
ABSTRACT
Using Illumina 450K arrays, 1.85% of all analyzed CpG sites were significantly hypermethylated and 0.31% hypomethylated in fetal Down syndrome (DS) cortex throughout the genome. The methylation changes on chromosome 21 appeared to be balanced between hypo- and hyper-methylation, whereas, consistent with prior reports, all other chromosomes showed 3–11 times more hyper- than hypo-methylated sites. Reduced NRSF/REST expression due to upregulation of DYRK1A (on chromosome 21q22.13) and methylation of REST binding sites during early developmental stages may contribute to this genome-wide excess of hypermethylated sites. Upregulation of DNMT3L (on chromosome 21q22.4) could lead to de novo methylation in neuroprogenitors, which then persists in the fetal DS brain where DNMT3A and DNMT3B become downregulated. The vast majority of differentially methylated promoters and genes was hypermethylated in DS and located outside chromosome 21, including the protocadherin gamma (PCDHG) cluster on chromosome 5q31, which is crucial for neural circuit formation in the developing brain. Bisulfite pyrosequencing and targeted RNA sequencing showed that several genes of PCDHG subfamilies A and B are hypermethylated and transcriptionally downregulated in fetal DS cortex. Decreased PCDHG expression is expected to reduce dendrite arborization and growth in cortical neurons. Since constitutive hypermethylation of PCDHG and other genes affects multiple tissues, including blood, it may provide useful biomarkers for DS brain development and pharmacologic targets for therapeutic interventions.
Abbreviations
| BA | = | Brodman area |
| ChIPSeq | = | chromatin immunoprecipitation sequencing |
| DMR | = | differentially methylated region |
| GEO | = | Gene Expression Omnibus |
| GO | = | gene ontology |
| DNMT | = | DNA methyltransferase |
| DS | = | Down syndrome |
| hESC | = | human embryonic stem cell |
| NRSF/REST | = | neuron-restrictive silencer factor/RE1-silencing transcription factor |
| OMIM | = | Online Mendelian Inheritance in Man |
| PCDHG | = | protocadherin gamma |
| ROS | = | reactive oxygen species |
| SNP | = | single nucleotide polymorphism |
| UCSC ENCODE | = | University of California at Santa Clara encyclopedia of DNA elements |
Introduction
With an incidence of approximately 1 in 700 to 1 in 1000 live births, Down syndrome (DS) or trisomy 21 (OMIM #190685; http://www.ncbi.nlm.nih.gov/omim) is the most common genetic disorder and the leading genetic cause of intellectual disability. DS is also associated with phenotypes outside the central nervous system, most importantly congenital heart defects, gastrointestinal malformations, facial and skeletal features, autoimmune disease, and signs of premature cellular aging. Although the clinical presentation of DS is highly variable, all affected individuals exhibit cognitive impairment. Intellectual disability can range from mild to moderate, with rare severe cases. Children with DS usually have learning difficulties, delayed language development, impaired memory, and neurobehavioral abnormalities.Citation1,2 In addition, essentially all adults with DS (in the fourth decade of life) develop dementia and neuropathological changes resembling Alzheimer disease. Cognitive impairment in DS has been attributed to progressive neuronal cell death and disruption of neuronal network formation with reduced dendrite branching and synaptic connectivity. Cortical neurons display abnormalities in the length of synaptic contact zones and synaptic density.Citation3-6 Organization of the synaptic network is a highly coordinated process and any perturbations of this network during fetal brain development can be expected to interfere with normal cognitive functions.
Although it has been known since 1959 that DS is caused by an extra copy of chromosome 21,Citation7 the mechanisms by which trisomy 21 disrupts development is still not well understood. The DS critical region hypothesis mainly relies on rare cases of partial trisomies, linking imbalance of a small number of genes in specific segments of chromosome 21 to the various clinical phenotypes.Citation8,9 Theoretically, trisomy 21 results in an 1.5-fold increased expression of chromosome 21 genes. However, genes do not function as autonomous units in the genome but are embedded in temporally and spatially highly coordinated regulatory networks. Transcriptome analyses in different human tissuesCitation10,11 and DS mouse modelsCitation12,13 did not reveal a strong (linear) correlation between genomic imbalance and gene expression levels. Expression changes were also observed in genes that are not on chromosome 21 and differed between cell types and tissues. Transcriptomes of fetal fibroblasts from a monozygotic twin pair discordant for DS suggest that differentially expressed genes are organized in large chromosomal domains.Citation14 In this context, it is important to emphasize that many genes show extensive expression variation and, even for genes that are dysregulated in DS, there is an extensive overlap of expression levels between normal and DS cohorts. It is plausible to assume that this expression variation significantly contributes to phenotypic variation, modulating the DS phenotypes.Citation15 Genetic variation may account for a substantial proportion of this gene expression variation.Citation16 However, the functional consequences of epigenetic changes, which occur at a much (one or 2 orders of magnitude) higher rate than DNA sequence changes,Citation17 are likely underestimated.
Epigenetic mechanisms control gene expression patterns without altering the DNA sequence. Epigenetic modifications, in particular in DNA methylation are transmitted to daughter cells during somatic cell division and, perhaps also from one generation to the next. CpG islands in promoter regions are usually demethylated. Promoter methylation during development or disease processes is associated with posttranslational histone modifications that lead to a locally condensed inactive chromatin structure and gene silencing.Citation18 In contrast, gene body methylation is positively correlated with transcription and may have functions in silencing transposable elements and regulating splicing.Citation19,20 The epigenome is highly plastic during development and susceptible to both internal and environmental cues.Citation21,22 Genome-wide methylation studies have identified epigenetic signatures of DS in several tissues, including leukocytes, skin fibroblasts, buccal cells, liver, placenta,Citation23-27 and brain,Citation28-30 adding another layer of complexity to the highly variable clinical features of DS. Interestingly, chromosome 21 carries several genes, most importantly DNA (cytosine-5)-methyltransferase 3-like (DNMT3L), that are key players in epigenetic regulation. Epigenetic dysregulation due to dosage imbalance of chromosome 21 during brain development may provide an important contribution to highly variable cognitive impairment.Citation31
Since methylation patterns are dependent on cell type/tissue, development and differentiation, it is crucial to study the appropriate target tissue and developmental stage for the phenotype of interest. Here we performed a genome-wide methylation analysis of DS fetal frontal cortex, which is essential for higher cognitive functions, such as attention, memory, complex actions, cognition, emotion and behavior.Citation32,33
Results
Hypermethylation in the developing DS brain
We used Infinium HumanMethylation450 (450K) BeadChips to analyze DNA methylation profiles of 16 DS and 27 control fetal frontal cortex, 8 DS and 8 control fetal temporal cortex, and 2 DS and 9 control adult frontal cortex samples (NCBI GEO accession no. GSE73747; Supplementary Table S1). Control fetuses were from spontaneous or induced abortions with amniotic infection, placental abruption, or other problems, which should not primarily interfere with brain development. To the extent possible, gestational age was matched (P = 0.35) between DS (median: 18 weeks, range: 12–42 weeks) and control (median: 20 weeks, range: 15–37 weeks) subjects. In an exploratory analysis based on a multivariate ordination approach, the strongest methylation difference was detected between fetal and adult brain samples, followed by brain region and gestational age. Finally, there was a clear difference between trisomy 21 and controls (Supplementary Fig. S1). We did not find significant effects of sex, postmortem time, or BeadChip.
To identify epigenetic signatures of DS on brain development, we focused our further analysis on the fetal frontal cortices. All together, 8,624 CpG sites (1.85% of all analyzed CpGs) were significantly (FDR-adjusted P < 0.05) hypermethylated and 1,447 (0.31%) hypomethylated, indicating a tendency toward hypermethylation in the DS brain. Global (average of all 465,572 analyzed CpG sites) methylation was 49.8% in DS and 49.5% in control samples, which is a trend difference (Welch T-test; P = 0.11). The differentially methylated sites were widespread throughout the genome (Supplementary Fig. S2, upper panel). Compared to other chromosomes, the effect sizes (β differences) of significant sites on chromosome 21 were rather small (Supplementary Fig. S2, lower panel). Chromosomes 19 (2.79%), 21 (2.58%), and 22 (2.25%) were enriched with hypermethylated CpGs (Supplementary Table S2). Compared to the rest of the genome, chromosome 21 was strongly enriched with hypomethylated sites (1.98%). The methylation profile of chromosome 21 in DS cortex appeared to be different from other chromosomes (). With exception of chromosome 21, all chromosomes displayed an excess (3–11 times) of hypermethylated sites with a maximum methylation change around β values of 0.5. The changes on chromosome 21 were more balanced between hyper- and hypo-methylation with stronger signals toward the extreme ends (0 and 1) of the β value distribution.
Figure 1. Plot of methylation changes (β differences) along the distribution of mean methylation levels (range of β values). Only significant changes (adjusted P < 0.05) were considered. The lowess smoother (red line) reveals hypermethylation of all chromosomes except chromosome 21 with a maximum change in the middle range of β values. The changes on chromosome 21 are balanced between hyper- and hypo-methylation with stronger signal toward extreme β values. The methylation data underlying this figure were normalized using the SWAN method; however, the observed genome-wide hypermethylation of all chromosomes except 21 remained stable under various normalization procedures (data not shown).
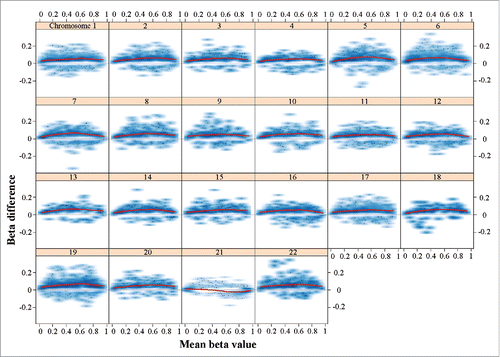
To further analyze the phenomenon of balanced hyper- and hypo-methylation on chromosome 21, we applied different normalization methods (Dasen with and without BMIQ, Funnorm)Citation34-36 in addition to SWAN,Citation37 which was used throughout the study. The observed genome-wide hypermethylation on all chromosomes except 21 remained stable under various normalization procedures when analyzing the entire data set, fetal cortex samples, or fetal frontal cortex samples (data not shown). Moreover, a similar decrease in the density of hypermethylated sites on chromosome 21 was also seen in a published DS blood data set.Citation26 In a genomic context, hypermethylated sites were enriched in CpG islands and adjacent north and south shores, whereas hypomethylated CpGs were enriched in north and south shelves (Supplementary Table S2).
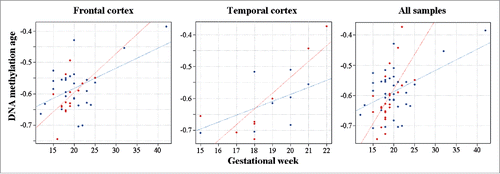
For comparison, 49,837 CpGs showed an increase and 25,268 sites a decrease in methylation during gestational development. However, DS and control brains displayed largely identical changes over time. Based on the interaction of chromosomal status (DS versus control) and gestational age in the regression model, only 83 sites were differentially (42 hyper- and 41 hypo-) methylated during development between DS and control brains. When we applied the DNA methylation age calculator,Citation38 which relies on only 353 CpG sites, to our fetal brain samples, it correctly predicted prenatal (negative) age. The gestational age was positively correlated with DNA methylation age in both frontal (Pearson's r = 0.51; P = 0.0005) and temporal cortex (r = 0.70; P = 0.002). The same was true for all samples (r = 0.49; P = 0.0001). Age acceleration during the fetal period appeared to differ between DS and control brains (). However, due to the narrow time window (most samples were from the second trimester) and limited sample size, a significant effect as defined by residual projectionCitation28 could not be demonstrated.
Figure 2. DNA methylation age in fetal DS brain. DS fetal cortices are indicated by red and control samples by blue dots. The gestational age of frontal, temporal and all cortex samples is positively correlated with DNA methylation age. The regression lines suggest accelerated DNA methylation aging in DS versus control brains.
A previous studyCitation29 identified 441 CpG sites with significant methylation (β > 0.15) changes between DS and control fetal brain samples. Despite differences in tissue (fetal cortex vs. cerebrum), there was a highly significant correlation (r = 0.58; P = 1E-10) of methylation changes between the reported sitesCitation29 and our study (Supplementary Fig. S3). In addition, enrichment of the reported sites in our set of significant sites was highly significant (P = 1E-10). A smaller subset of 47 pan-tissue DS sites was reported to be hypermethylated in multiple cell types (adult neurons, glia, T cells)/tissues (adult cortex, cerebellum, fetal brain) of DS individuals.Citation29 When applying the same filtering criteria (β difference > 0.15; P < 0.005) as for the definition of pan-tissue DS sites, we found a highly significant overlap (Fisher's exact test; OR 3407; P = 2.2E-15) of the sites shared between fetal brain (this study) and bloodCitation26 with the pan-tissue DS sites (Supplementary Fig. S4). Because the reported pan-tissue DS sites are enriched with CTCF binding motifs,Citation29 we similarly tested our subset of sites that are hypermethylated in fetal cortex and blood for enrichment with CTCF motifs. We found significant (OR 4.307, P = 0.017) enrichment with CTCF_ext and a trend (OR 1.11) enrichment with the CTCF motifs.
Differentially methylated regions (DMRs)
Throughout the genome, there were 15,490 promoter regions covered by 3 or more CpG sites on the array. Of the top 100 ranked differentially methylated promoters with at least 3 significant CpGs, 95 were hypermethylated and only 5 hypomethylated in DS (Supplementary Table S3). Only one of the 100 promoters belonged to a gene, adenosine deaminase, RNA-specific, B1 (ADARB1) on chromosome 21, compared to 6 on chromosome 19, 3 on chromosome 20, and 4 on chromosome 22 (). However, considering the low number (117) of analyzed gene promoters on chromosome 21, this is not a significant underrepresentation (Fisher's exact test). There were several clusters of differentially methylated promoters outside chromosome 21, most prominently the protocadherin gamma (PCDHG) cluster on chromosome 5q31 (with PCDHGA2, A3, A4, A5, A10, B1, and B2 among the top 100 promoters).
Figure 3. Chromosomal localization of differentially methylated promoters. Blue dots represent methylation β differences of 15,490 promoters (with >3 CpG sites on the array) between DS and control cortices. Promoters are mapped to their chromosomal position. Red dots represent the top 100 ranked differentially methylated promoters.
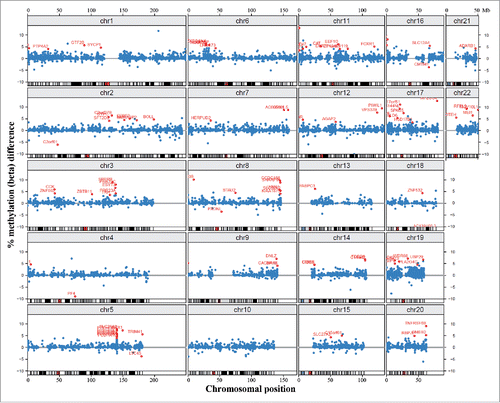
At the gene level (including promoter, exons, introns, 3′ and 5′ UTRs), we identified 69 differentially methylated genes containing at least 3 significant CpG sites with between-group (β > 0.1) methylation differences (). With notable exception of the hypomethylated platelet factor 4 (PF4), 68 genes were hypermethylated in DS cortex. When ranking genes according to the percentage of differentially methylated sites, our top candidate was tumor necrosis factor receptor superfamily, member 6B (TNFRSF6B). However, this gene was covered by only 3 CpGs on the array, which all were significantly hypermethylated. The second ranked gene, carnitine palmitoyltransferase 1B (CPT1B), was covered by 18 CpGs, 13 (72%) of which showed increased (β > 0.1) methylation (). CPT1B was hypermethylated throughout the gene body, but not in the promoter. Only one gene, the potassium channel, voltage gated subfamily E regulatory β subunit 1 (KCNE1), was on chromosome 21, which is not a significant underrepresentation. Consistent with the results of promoter methylation analysis, differentially methylated genes were clustered in several regions outside chromosome 21. The strongest cluster with 12 genes was the PCDHG cluster on chromosome 5q31 (). Interestingly, the cadherin, EGF LAG 7-pass G-type receptor 3 (CELSR3) on chromosome 3p21.31 was also hypermethylated (). Additional clusters were identified on chromosome 6p21.3 (DXO, STK19, TNXB, HLA-DQB2, and ZBTB22), 8q24.3 (GLI4, FAM83H, ZNF707, KIAA1875, LRRC14, and LRRC24), and 19q13.3 (NANOS, FKRP, CYTH2, LMTK3, and NR1H2). The distribution of differentially methylated genes was significantly different between chromosomes (Pearson's χ2 test; P = 2.15E-06).
Figure 4. Differential methylation of CPT1B and CELSR3. Red dots represent the methylation level of individual CpGs in DS cortex samples; the blue dots represent control samples. Light blue bars indicate transcribed sequence and orange bars CpG islands.
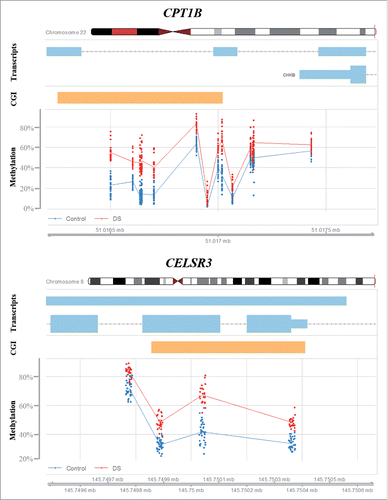
Table 1. Genes with at least 3 significant CpG sites (β difference > 0.1).
Recently, a number of genes with differential methylation in multiple DS cell types and tissues (Supplementary Table S4) and enrichment for CTCF sites has been identified.Citation29 Nine (C1orf35, CPT1B, DECR2, FAM83H, GLI4, LRRC14, LRRC24, STK19, and TNXB) of the 10 reported genes with pan-tissue DS DMRs (in T cells, neurons, glia, cerebellum, and fetal brain)Citation29 were also hypermethylated in fetal DS cortex. The 10th gene, CCDC144B, was hypermethylated in our data set as well but filtered out because it contained only 2 significant CpGs.
Gene ontology (GO) analysis was used for organizing the dysregulated candidate genes into known pathways/processes and predicting functional relevance. Single ranked GOrilla analysisCitation39 of promoter regions revealed enrichment for several GO terms (processes) associated with cell adhesion: homophilic cell adhesion via plasma membrane adhesion molecules, cell-cell adhesion, and cell-cell adhesion via plasma-membrane adhesion molecules (Supplementary Table S5). This enrichment was mainly due to genes from the PCDHG cluster (A1-A8, A10, A12, and B1-B4). GOrilla analysis of the 69 differentially methylated genes yielded similar results.
Since our data set contained only 2 adult DS brains, we performed an in silico analysis on a recently published 450K methylation array data set including 15 DS and 56 control adult cortices.Citation28 The top differentially methylated gene in adult brain was CPT1B. Consistent with our study, the PCDHG genes and CELSR3 were also hypermethylated. demonstrates genomic organization of the PCDHG genes and similar methylation changes in fetal and adult DS cortices. To test whether the observed methylation changes are brain specific, we re-analyzed the protocadherin gene cluster in another published 450K array data set from blood of 29 DS patients and their relatives.Citation26 Methylation of PCDHG genes was significantly higher in DS blood, compared to unaffected mothers and siblings (Supplementary Fig. S5).
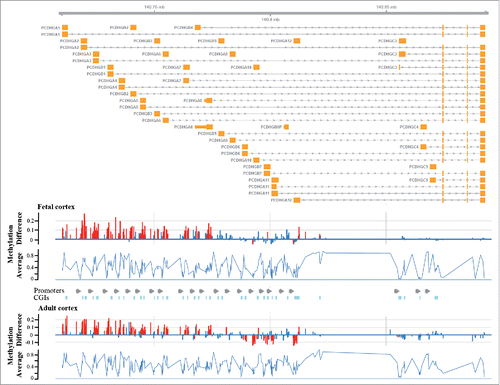
Figure 5. Differential methylation of the PCDHG gene cluster in DS brain. The upper part shows the genomic organization of the PCDHG gene cluster with exons depicted in orange. The bottom diagrams represent methylation β differences between DS and control samples and average methylation (of all samples) in fetal and adult frontal cortices. Significant methylation differences are indicated by red, non-significant changes by blue bars. Differential methylation is restricted to genes of the A and B but not the C subfamilies and very similar in fetal and adult brains.
Differential methylation is associated with expression changes
Differential methylation of the PCDHG gene cluster and CELSR3 was validated by bisulfite pyrosequencing. All 9 assays for different PCDHG genes included the promoter region, while the assay for CELSR3 targeted the first exon. Methylation was quantified in frontal (16 DS and 28 control), temporal (14 DS and 20 control), and occipital (15 DS and 20 control) fetal cortices as well as in fetal liver (14 DS and 38 controls). Consistent with methylation array results, there was a significant hypermethylation of all analyzed PCDHG promoters and CELSR3 exon 1 in all 4 analyzed tissues (Supplementary Table S6). The methylation values of PCDHG promoters were to some extent overlapping in DS and control samples, whereas the CELSR3 values were clearly separated between groups (). Adult frontal cortex (of 2 DS and 10 controls) showed the same hypermethylation of PCDHG promoters and CELSR3 as fetal samples. In addition, we sorted neuronal versus non-neuronal cells from fetal and adult cortices using NeuN-specific antibodies.Citation40 However, since the fetal brains were not always immunostaining positive, this was only reliable for adult cortex. All tested PCDHG genes as well as CELSR3 showed an increased methylation in neuronal cells from 2 adult DS cortices, compared to 10 controls (Supplementary Table S6). Similar to the previously published pan-tissue DMRs,Citation29 the PCDHG gene cluster and CELSR3 were constitutively hypermethylated in DS, affecting multiple tissues and developmental stages.
Figure 6. Hypermethylation and expression changes of PCDHG genes and CELSR3 in DS brain. Methylation was measured by bisulfite pyrosequencing. Each dot represents the mean methylation level of one individual DS or control fetal frontal cortex. Expression was measured by targeted RNA sequencing. Blue lines compare median methylation and expression, respectively, between DS and control fetal frontal cortices.
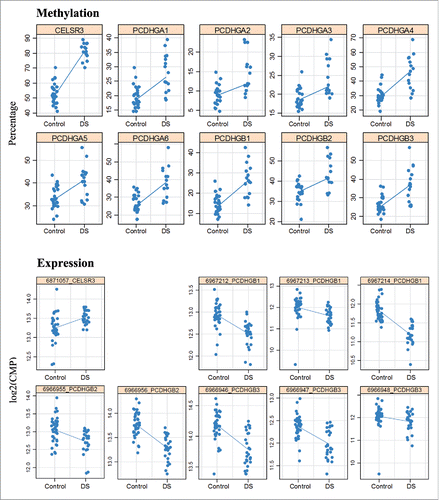
To determine whether the observed methylation changes affect gene expression, we performed targeted RNA sequencing with 15 DS and 20 control fetal frontal cortices. The top differentially methylated genes, in particular the PCDHG gene cluster on chromosome 5 were covered by multiple assays. Consistent with their promoter hypermethylation, several PCDHG genes (A1, A2, A5, A7, A8, B1-B4) showed significantly lower expression in DS frontal cortex (). Since many of these genes (A1, A5, B1, B2, and B3) exhibited similar changes in different assays, artifacts can be largely excluded. The strongest transcriptional downregulation was observed for PCDHGB1, B2, and B3 (). The hypermethylated NADPH oxidase EF-hand calcium binding domain 5 (NOX5) () and unc-45 myosin chaperone A (UNC45A) genes (Supplementary Table S3) on chromosome 15q23 and 15q26.1, respectively, were also transcriptionally downregulated in DS brain. In contrast, hypermethylation of the CPT1B gene body and CELSR3 exon 1 was associated with increased expression (; ). It is noteworthy that 6 targeted genes on chromosome 21, APP, BACE2, CBS, DYRK1A, PRDM15, and SOD all showed increased expression, as expected for trisomic genes. Consistent with overexpression of the dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1 A (DYRK1A), the neuron-restrictive silencer factor/RE1-silencing transcription factor (NRSF/REST) was downregulated in DS cortex (). Unfortunately, expression of DNMT3L, which also lies on chromosome 21, was too low for reliable quantification. The de novo methyltransferases DNMT3A and DNMT3B showed significantly reduced expression in DS brain.
Table 2. Expression differences of PCDHG and other genes between DS and control frontal cortex.
Comparison of promoter methylation β differences between fetal DS and control cortex (this study) and gene expression changes (logFC differences) in fetal fibroblasts of monozygotic twins discordant for DSCitation14 revealed a small negative correlation (Spearman's rank R = −0.03, P = 8.64E-04) for the 15,409 genes that were analyzed in both studies.
Effects of dosage imbalance of epigenetic regulators on chromosome 21
The DNMT3L gene on chromosome 21q22.4 stimulates de novo methylation by DNMT3A and DNMT3B.Citation41 To test whether an extra DNMT3L copy is associated with genome-wide methylation changes, we analyzed whole genome bisulfite sequencing data in human embryonic stem cells (hESCs) with disrupted DNMTs.Citation42 Hypomethylated CpG islands in DNMT3A (FDR-adjusted P = 0.02), DNMT3B (P = 0.02), and double knockout (P = 0.03) hESCs were significantly enriched with CpGs that are hypermethylated in DS fetal brains (Supplementary Table S7).
DYRK1A on chromosome 21q22.13 downregulates NRSF/REST.Citation43 Of 13,286 NRSF peaks in a previously published ChIP-Seq data set from hESCs (UCSC ENCODE), 2,700 were covered by probes on the 450K array. The top REST target sites (Supplementary Table S8) included CELSR3 (FDR-adjusted P = 2.16E-12) and the PCDHG gene cluster (P = 0.001 to 0.01 for different genes).
Discussion
The mechanisms underlying cognitive impairment in DS have not yet been fully elucidated. Accumulating evidence suggests that the distinctive behavioral and cognitive deficits, and neuropathological changes are caused by altered regulation and complex interrelations of many genes both on and outside chromosome 21. One strong candidate gene, exemplifying how dosage imbalance of chromosome 21 can lead to genome-wide disturbances is DYRK1A. Upregulation of this gene was associated with reduced REST mRNA levels in DS fetal cortex (this study) and neurospheres,Citation44 leading to dysregulation of genes in close proximity to REST binding sites.Citation43,45 Another key player on chromosome 21 involved in epigenetic regulation is DNMT3L. Although it lacks the catalytic domain, DNMT3L cooperates with de novo methyltransferases DNMT3A and DNMT3B.Citation41 DNMT3L is expressed in early developmental stages and at very low levels in some adult tissues, i.e., testis, ovary, and thymus.Citation46 As expected for a trisomic gene, Western blots showed significant overexpression of DNMT3L in DS fetal frontal cortex.Citation30 In our study, DNMT3L mRNA expression was too low to be quantified, whereas both DNMT3A and DNMT3B were significantly transcriptionally downregulated in DS cortices (mainly from the second trimester). We propose that DNMT3L upregulation in neuroprogenitorsCitation30 leads to increased de novo methylation. Following a wave of de novo methylation in early development, hypermethylation may persist in fetal DS brainCitation30 and other tissues,Citation24,27 although DNMT3A and DNMT3B become downregulated. CpG sites that are hypermethylated in DS brains are enriched in regions that are hypomethylated in DNMT3A and/or DNMT3B knockout hESCs, as well as in regions with REST binding sites. Methylation of REST binding sites in DS may facilitate genome-wide methylation. In mouse stem cells it was shown that REST binding is crucial to create or retain a low methylation state.Citation47
Consistent with other genome-wide expressionCitation10,11 and methylation studies,Citation23-26 we found an enrichment of differentially methylated CpGs on chromosome 21, with epigenetic dysregulation occurring throughout the genome. Hypermethylated sites (8,624) were 6 times more frequent in fetal DS cortex than hypomethylated sites (1,447). It is noteworthy that chromosome 21 was the only chromosome with a more or less balanced ratio (1.3) of hypomethylated and hypermethylated sites, whereas all other chromosomes showed an excess (3–11 times) of hypermethylated sites. Moreover the β value distribution differed between chromosome 21 and the rest of the genome. Although it is difficult to exclude the possibility that this unique methylation pattern of chromosome 21 is a bioinformatics artifact due to copy number variation, the difference between chromosome 21 and the rest of the genome was observed using various normalization methods in our data set as well as in blood from DS patients.Citation26 Thus, it may well have a biological basis. The high number of hypomethylated CpGs fits to our observation that 6 of 7 analyzed chromosome 21 genes were overexpressed in fetal DS cortex.
Arguably, our most interesting candidates for cognitive impairment are the PCDHG genes and CELSR3, which are hypermethylated in DS brain and enriched for REST binding sites. Protocadherins encode the largest group of the cadherin superfamily of cell-cell adhesion proteins. They are mainly expressed in the developing nervous system, where they play a major role in neural circuit formation via homophilic cell adhesion interactions.Citation48-50 The protocadherin family is subdivided into the clustered and non-clustered protocadherins, in addition to the atypical fat, dachsous, and 7-transmembrane (CELSR) cadherins. The α-, β-, and γ-protocadherins constitute a 1-Mb cluster with 60 genes on chromosome 5q31.Citation51 The most thoroughly studied is the gamma cluster, which contains 22 tandemly arranged genes of the A, B, and C subfamilies. Bisulfite pyrosequencing confirmed hypermethylation of several PCDHG and the 7-transmembrane cadherin CELSR3 in DS fetal frontal, temporal, and occipital cortices and liver. These genes were also hypermethylated in adult DS brain (specifically in neurons) and blood. This suggests that constitutive hypermethylation occurs early in development (during embryogenesis before separation of germ layers), affecting multiple cell types and tissues. In a mouse model, it has been shown that de novo methylation during early embryo development regulates the stochastic expression of different Pcdh isoforms at the individual cell level, thus generating single neuron diversity. The clustered protocadherins were all upregulated in Dnmt3 knockout mice.Citation52 Consistent with their increased promoter methylation, expression of the PCDHGA and B genes was decreased in fetal DS cortex. PCDHGC genes, which are ubiquitously expressed and not regulated in the same way as subfamilies A and B,Citation52 were neither differentially methylated nor differentially expressed in DS brain. Remarkably, CELSR3 was upregulated, suggesting that increased methylation in DS cortex abrogates a repressor activity or confers an enhancer-like activity.Citation53
PCDHG genes encode transmembrane receptors with an intracellular, a transmembrane, and an extracellular domain. The proteins are present in most neurons and localized at synapses but also in axons and dendrites.Citation54-56 Different PCDHG knockout mouse models suggest a role for gamma protocadherins in promoting dendritic self-avoidance, arborization, and synaptic development in cortical neurons.Citation55-58 In contrast, the CELSR3 cell surface protein appears to suppress dendrite outgrowth.Citation59 The opposing effects of gamma protocadherins and CELSR3 on neurite growth regulation imply that transcriptional downregulation of PCDHG genes and upregulation of CELSR3 act synergistically, both inhibiting dendrite arborization and growth in the developing DS cortex. Although neuronal density appears to be normal, DS fetal brains are characterized by reduced dendrite branching and impaired synaptosomal structure. Dendritic and axonal development is abnormal in fetal DS brains, including cortical lamination defects, reduced dendritic arborization, reduced number of synapses, and dendritic spine structural anomalies.Citation2,3,5
Strong epigenetic effects were also observed in CPT1B, which showed increased gene body methylation and mRNA expression in DS fetal cortex. A published data setCitation28 revealed that this hypermethylation persists in the adult DS brain. Moreover, differential CPT1B methylation was reported in blood leukocytes, buccal epithelial cells, placenta, and brain of DS patients,Citation23-25,29 consistent with constitutive methylation changes. CPT1B on chromosome 22 is one of 3 carnitine palmitoyltransferase 1 genes that is mainly expressed in heart and skeletal muscle.Citation60 It encodes a mitochondrial enzyme, regulating entry of long chain fatty acids into the mitochondria. The NOX5 gene on chromosome 15 exhibited gene body methylation and decreased expression in DS brain. NOX5 is a calcium-dependent NADPH oxidase family protein that produces superoxide and functions as a calcium-dependent proton channel.Citation61 Dysregulation of CPT1B and NOX5 may be involved in mitochondrial dysfunction and oxidative stress in DS brains. The link between reactive oxygen species (ROS) and DS neuropathology dates back to early observations that superoxide dismutase 1 (SOD1), a key enzyme in the free radical metabolism, is overexpressed from the DS critical region on chromosome 21.Citation62 Accumulating evidence from more than 2 decades suggests that increased ROS production and/or deficient antioxidant capacity contribute to brain damage (cell death) and cognitive impairment in DS and Alzheimer disease.Citation4,63,64 More recently, mitochondrial dysfunction has been found in the brains of patients with different psychiatric disorders.Citation65 CPT1B expression was altered in brain (amygdala) and blood of a rat stress model as well as in blood of patients with posttraumatic stress disorder.Citation66
Because of legal and ethical restrictions (which differ between countries), there is only limited access to fetal brain samples and the quality of tissue samples is often not optimum. Our control samples were from spontaneous or induced abortions, mainly due to amniotic infection or placental problems. Although we cannot exclude that the various pathologies (of the control group) and postmortem times (<24 h to 72 h) affect methylation patterns in individual samples, this does not explain the observed methylation differences between DS and control cortices. The majority of DS samples were from induced abortions, representing the whole spectrum of DS pathologies. One advantage of our study is that all fetuses underwent autopsy by an experienced pediatric pathologist and frontal cortex tissue was dissected from a well-defined area (BA10), compared to a more comprehensive data setCitation29 using undissected fetal brain tissue (cerebrum) as well as adult frontal and cerebellar cortex. Unfortunately, we could not reliably sort neuronal cells and non-neuronal cells from frozen fetal cortex samples. However, hypermethylation of the PCDHG cluster was validated in adult DS neurons and is consistent with the observed expression changes. In addition, some of our results were validated in fetal and adult DS brain,Citation28,29 using publicly available data. Although studies on a limited number of fetal brain samples are likely polluted with false negatives and false positives, it is reassuring that there is a significant overlap between DS-specific changes observed in different studies.
Outlook
This study presents a thorough analysis of methylation abnormalities in the developing DS brain. Our results suggest that accelerated DNA methylation aging, which has been reported in adult DS brain,Citation28 already starts in utero. Methylation-dependent downregulation of PCDHG subfamily A and B genes is expected to affect wiring processes in the developing cortex and, consequently, contribute to cognitive impairment in DS. Epigenetic dysregulation of CPT1B and other genes may perturb mitochondrial functions, leading to brain cell damage. Constitutive hypermethylation of PCDHG and CPT1B in brain and blood can be exploited as epigenetic biomarkers. It will be interesting to study the correlation between PCDHG blood methylation levels and highly variable DS phenotypes, in particular cognitive impairment.
Blood methylation changes of PCDHG genes were also reported in patients with Williams Beuren and 7q11.23 duplication syndrome.Citation67 The observed dysregulation of PCDHG genes in different chromosome disorders with cognitive deficits indicates novel avenues for clinical management and maybe even therapeutic interventions of patients with cognitive impairment. In contrast to the trisomy 21, epigenetic marks are in principle reversible. It has been shown that PCDHG genesCitation68 and CELSR3Citation59 can be pharmacologically modulated by protein kinase C and calcineurin inhibitors. Thus, similar to epigenetic drugs for cancer treatment, it may be feasible to develop epigenetic therapies for enhancing cognition and/or phenotypes in DS.Citation31
Conditional knockout mice with PCDHG−/− cortex showed reduction in the dendrite arborization complexity of pyramidal neurons.Citation56 Similar dendritic abnormalities in cortical neurons have been described in DS and DS mouse models.Citation5 Unfortunately, the cortical phenotype has not been examined in mice lacking only subsets (A1-A3 and C3-C5, respectively) of the PCDHG cluster.Citation57 It will be interesting to analyze whether and to which extent the different PCDHG knockout mouse models show cortical and other physical abnormalities of DS. If so, these mouse models may become valuable for the development of new drugs and maybe prenatal interventions preventing the negative effects of PCDHG dysregulation in DS.
Materials and methods
Samples
Tissue samples (Supplementary Table S1) were obtained from fetuses after spontaneous and induced abortions and underwent diagnostic examination at the Department of Pathology at Mainz University Medical Center. Use of excess tissue materials for scientific analyses was approved by the ethics committees of the Landesärztekammer Rheinland-Pfalz (no. 837.103.04_4261) and the Julius Maximilians Universität Würzburg (no. 262/14). Gestational age of each fetus was determined by foot length measurements (in mm) and last menstrual period. Following autopsy, the fetuses were photo documented, measured, weighed, and X-rayed in 2 levels. The postmortem time was determined using anamnestic data and autolytic processes. Chromosome (GTG banding) analyses were performed on primary fibroblast cultures from Achilles tendon. All DS samples included in this study displayed a complete extra copy of chromosome 21 (47,XX,+21 or 47,XY,+21). All controls showed normal karyotypes. The causes for abortion were vastly different, ranging from amniotic infections and placental abnormalities to various syndromes. Cortex tissues were dissected from the frontal lobe, Brodmann area BA10 (16 DS and 28 controls), the temporal lobe, BA38 (14 DS and 21 controls), and the occipital lobe, BA17/18 (15 DS and 21 controls). Liver tissue samples (14 DS and 38 controls) were obtained after an upper median incision to the abdomen. Adult cortices were obtained from 2 DS patients (from the Neurobiobank Munich and 12 suicide completers/sudden-death controls. Cortex tissue was dissected from the frontal pole, Brodman area BA10. Adult frontal cortex tissues of 2 DS and 10 controls were sorted into neuronal and non-neuronal cell using a NeuN-specific antibody, as described previously.Citation40
For DNA and RNA preparation, tissue samples were disrupted using a Precellys24 high-throughput homogenizer (Peqlab, Erlangen, Germany). Genomic DNA was isolated with the DNeasy blood and tissue kit (Qiagen, Hilden, Germany). Bisulfite conversion was performed with 1 μg DNA each using the EZ-96 DNA methylation kit (Zymo Research, Irvine, CA, USA). Amount and quality of DNA were determined with a Nanodrop spectrophotometer (NanoDrop, Wilmington, DEL, USA). The ratio of absorbance at 260 vs. 280 nm was around 1.8 for all samples, indicative of pure DNA. Total RNA was isolated with the RNeasy lipid and tissue mini kit (Qiagen). Amount and quality of DNA and RNA were analyzed with an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) system using the RNA 6000 Nano kit (Agilent).
Methylation array analysis
After bisulfite conversion, 72 samples (16 DS and 27 control fetal frontal, 8 DS and 8 control fetal temporal, 2 DS and 9 control adult frontal cortices) including some duplicates were whole-genome amplified, enzymatically fragmented, and analyzed with 6 HumanMethylation450 BeadChips (Illumina, San Diego, CA, USA), according to the manufacturer's protocol. To avoid batch effects, the 12 arrays on each chip were hybridized with matched DS and control samples as well as samples from different brain regions and all chips were processed together. The arrays were scanned with an Illumina iScan.
Resulting array data (NCBI GEO accession number GSE73747) were preprocessed using the RnBeads pipelineCitation69 with default settings. Sites overlapping SNPs, sites not in the CpG context and probes flagged as unreliable based on the corresponding detection P value were removed. Furthermore, 11,169 probes on the sex chromosomes were excluded, leaving a total number of 465,572 probes for subsequent analyses (of > 485,000 CpGs covering 99% of RefSeq genes with promoter, first exon, gene body, 5′ and 3′ UTRs, and 96% of CpG islands). The signal intensity values were normalized using the SWAN normalization method,Citation37 as implemented in the minfi package.Citation70 Different normalization methods were used to confirm the unique methylation profile of chromosome 21. In addition to SWAN,Citation37 we applied dasen normalizationCitation34 with and without subsequent Type II probe bias correction with the BMIQ methodCitation35 as well as a novel functional normalization approach (Funnorm) for situations where substantial global differences in methylation can be expected.Citation36
Differential methylation analysis has been performed using the moderated T-test model as implemented in the limma packageCitation71 based on β values of the fetal samples. A site-wise analysis was conducted based on a model with brain region, chromosomal status, and gestational age as covariates, including the first order interaction between the covariates. Reported P values are based on the moderated T statistics of the main effects or interaction terms. A set of pan-tissue hypermethylated sites has been created based on our fetal cortex data set and methylation profiles from human blood samples.Citation26 Analogously to the approach described,Citation29 we selected all sites hypermethylated in both tissues with a β difference > 0.15 and P < 0.005. The set of CTCF sites for the motif analysis was obtained from the authors of a conceptually related study.Citation29
For subsequent gene-based analyses the GENCODE catalog (Version 19, GRCh37 genome build) was used. The catalog comprises a total of 57,820 genes from which only the subset of 19,430 protein coding genes was included in the analyses. Promoters were defined as regions flanking the transcription start sites (2,000 bp upstream and 200 bp downstream). To determine the significance of methylation changes over the promoter regions, the P values from the site-wise analysis were aggregated using the RnBeads function “combineTestPvalsMeth,” which is based on the weighted inverse χ2 method for correlated significance tests.Citation72 The site-wise methylation differences have been averaged over the promoter regions, and the promoters have been ranked using a combined rank approach. This combines the effect size (β difference) and statistical significance (P values) ranking the regions according the maximum rank of P value and negative absolute difference of β values. The Gene ontology enrichment analysis and visualization (GOrilla) tool was used to identify enriched GO terms in ranked lists of genes.Citation39
Bisulfite pyrosequencing
PCR and sequencing primers (Supplementary Table S9) were designed using the PyroMark Assay Design 2.0 software (Qiagen). PCR reactions were performed in a total volume of 25 µl using the FastStart Taq DNA Polymerase system (Roche Diagnostics, Mannheim, Germany). The 25-μl reaction consisted of 2.5 μl 10x PCR buffer, 20 mM MgCl2, 0.5 μl dNTP (10 mM) mix, 10 pmol of forward and reverse primer, 1 IU of FastStart Polymerase (Roche Diagnostics), 1 μl (approximately 100 ng) bisulfite converted template DNA, and 18.8 μl PCR-grade water. To reduce technical noise (batch effects), bisulfite conversion and PCR (of matched DS and control samples) were performed in 96-well microtiter plates. Pyrosequencing was performed on a PyroMark Q96 MD system with PyroMark Gold Q96 CDT reagents (Qiagen). Methylation values were quantified using the Pyro Q-CpG software. The average methylation difference between technical replicates was approximately one percentage point. To obtain a common value for each gene the β values of CpG sites have been averaged over the gene for each tissue separately. To account for a potential effect of gestational age a linear model was subsequently fitted to each gene and tissue separately including chromosomal status and gestational age as covariates. All P values have been corrected for multiple testing using the method of Bonferroni-Holm.Citation73
Targeted RNA sequencing
A customized TruSeq RNA expression panel with 135 assays targeting 38 genes, in particular the PCDHG gene cluster on chromosome 5, CELSR3, CPT1B, NOTCH4, NOX5, 3 genes (BACE, CBS, and PRDM15) on chromosome 21, and 5 internal control genes, was designed by Illumina DesignStudio. A smaller panel with 25 assays targeted 4 differentially methylated genes (DND1, EFCAB4A, TNFRSF6B, and UNC45A), 6 epigenetic modifiers (CHAF1B, CBS, DNMT3L, DYRK1A, MIR155, and MIR802) and 2 other genes (APP, SOD1) on chromosome 21, DNMT3A, DNMT3B, NRSF/REST, and 4 internal control genes. cDNA was synthesized from 15 DS and 20 control fetal frontal cortex samples using ProtoScript II Reverse Transcriptase. Subsequent steps were performed according to the TruSeq targeted RNA expression guide. All RNA samples were analyzed in technical duplicates. FirstChoice Human Brain Reference RNA (ThermoFisher Scientific, Waltham, MA, USA) was used as positive control, as recommended in the Illumina protocol. Extension-ligation products of all samples were amplified with a 96 indexing combination of adapters (A501-A508 and R701-R712) in the TruSeq targeted RNA index kit A. PCR products were purified with AMPure XP beads (Beckman Coulter, Fullerton, CA, USA), pooled, and quantified with a 2100 Bioanalyzer and the Agilent DNA 1000 Kit. The TruSeq RNA library was sequenced (single reads) for 50 cycles and dual-index 6 and 8 cycles using Illumina MiSeq and the MiSeq Reagent Kit v3.
Mapping and counting were performed with Illumina GenomeStudio software according to the manufacturer's protocol. Assays comprising < 100 reads in total and samples with < 1,000 reads in total were excluded. Differential expression analysis was performed using the limma modeling frameworkCitation71 in combination with the “voom” method,Citation74 which has been specifically designed for the analysis of count data in RNASeq experiments. The correlation of technical replicates was estimated with the “duplicateCorrelation” function and modeled by a mixed linear model implemented in the limma package. Chromosomal status and gestational age were incorporated as covariates in the model to obtain estimates of DS corrected for gestational age. All P values have been corrected for multiple testing using the Benjamini-Hochberg method.Citation75
Bioinformatics analyses of published data sets
Genomic coordinates of DMRs in DNMT3A, DNMT3B, and DNMT3A/DNMT3B knockout cell lines (Supplementary Table S7) have been obtained from a recently published data set.Citation42 Probes on HumanMethylation450 BeadChip were mapped to these regions. Counting only CpGs falling within these regions resulted in 7,508 probes for the enrichment analysis. To test for the enrichment of DNMT binding regions among the differentially methylated sites the “camera” algorithm of the limma package has been applied.Citation76 This method has originally been designed as a competitive gene set enrichment method. However, it can also be used to test whether a set of CpG sites is highly ranked relative to other sites in terms of differential methylation. Here, the CpG sites of the DNMT knockout DMRs were used as the input set for the “camera” analysis. As adjacent sites usually show a correlation due to spatial proximity, this technique is particularly adequate as it accounts for potential inter-site correlation.
A list of potential REST binding sites was generated from the UCSC ENCODE transcription factor ChIPseq Uniform Peaks (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeAwgTfbsUniform/). These data sets were generated by the ENCODE TFBS ChIP-Seq production groups and comprise 91 different human cell types under diverse conditions. These include REST antibody enrichment for 10 cell types, of which the human embryonic stem cell (H1-hESC) data was used for analysis. CpGs on the HumanMethylation450 BeadChip were mapped to REST binding regions and only CpGs within the ChIPSeq peak boundaries were considered as defining an overlap. Differential methylation for each binding region was tested by the self-contained enrichment method as implemented in the “fry” function, which can be regarded as a faster version of the “mroast” method in the limma package.Citation77
Methylation profiles of adult blood were downloaded from the NCBI GEO database (accession no. GSE52588). The data set comprises 450K methylation array profiles of whole blood samples from 29 Down syndrome patients (DSP), using their unaffected mothers (DSM) and siblings (DSS) as controls.Citation26 The data has been processed using the minfi pipepline.Citation70 After removal of SNP-containing probes, the 467,971 remaining β values were normalized using the dasen method as implemented in the wateRmelon package.Citation34 Differential methylation analysis was performed using the linear modeling approach of limma as described above.Citation71 To account for the relationship structure the family identifier of the trios has been included as a factor covariate in the model. Methylation profiles of adult brains were downloaded from the NCBI GEO database (accession number GSE63347). The data set included 71 samples from multiple brain regions (cerebellum, temporal, occipital and frontal cortex).Citation28 Preprocessing and differential methylation analysis were performed as described above focusing on the contrast between male DS and normal male control cortices.
DNA methylation age was calculated, as described previously.Citation38 A measure of age acceleration was defined as the residual of a linear model regressing DNA methylation age on chronological age in controls.Citation28 Samples with a DNA methylation age higher than expected have positive residuals, indicating accelerated aging. The significance of the age acceleration effect has been assessed using a Wilcox rank sum test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Declarations
Ethical approval references are listed in the methods. All authors read and approved the manuscript.
Supplemental_Data.doc
Download MS Word (2.1 MB)Acknowledgments
We thank the donors and families who made this research possible. We thank Dr. Camelia-Maria Monoranu, Dr. Thomas Arzberger and the Neurobiobank Munich for providing adult DS brain samples. We thank Dr. Catherine Do for providing us with a list of pan-tissue DS sites and CTCF motifs. The support of Prof. Heinrich Leonhardt and Dr. Sebastian Bultmann for performing FACS sorting is highly appreciated.
References
- Nelson L, Johnson JK, Freedman M, Lott I, Groot J, Chang M, Milgram NW, Head E. Learning and memory as a function of age in Down syndrome: a study using animal-based tasks. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29:443-53; PMID:15795053; http://dx.doi.org/10.1016/j.pnpbp.2004.12.009
- Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down syndrome. Lancet Neurol 2010; 9:623-33; PMID:20494326; http://dx.doi.org/10.1016/S1474-4422(10)70112-5
- Weitzdoerfer R, Dierssen M, Fountoulakis M, Lubec G. Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. J Neural Transm Suppl 2001; 61:59-70; PMID:11771761; http://dx.doi.org/10.1007/978-3-7091-6262-0_5
- Lubec G, Engidawork E. The brain in Down syndrome (trisomy 21). J Neurol 2002; 249:1347-56; PMID:12382149; http://dx.doi.org/10.1007/s00415-002-0799-9
- Benavides-Piccione R, Ballesteros-Yáñez I, de Lagrán MM, Elston G, Estivill X, Fillat C, Defelipe J, Dierssen M. On dendrites in Down syndrome and DS murine models: a spiny way to learn. Prog Neurobiol 2004; 74:111-52; PMID:15518956; http://dx.doi.org/10.1016/j.pneurobio.2004.08.001
- Kleschevnikov AM, Belichenko PV, Salehi A, Wu C. Discoveries in Down syndrome: moving basic science to clinical care. Prog Brain Res 2012; 197:199-221; PMID:22541294; http://dx.doi.org/10.1016/B978-0-444-54299-1.00010-8
- Lejeune J, Gautier M, Turpin R. Etude des chromosomes somatiques de neuf enfants mongoliens. CR Hebd Seances Acad Sci 1959; 248:1721-2; PMID:13639368
- Korenberg JR, Kawashima H, Pulst SM, Ikeuchi T, Ogasawara N, Yamamoto K, Schonberg SA, West R, Allen L, Magenis E, et al. Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am J Hum Genet 1990; 47:236-46; PMID:2143053
- Lana-Elola E, Watson-Scales SD, Fisher EM, Tybulewicz VL. Down syndrome: searching for the genetic culprits. Dis Model Mech 2011; 4:586-95; PMID:21878459; http://dx.doi.org/10.1242/dmm.008078
- FitzPatrick DR, Ramsay J, McGill NI, Shade M, Carothers AD, Hastie D. Transcriptome analysis of human autosomal trisomy. Hum Mol Genet 2002; 11:3249-56; PMID:12471051; http://dx.doi.org/10.1093/hmg/11.26.3249
- Aït Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, Golfier G, Rossier J, Personnaz L, Creau N, Bléhaut H, et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet 2007; 81:475-91; PMID:17701894; http://dx.doi.org/10.1086/520000
- Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, et al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of Down syndrome. Genome Res 2004; 14:1258-67; PMID:15231742; http://dx.doi.org/10.1101/gr.1951304
- Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis S. Gene expression from the aneuploid chromosome in a trisomy mouse model of Down syndrome. Genome Res 2004; 14:1268-74; PMID:15231743; http://dx.doi.org/10.1101/gr.2090904
- Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom RS, Hibaoui Y, et al. Domains of genome-wide gene expression dysregulation in Down syndrome. Nature 2014; 508:345-50; PMID:24740065; http://dx.doi.org/10.1038/nature13200
- Prandini P, Deutsch S, Lyle R, Gagnebin M, Delucinge Vivier C, Delorenzi M, Gehrig C, Descombes P, Sherman S, Dagna Bricarelli F, et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet 2007; 81:2522-63; PMID:17668376; http://dx.doi.org/10.1086/519248
- Cheung VG, Spielman RS. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat Rev Genet 2009; 10:595-604; PMID:19636342; http://dx.doi.org/10.1038/nrg2630
- Bennett-Baker PE, Wilkowski J, Burke DT. Age-associated activation of epigenetically repressed genes in the mouse. Genetics 2003; 165:2055-62; PMID:14704185
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33:245-54; PMID:12610534; http://dx.doi.org/10.1038/ng1089
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 1997; 13:335-40; PMID:9260521; http://dx.doi.org/10.1016/S0168-9525(97)01181-5
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; http://dx.doi.org/10.1038/nrg3230
- Haaf T. Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol 2006; 310:13-22; PMID:16909904; http://dx.doi.org/10.1007/3-540-31181-5_2
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 2012; 13:97-109; PMID:22215131; http://dx.doi.org/10.1038/nrg3142
- Kerkel K, Schupf N, Hatta K, Pang D, Salas M, Kratz A, Minden M, Murty V, Zigman WB, Mayeux RP, et al. Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet 2010; 6:e1001212; PMID:21124956; http://dx.doi.org/10.1371/journal.pgen.1001212
- Jin S, Lee YK, Lim YC, Zheng Z, Lin XM, Ng DP, Holbrook JD, Law HY, Kwek KY, Yeo GS, et al. Global DNA hypermethylation in Down syndrome placenta. PLoS Genet 2013; 9:e1003515; PMID:23754950; http://dx.doi.org/10.1371/journal.pgen.1003515
- Jones MJ, Farré P, McEwen LM, Macisaac JL, Watt K, Neumann SM, Emberly E, Cynader MS, Virji-Babul N, Kobor MS. Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome. BMC Med Genomics 2013; 6:58; PMID:24373378; http://dx.doi.org/10.1186/1755-8794-6-58
- Bacalini MG, Gentilini D, Boattini A, Giampieri E, Pirazzini C, Giuliani C, Fontanesi E, Scurti M, Remondini D, Capri M, et al. Identification of a DNA methylation signature in blood cells from persons with Down syndrome. Aging (Albany NY) 2015; 7:82-96; PMID:25701644; http://dx.doi.org/10.18632/aging.100715
- Sailani MR, Santoni FA, Letourneau A, Borel C, Makrythanasis P, Hibaoui Y, Popadin K, Bonilla X, Guipponi M, Gehrig C, et al. DNA-methylation patterns in trisomy 21 using cells from monozygotic twins. PLoS One 2015; 10:e0135555; PMID:26317209; http://dx.doi.org/10.1371/journal.pone.0135555
- Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, di Blasio AM, Giuliani C, Tung S, Vinters HV, et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015; 14:491-5; PMID:25678027; http://dx.doi.org/10.1111/acel.12325
- Mendioroz M, Do C, Jiang X, Liu C, Darbary HK, Lang CF, Lin J, Thomas A, Abu-Amero S, Stanier P, et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol 2015; 16:263; PMID:26607552; http://dx.doi.org/10.1186/s13059-015-0827-6
- Lu J, McCarter M, Lian G, Esposito G, Capoccia E, Delli-Bovi LC, Hecht J, Sheen V. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum Mol Genet 2016; Epub ahead of print; PMID:26911678; http://dx.doi.org/10.1093/hmg/ddw043
- Dekker AD, de Deyn PP, Rots MG. Epigenetics: the neglected key to minimize learning and memory deficits in Down syndrome. Neurosci Biobehav Rev 2014; 45:72-84; PMID:24858130; http://dx.doi.org/10.1016/j.neubiorev.2014.05.004
- Courtney SM, Petit L, Haxby JV, Ungerleider LG. The role of prefrontal cortex in working memory: examining the contents of consciousness. Philos Trans R Soc Lond B Biol Sci 1998; 353:1819-28; PMID:9854254; http://dx.doi.org/10.1098/rstb.1998.0334
- Faw B. Pre-frontal executive committee for perception, working memory, attention, long-term memory, motor control, and thinking: a tutorial review. Conscious Cogn 2003; 12:83-139; PMID:12617864; http://dx.doi.org/10.1016/S1053-8100(02)00030-2
- Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450 K methylation array data. BMC Genomics 2013; 14:293; PMID:23631413; http://dx.doi.org/10.1186/1471-2164-14-293
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A β-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013; 29:189-96; PMID:23175756; http://dx.doi.org/10.1093/bioinformatics/bts680
- Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol 2014; 15:503; PMID:25599564; http://dx.doi.org/10.1186/s13059-014-0503-2
- Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol 2012; 13:R44; PMID:22703947; http://dx.doi.org/10.1186/gb-2012-13-6-r44
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013; 14:R115; PMID:24138928; http://dx.doi.org/10.1186/gb-2013-14-10-r115
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009; 10:48; PMID:19192299; http://dx.doi.org/10.1186/1471-2105-10-48
- Wagner M, Steinbacher J, Kraus TF, Michalakis S, Hackner B, Pfaffeneder T, Perera A, Müller M, Giese A, Kretzschmar HA, et al. Age-dependent levels of 5-methyl-, 5-hydroxymethyl-, and 5-formylcytosine in human and mouse brain tissues. Angew Chem Int Ed Engl 2015; 54:12511-4; PMID:26137924; http://dx.doi.org/10.1002/anie.201502722
- Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem 2004; 279:27816-23; PMID:15105426; http://dx.doi.org/10.1074/jbc.M400181200
- Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, Akopian V, Gifford CA, Donaghey J, Galonska C, et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet 2015; 47:469-78; PMID:25822089; http://dx.doi.org/10.1038/ng.3258
- Canzonetta C, Mulligan C, Deutsch S, Ruf S, O'Doherty A, Lyle R, Borel C, Lin-Marq N, Delom F, Groet J, et al. DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in Down syndrome. Am J Hum Genet 2008; 83:388-400; PMID:18771760; http://dx.doi.org/10.1016/j.ajhg.2008.08.012
- Bahn S, Mimmack M, Ryan M, Caldwell MA, Jauniaux E, Starkey M, Svendsen CN, Emson P. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with Down syndrome: a gene expression study. Lancet 2002; 359:310-5; PMID:11830198; http://dx.doi.org/10.1016/S0140-6736(02)07497-4
- Lepagnol-Bestel AM, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N, Imbeaud S, Loe-Mie Y, Benihoud K, Agier N, et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum Mol Genet 2009; 18:1405-14; PMID:19218269; http://dx.doi.org/10.1093/hmg/ddp047
- Aapola U, Kawasaki K, Scott HS, Ollila J, Vihinen M, Heino M, Shintani A, Kawasaki K, Minoshima S, Krohn K, et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000; 65:293-8; PMID:10857753; http://dx.doi.org/10.1006/geno.2000.6168
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011; 480:490-5; PMID:22170606; http://dx.doi.org/10.1038/nature10716
- Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci 2007; 8:11-20; PMID:17133224; http://dx.doi.org/10.1038/nrn2043
- Weiner JA, Jontes JD. Protocadherins, not prototypical: a complex tale of their interactions, expression, and functions. Front Mol Neurosci 2013; 6:4; PMID:23515683; http://dx.doi.org/10.3389/fnmol.2013.00004
- Keeler AB, Molumby MJ, Weiner JA. Protocadherins branch out: multiple roles in dendrite development. Cell Adh Migr 2015; 9:214-26; PMID:25869446; http://dx.doi.org/10.1080/19336918.2014.1000069
- Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 1999; 97:779-90; PMID:10380929; http://dx.doi.org/10.1016/S0092-8674(00)80789-8
- Toyoda S, Kawaguchi M, Kobayashi T, Tarusawa E, Toyama T, Okano M, Oda M, Nakauchi H, Yoshimura Y, Sanbo M, et al. Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 2014; 82:94-108; PMID:24698270; http://dx.doi.org/10.1016/j.neuron.2014.02.005
- Farcas R, Schneider E, Frauenknecht K, Kondova I, Bontrop R, Bohl J, Navarro B, Metzler M, Zischler H, Zechner U, et al. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol Biol Evol 2009; 26:1379-89; PMID:19282513; http://dx.doi.org/10.1093/molbev/msp046
- Wang X, Weiner JA, Levi S, Craig AM, Bradley A, Sanes JR. Gamma protocadherins are required for survival of spinal interneurons. Neuron 2002; 36:843-54; PMID:12467588; http://dx.doi.org/10.1016/S0896-6273(02)01090-5
- Garrett AM, Weiner JA. Control of CNS synapse development by (gamma)-protocadherin-mediated astrocyte-neuron contact. J Neurosci 2009; 29:11723-31; PMID:19776259; http://dx.doi.org/10.1523/JNEUROSCI.2818-09.2009
- Garrett AM, Schreiner D, Lobas MA, Weiner JA. γ-protocadherins control cortical dendrite arborization by regulating the activity of a FAK/PKC/MARCKS signaling pathway. Neuron 2012; 74:269-76; PMID:22542181; http://dx.doi.org/10.1016/j.neuron.2012.01.028
- Chen WV, Alvarez FJ, Lefebvre JL, Friedman B, Nwakeze C, Geiman E, Smith C, Thu CA, Tapia JC, Tasic B, et al. Functional significance of isoform diversification in the protocadherin gamma gene cluster. Neuron 2012; 75:402-9; PMID:22884324; http://dx.doi.org/10.1016/j.neuron.2012.06.039
- Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, Sanes JR. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 2012; 488:517-21; PMID:22842903; http://dx.doi.org/10.1038/nature11305
- Shima Y, Kawaguchi SY, Kosaka K, Nakayama M, Hoshino M, Nabeshima Y, Hirano T, Uemura T. Opposing roles in neurite growth control by two seven-pass transmembrane cadherins. Nat Neurosci 2007; 10:963-9; PMID:17618280; http://dx.doi.org/10.1038/nn1933
- Brown NF, Weis BC, Husti JE, Foster DW, McGarry JD. Mitochondrial carnitine palmitoyltransferase I isoform switching in the developing rat heart. J Biol Chem 1995; 270:8952-7; PMID:7721804; http://dx.doi.org/10.1074/jbc.270.15.8952
- Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 2007; 43:332-47; PMID:17602948; http://dx.doi.org/10.1016/j.freeradbiomed.2007.03.027
- Brugge KL, Nichols S, Delis D, Saitoh T, Truaner D. The role of alterations in free radical metabolism in mediating cognitive impairments in Down syndrome. EXS 1992; 62:190-8; PMID:1450586; http://dx.doi.org/10.1007/978-3-0348-7460-1_19
- Pagano G, Castello G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol 2012; 724:291-9; PMID:22411251; http://dx.doi.org/10.1007/978-1-4614-0653-2_22
- Friedland-Leuner K, Stockburger C, Denzer I, Eckert GP, Müller WE. Mitochondrial dysfunction: cause and consequence of Alzheimer disease. Prog Mol Biol Transl Sci 2014; 127:183-210; PMID:25149218; http://dx.doi.org/10.1016/B978-0-12-394625-6.00007-6
- Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, Bunney WE, Vawter MP. Mitochondrial involvement in psychiatric disorders. Ann Med 2008; 40:281-95; PMID:18428021; http://dx.doi.org/10.1080/07853890801923753
- Zhang L, Li H, Hu X, Benedek DM, Fullerton CS, Forsten RD, Naifeh JA, Li X, Wu H, Benevides KN, et al. Mitochondria-focused gene expression profile reveals common pathways and CPT1B dysregulation in both rodent stress model and human subjects with PTSD. Transl Psychiatry 2015; 5:e580; PMID:26080315; http://dx.doi.org/10.1038/tp.2015.65
- Strong E, Butcher DT, Singhania R, Mervis CB, Morris CA, de Carvalho D, Weksberg R, Osborne LR. Symmetrical dose-dependent DNA-methylation profiles in children with deletion or duplication of 7q11.23. Am J Hum Genet 2015; 97:216-27; PMID:26166478; http://dx.doi.org/10.1016/j.ajhg.2015.05.019
- Keeler AB, Schreiner D, Weiner JA. Protein Kinase C Phosphorylation of a γ-protocadherin C-terminal lipid binding domain regulates focal adhesion kinase inhibition and dendrite arborization. J Biol Chem 2015; 290:20674-86; PMID:26139604; http://dx.doi.org/10.1074/jbc.M115.642306
- Assenov Y, Muller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods 2014; 11:1138-40; PMID:25262207; http://dx.doi.org/10.1038/nmeth.3115
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30:1363-9; PMID:24478339; http://dx.doi.org/10.1093/bioinformatics/btu049
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47; PMID:25605792; http://dx.doi.org/10.1093/nar/gkv007
- Makambi KH. Weighted inverse chi-square method for correlated significance tests. J Appl Stat 2003; 30:225-34; http://dx.doi.org/10.1080/0266476022000023767
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat 1979; 16:65-70; http://dx.doi.org/10.2307/4615733
- Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15:R29; PMID:24485249; http://dx.doi.org/10.1186/gb-2014-15-2-r29
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. JR Statist Soc B 1995; 57:289-300; http://dx.doi.org/10.2307/2346101
- Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res 2012; 40:e133; PMID:22638577; http://dx.doi.org/10.1093/nar/gks461
- Phipson B, Smyth GK. Permutation p-values should never be zero: calculating exact p-values when permutations are randomly drawn. Stat Appl Genet Mol Biol 2010; 9:39; PMID:21044043; http://dx.doi.org/10.2202/1544-6115.1585.