognosis in women with early-stage breast cancer. PLoS One 2013; 8:6; PMID:23405268; http://dx.doi.org/10.1371/journal.pone.0056256
- Fackler MJ, McVeigh M, Evron E, Garrett E, Mehrotra J, Polyak K, Sukumar S, Argani P. DNA methylation of RASSF1A, HIN-1, RAR-β, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Int J Cancer 2003; 107:970-5; PMID:14601057; http://dx.doi.org/10.1002/ijc.11508
- Li S, Rong M, Iacopetta B. DNA hypermethylation in breast cancer and its association with clinicopathological features. Cancer Lett 2006; 237:272-80; PMID:16029926; http://dx.doi.org/10.1016/j.canlet.2005.06.011
- Xu X, Gammon MD, Jefferson E, Zhang Y, Cho YH, Wetmur JG, Teitelbaum SL, Bradshaw PT, Terry MB, Garbowski G, et al. The influence of one-carbon metabolism on gene promoter methylation in a population-based breast cancer study. Epigenetics 2011; 6:1276-83; PMID:22048254; http://dx.doi.org/10.4161/epi.6.11.17744
- Verschuur-Maes AH, de Bruin PC, van Diest PJ. Epigenetic progression of columnar cell lesions of the breast to invasive breast cancer. Breast Cancer Res Treat 2012; 136:705-15; PMID:23104224; http://dx.doi.org/10.1007/s10549-012-2301-4
- Cheol Kim D, Thorat MA, Lee MR, Cho SH, Vasiljevic N, Scibior-Bentkowska D, Wu K, Ahmad AS, Duffy S, Cuzick JM, et al. Quantitative DNA methylation and recurrence of breast cancer: a study of 30 candidate genes. Cancer Biomark 2012; 11:75-88; PMID:23011154; http://dx.doi.org/10.3233/CBM-2012-0266
- Wang S, Dorsey TH, Terunuma A, Kittles RA, Ambs S, Kwabi-Addo B. Relationship between tumor DNA methylation status and patient characteristics in African-American and European-American women with breast cancer. PLoS One 2012; 7:31; PMID:22701537; http://dx.doi.org/10.1371/journal.pone.0037928
- Arai T, Miyoshi Y, Kim SJ, Taguchi T, Tamaki Y, Noguchi S. Association of GSTP1 CpG islands hypermethylation with poor prognosis in human breast cancers. Breast Cancer Res Treat 2006; 100:169-76; PMID:16791478; http://dx.doi.org/10.1007/s10549-006-9241-9
- Miyake T, Nakayama T, Kagara N, Yamamoto N, Nakamura Y, Otani Y, Uji K, Naoi Y, Shimoda M, Maruyama N, et al. Association of GSTP1 Methylation with Aggressive Phenotype in ER-positive Breast Cancer. Anticancer Res 2013; 33:5617-23; PMID:24324107
- Cho YH, Shen J, Gammon MD, Zhang YJ, Wang Q, Gonzalez K, Xu X, Bradshaw PT, Teitelbaum SL, Garbowski G, et al. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast Cancer Res Treat 2012; 131:197-205; PMID:21837480; http://dx.doi.org/10.1007/s10549-011-1712-y
- Sharma G, Mirza S, Parshad R, Srivastava A, Gupta SD, Pandya P, Ralhan R. Clinical significance of promoter hypermethylation of DNA repair genes in tumor and serum DNA in invasive ductal breast carcinoma patients. Life Sci 2010; 87:83-91; PMID:20470789; http://dx.doi.org/10.1016/j.lfs.2010.05.001
- Alnaes GI, Ronneberg JA, Kristensen VN, Tost J. Heterogeneous DNA Methylation Patterns in the GSTP1 Promoter Lead to Discordant Results between Assay Technologies and Impede Its Implementation as Epigenetic Biomarkers in Breast Cancer. Genes 2015; 6:878-900; PMID:26393654; http://dx.doi.org/10.3390/genes6030878
- Quillien V, Lavenu A, Karayan-Tapon L, Carpentier C, Labussiere M, Lesimple T, Chinot O, Wager M, Honnorat J, Saikali S, et al. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 2012; 118:4201-11; PMID:22294349; http://dx.doi.org/10.1002/cncr.27392
- Han D, Nie J, Bonner MR, McCann SE, Muti P, Trevisan M, Ramirez-Marrero FA, Vito D, Freudenheim JL. Lifetime adult weight gain, central adiposity, and the risk of pre- and postmenopausal breast cancer in the Western New York exposures and breast cancer study. Int J Cancer 2006; 119:2931-7; PMID:17016824; http://dx.doi.org/10.1002/ijc.22236
- Johnson KC, Koestler DC, Fleischer T, Chen P, Jenson EG, Marotti JD, Onega T, Kristensen VN, Christensen BC. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin Epigenetics 2015; 7:015-0094; PMID:26213588; http://dx.doi.org/10.1186/s13148-015-0094-0
- Muggerud AA, Ronneberg JA, Warnberg F, Botling J, Busato F, Jovanovic J, Solvang H, Bukholm I, Borresen-Dale AL, Kristensen VN, et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res 2010; 12:7; PMID:20056007; http://dx.doi.org/10.1186/bcr2466
- Brasky TM, Bonner MR, Moysich KB, Ambrosone CB, Nie J, Tao MH, Edge SB, Kallakury BV, Marian C, Goerlitz DS, et al. Non-steroidal anti-inflammatory drugs (NSAIDs) and breast cancer risk: differences by molecular subtype. Cancer Causes Control 2011; 22:965-75; PMID:21516318; http://dx.doi.org/10.1007/s10552-011-9769-9
- Zhang X, Shrikhande U, Alicie BM, Zhou Q, Geahlen RL. Role of the protein tyrosine kinase Syk in regulating cell-cell adhesion and motility in breast cancer cells. Mol Cancer Res 2009; 7:634-44; PMID:19435818; http://dx.doi.org/10.1158/1541-7786.MCR-08-0371
- Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc 2007; 2:2265-75; PMID:17853883; http://dx.doi.org/10.1038/nprot.2007.314
- Anderson WF, Rosenberg PS, Prat A, Perou CM, Sherman ME. How Many Etiological Subtypes of Breast Cancer: Two, Three, Four, Or More? J Natl Cancer Institute 2014; 106:1-11; PMID:25118203; http://dx.doi.org/10.1093/jnci/dju165.
- Moelans CB, Verschuur-Maes AH, van Diest PJ. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. J Pathol 2011; 225:222-31; PMID:21710692; http://dx.doi.org/10.1002/path.2930
- Coopman PJP, Do MTH, Barth M, Bowden ET, Hayes AJ, Basyuk E, Blancato JK, Vezza PR, McLeskey SW, Mangeat PH, et al. The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature 2000; 406:742-7; PMID:10963601; http://dx.doi.org/10.1038/35021086
- Yuan Y, Liu H, Sahin A, Dai JL. Reactivation of SYK expression by inhibition of DNA methylation suppresses breast cancer cell invasiveness. Int J Cancer 2005; 113:654-9; PMID:15455373; http://dx.doi.org/10.1002/ijc.20628
- Szyf M, Pakneshan P, Rabbani SA. DNA methylation and breast cancer. Biochem Pharmacol 2004; 68:1187-97; PMID:15313416; http://dx.doi.org/10.1016/j.bcp.2004.04.030
- Chavez-Munoz C, Hartwell R, Jalili RB, Jafarnejad SM, Lai A, Nabai L, Ghaffari A, Hojabrpour P, Kanaan N, Duronio V, et al. SPARC/SFN interaction, suppresses type I collagen in dermal fibroblasts. J Cell Biochem 2012; 113:2622-32; PMID:22422640; http://dx.doi.org/10.1002/jcb.24137
- Mirza S, Sharma G, Parshad R, Srivastava A, Gupta SD, Ralhan R. Clinical significance of Stratifin, ERα and PR promoter methylation in tumor and serum DNA in Indian breast cancer patients. Clin Biochem 2010; 43:380-6; PMID:19961842; http://dx.doi.org/10.1016/j.clinbiochem.2009.11.016
- Steiner M, Clark B, Tang J-Z, Zhu T, Lobie PE. Fourteen-3-3σ mediates G2–M arrest produced by 5-aza-2′-deoxycytidine and possesses a tumor suppressor role in endometrial carcinoma cells. Gynecologic Oncol 2012; 127:231-40; PMID:22772061; http://dx.doi.org/10.1016/j.ygyno.2012.06.039