
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Curcumin and its analogs exhibited antileukemic activity either as single agent or in combination therapy. Dimethoxycurcumin (DMC) is a more metabolically stable curcumin analog that was shown to induce the expression of promoter-methylated genes without reversing DNA methylation. Accordingly, co-treatment with DMC and DNA methyltransferase (DNMT) inhibitors could hypothetically enhance the re-expression of promoter-methylated tumor suppressor genes. In this study, we investigated the cytotoxic effects and epigenetic changes associated with the combination of DMC and the DNMT inhibitor decitabine (DAC) in primary leukemia samples and cell lines. The combination demonstrated antagonistic cytotoxic effects and was minimally cytotoxic to primary leukemia cells. The combination did not affect the metabolic stability of DMC. Although the combination enhanced the downregulation of nuclear DNMT proteins, the hypomethylating activity of the combination was not increased significantly compared to DAC alone. On the other hand, the combination significantly increased H3K27 acetylation (H3K27Ac) compared to the single agents near the promoter region of promoter-methylated genes. Furthermore, sequential chromatin immunoprecipitation (ChIP) and DNA pyrosequencing of the chromatin-enriched H3K27Ac did not show any significant decrease in DNA methylation compared to other regions. Consequently, the enhanced induction of promoter-methylated genes by the combination compared to DAC alone is mediated by a mechanism that involves increased histone acetylation and not through potentiation of the DNA hypomethylating activity of DAC. Collectively, our results provide the mechanistic basis for further characterization of this combination in leukemia animal models and early phase clinical trials.
Introduction
The role of epigenetic changes in health and disease is well established.Citation1 Recent discoveries of the role of epigenetic modifications in the development and progression of different diseases such as cancer, diabetes, chronic kidney disease, and neurodegeneration urged the parallel development of drugs that modulate these modifications. DNA methyltransferase (DNMT) inhibitors and histone deacetylase (HDAC) inhibitors were the first categories of drugs tested in clinical trials as epigenetic modifiers and FDA-approved for treatment of different hematologic malignancies.Citation2,3 Nutraceuticals like genistein, epigallocatechin-3-gallate, and curcumin demonstrated activity as epigenetic modifiers and a long list of other nutraceuticals is awaiting validation of their activity.Citation4 Curcumin, a hydrophobic polyphenol derived from the rhizome of the herb Curcuma longa (turmeric), exhibits a wide spectrum of pharmacological activities.Citation5 Curcumin demonstrated anti-inflammatory, antimicrobial, antiviral, antioxidant, and antitumor activity in several studies.Citation6-8 Curcumin is safe when administered at high doses; however, its low bioavailability due to poor absorption and rapid metabolism is a major drawback.
Several structural analogs like dimethoxycurcumin (DMC) were synthesized to improve the bioavailability and stability of curcumin.Citation9 Curcumin and its analogs were shown to induce epigenetic changes through different mechanisms like inhibition of (HDAC) and histone acetyltransferase (HAT) enzymes, modulating miRNAs expression and DNA methylation reversal. For instance, curcumin was shown to inhibit or downregulate the expression of DNMT enzymes with consequent reversal of DNA methylation.Citation10,11 On the contrary, curcumin and DMC were shown to induce the expression of promoter-methylated genes without reversing DNA methylation.Citation12,13
In this study, we hypothesized that the combination of DMC with the DNMT inhibitor decitabine (DAC) could enhance the induction of promoter-methylated tumor suppressor genes in leukemia cells through a mechanism that involves potentiation of the DNA hypomethylating activity of DAC and/or by inducing histone modifications such as acetylation and methylation. The combination demonstrated antagonistic cytotoxic effects and induced minimal apoptosis in primary leukemia cells. The combination repressed gene expression and downregulated the nuclear protein levels of DNMT enzymes. However, the DNA hypomethylating activity of the combination did not significantly increase compared to the use of DAC as a single agent. On the other hand, the combination enhanced the induction of promoter-methylated genes such as p15 and CDH-1 compared to single agents. Analysis of the histone marks associated with actively transcribed genes such as acetylated H3K27 (H3K27Ac) and trimethylated H3K36 (H3K36me3) near the promoter region of both genes demonstrated significant increase in the H3K27Ac mark. Furthermore, DNA pyrosequencing of the chromatin enriched H3K27Ac did not show significant decrease in DNA methylation compared to other regions, indicating that the chromatin enriched H3K27Ac was not preferentially demethylated. Our results provide mechanistic insight and demonstrate the utility of the DMC-DAC combination as a non-cytotoxic combinatorial epigenetic therapy for the treatment of leukemia.
Materials and methods
Patient samples
Frozen acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) bone marrow mononuclear cells (BMNC) were purchased from Allcells (Alameda, CA). BMNC were obtained from newly diagnosed and untreated patients. The viability of the cells was tested using the Guava ViaCount Reagent (Millipore, Hayward, CA), which distinguishes between viable and non-viable cells based on the differential permeability of DNA-binding dyes in the ViaCount reagent. The thawed BMNC showed at least 90% viability in all the samples. Supplementary Fig. 1 shows the viability assessment from one of the samples.
Cell culture and chemicals
Leukemia cell lines (CEM and Jurkat) were grown in RPMI medium supplemented with 10% fetal bovine serum (Life Technologies, CA) in a humidified atmosphere containing 5% CO2 at 37°C. DMC was purchased from Cayman (Ann Arbor, MI) and dissolved in DMSO to prepare a 10 mM stock. Decitabine was purchased from Sigma (Madison, WI) and dissolved in PBS to prepare a 10 mM stock. PCR primers were purchased from Integrated DNA Technologies (Coralville, IA).
Apoptosis quantitation
Apoptosis quantitation was performed by double staining with both annexin V-phosphatidylethanolamine (PE) and the cell impermeant dye, 7-aminoactinomycin D (7-AAD). Fluorescence detection of apoptotic cells was done using flow cytometry (Guava easyCyte 5 system) as described previously.Citation12
Immunoblotting and nuclear protein extraction
Western blotting was performed as described previously.Citation14 Briefly, RIPA lysis buffer (Millipore) was used for total protein extraction. Protein concentration was estimated using the BCA protein assay kit (Pierce, IL). SDS-PAGE using 10 or 12% gels was used to separate proteins and transfer to PVDF membrane (Pierce) was done using semi-dry transfer method. Incubation with 1:250 dilutions of mouse monoclonal CDH-1 primary antibody (SC-21791) from Santa Cruz (Dallas, TX) and rabbit polyclonal p15 primary antibody (PIPA124258) from ThermoFisher Scientific (Waltham, MA) overnight at 4°C was performed followed by 1 h incubation with HRP conjugated secondary antibodies and chemiluminescence detection using ECL Western blotting substrate (Pierce). Image acquisition and band quantitation was done using a CCD camera and a GeneSys image acquisition software (Syngene, MD). Nuclear proteins were extracted using a hypotonic buffer solution (ThermoFisher) and incubated with monoclonal antibodies against the 3 isoforms of DNMT enzymes (DNMT1, Catalog No. 39204; DNMT3a, Catalog No. 39206; DNMT3b, Catalog No. 39207) purchased from Active Motif (Carlsbad, CA) for Western blotting.
Metabolism of DMC and DAC by human liver microsomal enzymes
The time course metabolism of 500 nM DMC alone and in presence of 250 nM DAC (ratio 2:1) was evaluated using human pooled liver microsomes followed by HPLC analysis as described previously.Citation12 Briefly, the metabolism studies were conducted in triplicates using human pooled liver microsomes (0.5 mg/ml). The reaction mixture consisted of NADPH-regenerating system (1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride) and 10 mM potassium phosphate buffer (pH 7.4). The reactions were initiated by adding the drug to the pre-warmed reaction mixture. After 0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, and 60 min of incubation at 37°C, 100 µL of the reaction was sampled, immediately mixed with 100 µL of acetonitrile to terminate the reaction and centrifuged at 14,000 rpm for 20 min. Aliquots of the supernatant were collected and analyzed using validated HPLC quantification method. The intrinsic clearance values were calculated using the formula:
Where CLint indicates intrinsic clearance, Vmax indicates the maximum rate achieved at maximum substrate concentration, Km indicates the substrate concentration when the reaction rate is half of Vmax, V0 indicates the initial metabolic rate, and C0 is the substrate concentration at time 0. The intrinsic clearance values were calculated separately from each of the replicates. CLint values are presented as mean ± standard deviation (SD) from 3 replicates performed for each reaction.
Quantitative real-time RT-PCR analysis of gene expression
RNA was extracted using RNeasy kit according to the manufacturer's instructions (Qiagen, CA). RNA was treated with DNase enzyme to remove any DNA contamination associated with the process of RNA extraction using the Turbo DNA-free kit (Ambion-Life Technologies, NY) according to the manufacturer's instructions. cDNA was generated using the Verso cDNA synthesis kit (ThermoScientific). Real-time PCR was performed using a RealPlex II thermal cycler (Eppendorf, NY) and Dynamo HS SYBR Green qPCR kit (ThermoScientific). A three-step cycling protocol with annealing/extension temperature of 60°C was used. Supplementary Table 1 shows the nucleotide sequences of the primers used for DNMT1, DNMT3a, DNMT3b, p15, and CDH-1 genes.
DNA pyrosequencing
Quick-gDNA microprep kit (Zymo Research, CA) was used to extract genomic DNA. Bisulfite treatment of DNA (500 ng) was performed using the EZ DNA Methylation-Gold kit (Zymo Research). Amplification of template DNA for pyrosequencing was performed using a PyroMark PCR kit (Qiagen). Primers for CDH-1 and p15 genes were designed using the PyroMark Assay Design Software (Qiagen) and the reverse primers were biotin-labeled. The amplicon sizes were 170 and 207 bp for the p15 and the CDH-1, respectively. Supplementary Fig. 2 shows the verification of the amplicon size by agarose gel electrophoresis. The primer sequence to analyze the p15 gene was 5′CGGGCCGCTGCGCGTCTGGGGGCTGCGGAATGCGCGA3′ and included 7 CpG sites (underlined). The primer sequence to analyze for the CDH-1 gene was 5′CGGCAGCGCGCCCTCACCTCTGCCCAGGACGCGGC3′ and included 5 CpG sites (underlined). Pyrosequencing was performed using a PyroMark Q24 instrument (Qiagen).
Chromatin immunoprecipitation (ChIP) and sequential ChIP-DNA pyrosequencing
ChIP was performed using the EZ-Magna ChIP G kit (Millipore). Cells (2 × 106) were fixed using non-methanol formaldehyde ampules (ThermoScientific). The cells were incubated in a final concentration of 1% formaldehyde for 4 min at room temperature with shaking. The cross-linked DNA was sheared using a Covaris S2 Focused-ultrasonicator (Covaris, MA). The shearing conditions were: duty cycle: 4%; intensity: 4; and cycles per burst: 200 for 12 min using 1 ml Covaris tubes (12×12 mm). The sheared DNA was analyzed by Agilent 2100 Bioanalyzer to determine the average size of the sheared DNA. Supplementary Fig. 3 shows the average size of the input DNA after Covaris shearing in CEM and Jurkat cells. Immunoprecipitation of the cross-linked DNA was performed using overnight incubation with a ChIP grade rabbit monoclonal anti-H3K36me3 (Millipore, Cat#17-10032) and rabbit polyclonal anti-H3K27Ac (Millipore, Cat#07-360-S) in the presence of protein G magnetic beads. Normal rabbit IgG was used as a negative control (Millipore). The immunoprecipitated cross-linked DNA was pulled down using a magnetic separator followed by reversal of the cross linking to free DNA. Real-time PCR analysis of the pulled down DNA was performed. The ChIP primers for the p15 gene were designed to amplify a 155 bp amplicon within the CpG island associated with exon 1. The forward primer was 5′ AGGGTAATGAAGCTGAGCCC3′ and the reverse primer was 5′CTTGTTCTCCTCGCGCATTC3′. The ChIP primers for the CDH-1 gene amplified a 207 bp amplicon within the CpG island associated with exon 1 and the forward primer was 5′GGGTGTGGAGAAGGGGTG3′ while the reverse primer was 5′GGAATGCACCACTCCTCAGA3′. The input DNA was amplified using GAPDH primers; forward primer 5′tACTAGCGGTTTTACGGGCG3′ and reverse primer 5′tCGAACAGGAGGAGCAGAGAGCGA3′. The cycle threshold (Ct) for the ChIP antibodies and the negative control (IgG) were normalized to the input Ct using the equation:
Where IP indicates the Ct for the ChIP antibodies or the IgG and DF indicates the input dilution factor. The ChIP yield was calculated as a percentage of the input using the equation:
Sequential ChIP-DNA pyrosequencing is a technique that directly interrogates the genomic relationship between allele-specific DNA methylation and histone modifications.Citation15,16 To directly analyze DNA methylation associated with the histone mark H3K27Ac, 50 ng of DNA derived from ChIP experiments using H3K27Ac and rabbit IgG antibodies were bisulfite treated, PCR amplified and pyrosequencing analyzed as described above.
Statistical analyses
Data are represented as the mean ± SD. Statistical difference between the control and drug-treated samples was calculated using Student's t-test or ANOVA followed by Bonferroni post-hoc test where appropriate. P < 0.05 was considered statistically different.
Results
DMC antagonizes the cytotoxicity of DAC in primary leukemia patient samples and cell lines
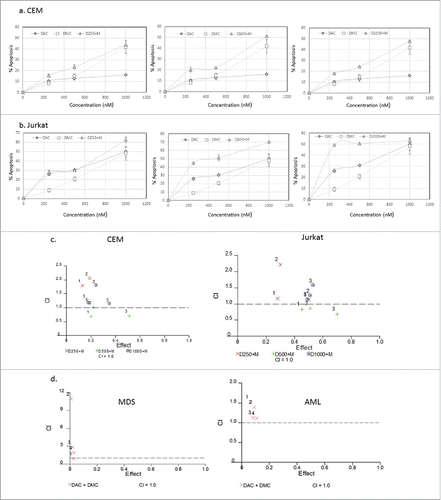
The combination of DAC or 5-azacytidine with histone deacetylase inhibitors or chemotherapeutic agents was previously reported.Citation17-20 The combination of DAC and DMC could induce synergistic cytotoxicity and abrogate their induced epigenetic changes. In order to select the combination concentrations that induce minimal cytotoxicity, we studied the effect of the combination of DMC and DAC on apoptosis induction in leukemia cell lines and primary leukemia patient samples. The combination regimen included fixed concentration of DAC with different concentrations of DMC. shows the effect of the combination and single drugs on apoptosis induction in CEM cells () and Jurkat cells (). The combinations of DMC (250 and 500 nM) with DAC (250 nM) were the least cytotoxic in both cell lines. Analysis of the synergistic and antagonistic effects of the drug combinations by the median-effect equation to calculate the combination index (CI) was performed using CalcuSyn software.Citation21 DAC (250 and 1000 nM) combined with different concentrations of DMC (250, 500, or 1000 nM) demonstrated antagonistic effects on apoptosis induction in both cell lines as indicated by a CI value > 1 (). On the other hand, DAC (500 nM) demonstrated slightly synergistic or additive effects when combined with different DMC concentrations in both cell lines (). To study the cytotoxicity of the combination in primary AML and MDS BMNC samples, we selected the lower concentration of DAC (250 nM) that showed antagonistic effect in addition to a lower concentration (100 nM) and combined them with DMC (250 and 500 nM). The combination induced minimal cytotoxicity in both AML and MDS BMNC (Supplementary Fig. 4) and demonstrated antagonistic effects on apoptosis induction (). Supplementary Table 2 summarizes the CI values for the cell lines and the MDS and AML primary samples. Accordingly, we selected DAC concentrations (100 and 250 nM) and DMC concentrations (250 and 500 nM) to study their effect on gene expression and DNA methylation as they demonstrated antagonistic effects and induced minimal cytotoxicity.
Figure 1. Effect of simultaneous treatment with DAC and DMC on apoptosis induction in leukemia cells. a. CEM leukemia cells were treated with different concentrations of DAC, DMC, and the combination for 48 h and apoptosis induction was quantified as described under methods. The left, middle, and right panels: we used 250 nM, 500 nM, and 1000 nM of DAC in combination with different concentrations of DMC, respectively. b. Jurkat leukemia cells were treated with DAC, DMC, and the combination, similar to the treatment to CEM cells. In all experiments, data represent the mean ± SD for 3 replicates. c. Analysis of the synergistic and antagonistic effects of the combination on apoptosis induction. The combination index (CI) was calculated by CalcuSyn software for dose-effect analysis in CEM cells (left panel) and Jurkat cells (right panel). The combinations used were fixed concentrations of DAC with variable concentrations of DMC (250, 500, 1000 nM). d. Analysis of the synergistic and antagonistic effects of the combination on apoptosis induction in BMNC derived from MDS (left panel) and AML patients (right panel). ‘1′ indicates 100 nM DAC + 250 nM DMC, ‘2′ indicates 100 nM DAC + 500 nM DMC, ‘3′ indicates 250 nM DAC + 250 DMC, and ‘4′ indicates 250 nM DAC + 500 nM DMC. CI values equal to 1 are represented by the dashed line and considered additive, values greater than 1 are antagonistic, and values lower than 1 are synergistic.
The combination of DAC and DMC represses the expression of DNMT enzymes
Curcumin and its synthetic analogs were reported to modulate the expression of the enzymes that catalyze DNA methylation.Citation22 We hypothesized that DMC could potentiate the DNA hypomethylating effect of DAC by repressing the gene expression of the enzymes involved in DNA methylation. To test this hypothesis, leukemia cells were treated with the single drugs and the combination followed by quantifying the gene expression of the DNMT isoforms that catalyze DNA methylation (DNMT1, DNMT3a and DNMT3b) as described under methods. The single agents did not significantly decrease DNMT1 expression except for DMC when used at 500 nM in both cell lines (). On the other hand, the different combinations decreased DNMT1 expression in both cell lines but to a different extent, where the combination of DAC (100 nM) and DMC (250 nM) induced a 2-fold decrease in Jurkat cells and the combination of DAC (250 nM) and DMC (500 nM) induced a 7-fold decrease in CEM cells (). Similar to DNMT1, DAC did not affect the expression of DNMT3a and DMC decreased its expression in CEM cells only (). The different combinations decreased the expression of DNMT3a with different extents ranging between 2- and 7-folds in both cell lines (). Contrary to DNMT1 and DNMT3a, DAC slightly induced the expression of DNMT3b, while DMC decreased its expression at the higher concentration (). Similar to DNMT1 and DNMT3a, the combination decreased the expression of DNMT3b in CEM cells 2–3 folds when using 250 nM DAC (). Collectively, the combination was more effective than the single drugs in decreasing the expression of the 3 isoforms of DNMT and the fold decrease was both concentration and cell line dependent.
Figure 2. The combination therapy represses DNMT isoforms gene expression. CEM and Jurkat cell lines were treated with the single agents and the combination for 48 h followed by RNA extraction and quantitative real time RT-PCR using gene specific primers for the 3 DNMT isoforms DNMT1 (a), DNMT3a (b), and DNMT3b (c). The alphanumeric characters represent the name of the drug and the concentration in nM, where ‘D’ represents DAC and ‘M’ represents DMC. For instance, D100 represents DAC 100 nM and D250 + M500 represents the combination of DAC 250 nM and DMC 500 nM. Data represent the mean ± SD for 3 replicates. d. CEM cells treated with single agents and the combination for 48 h followed by nuclear protein extraction and protein gel blotting.
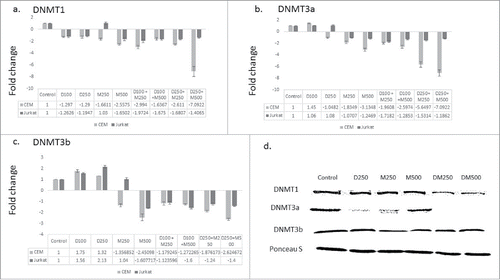
To further test our hypothesis, we quantified the nuclear protein level of the 3 DNMT isoforms after treatment with the single drugs and the combination in CEM cells, which demonstrated higher gene repression of DNMT enzymes. The combination decreased the nuclear protein expression of both DNMT1 and DNMT3a isoforms but did not affect DNMT3b (). Collectively, the combination repressed the gene expression and decreased the nuclear protein level of the enzymes involved in DNA methylation.
The combination of DMC and DAC enhances the gene expression induction of promoter-methylated genes
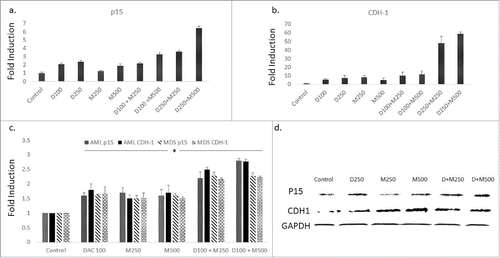
p15 and CDH-1 are tumor suppressor genes and were shown to be silenced in leukemia cells by promoter DNA methylation.Citation12 We hypothesized that the combination of DMC and DAC will enhance the re-expression of genes silenced by promoter methylation based on the effect of the combination on the expression of the nuclear DNMT enzymes. Although the single agents induced the expression of both genes, the combination of DAC (250 nM) with DMC (250 or 500 nM) further enhanced the induction of p15 and CDH-1 compared to the single drugs in CEM cells () and Jurkat cells (Supplementary Fig. 5). The combination also enhanced the induction of expression of both genes in BMNC derived from AML and MDS patients (). Furthermore, both the single agents and the combination increased the protein expression of p15 and CDH-1 in CEM cells () and Jurkat cells (Supplementary Fig. 6). Taken together, the combination of DMC and DAC enhances the induction of expression of promoter-methylated genes compared to single agents and upregulated their protein expression.
Figure 3. The combination of DMC and DAC enhances gene expression of promoter-methylated genes. CEM cells treated with the single drugs and the combination for 24 h and gene expression of p15 (a) and CDH-1 (b) was quantified by RT-PCR as described under Materials and methods. c. BMNC from AML and MDS patients were treated with the single drugs and the combination for 24 h and gene expression of p15 and CDH-1 was quantified. The alphanumeric characters represent the name of the drug and the concentration in nM, where ‘D’ represents DAC and ‘M’ represents DMC. For instance, D100 represents DAC 100 nM and D250 + M500 represents the combination of DAC 250 nM and DMC 500 nM. Data represent the mean ± SD for 3 replicates. * indicates a significant difference from the control at P < 0.05. d. CEM cells treated with the single drugs and the combination for 24 h and the protein expression of p15 and CDH-1 was quantified by Western blotting. D + M250 indicates the combination of 250 nM DAC and 250 nM of DMC. D + M500 indicates the combination of 250 nM DAC and 500 nM DMC.
DMC does not reverse promoter DNA methylation or enhance DAC hypomethylating activity
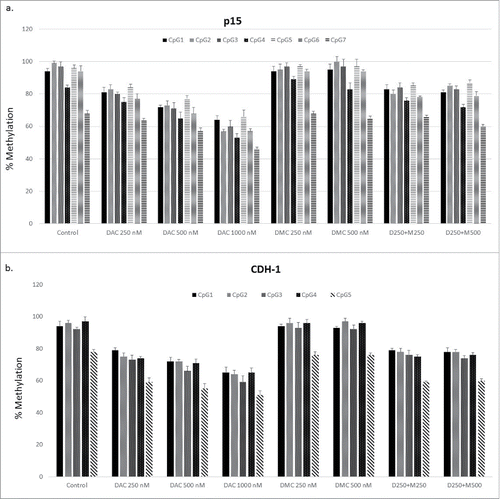
The decreased nuclear protein expression of DNMT1 and DNMT3a after treatment with the combination compared to DAC alone suggests an enhanced DNA hypomethylating effect of the combination. CEM cells were treated with different concentrations of DAC, DMC, and the combination followed by quantitation of DNA methylation reversal by DNA pyrosequencing in the CpG island associated with exon 1 of p15 (7 CpG sites) and CDH-1 (5 CpG sites) genes. All the CpG sites analyzed in both genes were heavily methylated (>90 %), except for CpG 7 in p15 (68 ± 2.8%) and CpG 5 in CDH-1 (78 ± 1.4%) (). Treatment with increasing DAC concentrations induced dose-dependent methylation reversal in all the CpG sites in both genes. On the other hand, treatment with increasing concentrations of DMC alone did not reverse methylation in any of the CpG sites in both genes (). The combination of DAC with different concentrations of DMC did not significantly increase the DNA hypomethylating activity of DAC in any of the CpG sites in both genes (). Collectively, DMC lacks DNA hypomethylating activity and does not enhance the hypomethylating activity of DAC despite decreasing the gene expression of the different DNMT isoforms when combined with DAC.
Figure 4. DMC does not enhance the hypomethylating activity of DAC. a. CEM cells treated with single drugs, and the combination for 48 h followed by DNA extraction, bisulfite treatment, and DNA pyrosequencing for 7 CpG sites within the CpG island associated with exon 1 of the p15 gene. b. CEM cells treated with single drugs and the combination for 48 h followed by DNA extraction, bisulfite treatment, and DNA pyrosequencing for 5 CpG sites within the CpG island associated with exon 1 of the CDH-1 gene. Data represent the mean ± SD for duplicates. ‘D’ represents DAC, ‘M’ represents DMC, and D250 + M250 indicates the combination of DAC (250 nM) and DMC (250 nM).
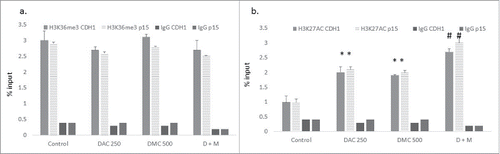
The combination of DAC and DMC increases histone acetylation
Curcumin and its analogs were shown to inhibit isoforms of both histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes.Citation23,24 On the other hand, the effect of curcumin and its analogs on histone methylation is not clear. H3K36me3 and H3K27Ac are epigenetic marks associated with active gene transcription.Citation25 We hypothesized that the combination could affect the expression of these marks and consequently enhance the induction of promoter-methylated genes. Although the combination of DAC and DMC did not significantly increase H3K36me3 (), it significantly increased the H3K27Ac mark within the putative CpG islands associated with exon 1 of both p15 and CDH-1 compared to the single agents (). To investigate the status of DNA methylation in the chromatin enriched with H3K27Ac, we performed sequential ChIP- DNA pyrosequencing using H3K27Ac antibody and an IgG antibody as a negative control. DNA pyrosequencing of 7 CpG sites within the CpG island associated with exon 1 of the p15 gene was performed. The DNA immunoprecipitated from IgG (negative control) did not show any significant difference in DNA methylation from that immunoprecipitated by H3K27Ac antibody (Supplementary Fig. 7), indicating that the chromatin enriched with the H3K27Ac epigenetic mark in the CpG island of the p15 gene was not preferentially demethylated.
Figure 5. The combination of DAC and DMC increases H3K27 acetylation. ChIP analysis of CEM cells treated with single agents or the combination for 24 h was performed using antibodies against H3K36me3 (a) and H3K27Ac (b) followed by real-time quantitative PCR using primers for p15 and CDH-1. IgG was used as a negative control. DAC 250 indicates 250 nM DAC and DMC 500 indicates 500 nM of DMC. D + M indicates the combination of 250 nM DAC and 500 nM DMC. Data represent the mean ± SD of duplicate ChIP reactions and triplicate PCR reactions. * indicates a significant difference from the control at P < 0.05 and# indicates significant difference from the control and single agents using one way ANOVA followed by Bonferroni post-hoc test.
Metabolic stability of the combination of DAC and DMC
DMC was previously reported as a more metabolically stable curcumin analog.Citation9,12 The combination of DMC with DAC could affect the metabolic stability of DMC. Incubating DMC and DAC in the ratio of 2:1 with human liver microsomes to mimic the ratio used in other experiments was performed and the intrinsic clearance of DMC alone and the combination was estimated. The intrinsic clearance of DMC was 41.5 ± 1.66 ul/min/mg protein and when combined with DAC was 47.6 ± 7.03 ul/min/mg protein. There was no significant difference between the single agent and the combination indicating that the combination did not affect DMC metabolism by the liver microsomal enzymes.
Discussion
Curcumin is a powerful chemopreventive and anticancer agent that mediates its therapeutic action by inducing epigenetic changes.Citation26 Curcumin and its analogs demonstrated antileukemic activity either as single agent or in combination therapy.Citation27-30 DMC is a more metabolically stable curcumin analog that was shown to induce the gene expression of promoter-methylated genes without reversing DNA methylation.Citation12 Accordingly, the combination of DMC with DNMT inhibitors could hypothetically enhance the re-expression of epigenetically silenced tumor suppressor genes. In this study, we investigated the effect of combining DMC with the DNMT inhibitor DAC on gene expression, cytotoxicity, and epigenetic changes in primary leukemia cells and leukemia cell lines. The combination demonstrated antagonistic cytotoxic effects, transcription repression, and nuclear protein downregulation of DNMT enzymes and enhanced induction of gene expression of promoter-methylated genes compared to single agents. Although the combination downregulated the expression of nuclear DNMT proteins, the hypomethylating activity of the combination was not significantly increased compared to DAC as a single agent. On the other hand, the combination significantly increased H3K27 acetylation compared to the single agents within the CpG island associated with exon 1 of promoter-methylated genes. Consequently, the enhanced induction of promoter-methylated genes by the combination is mediated by a mechanism that involves increased histone acetylation and not through the potentiation of the DNA hypomethylating activity of DAC.
The objective of epigenetic therapy is to induce the re-expression of silenced tumor suppressor genes with minimal cytotoxic effects.Citation31 Indeed, the FDA-approved epigenetic modifiers like the DNMT inhibitor 5-azacytidine and DAC are used in doses that are substantially lower than their maximum tolerated dose to avoid their cytotoxic effects.Citation32 The combination of DMC with DAC did not synergize or enhance the cytotoxicity of the single agents, which is consistent with the aim of using epigenetic therapy. The observed antagonistic effect and the minimal cytotoxicity of the combination observed in primary MDS and AML patient samples demonstrate the utility of the combination as a non-cytotoxic epigenetic therapy.
We previously demonstrated that both curcumin and DMC lack DNA hypomethylating activity.Citation12 DAC is known to deplete nuclear DNMT enzymes by incorporating into DNA and trapping the enzymes by forming a covalent complex with consequent enzymes degradation.Citation33 In this study, the combination of DMC and DAC repressed the gene expression of the 3 DNMT isoforms at different concentrations. Although there is not always a direct correlation between levels of RNA and their corresponding proteins,Citation34,35 we observed higher downregulation of both nuclear DNMT1 and DNMT3a proteins but not DNMT3b by the combination compared to DAC alone. Accordingly, we expected DMC to potentiate the DNA hypomethylating activity of DAC as a consequence of this downregulation. However, no significant difference between DAC and the combination in reversing DNA methylation was observed. An explanation for this is that the observed difference in downregulation of DNMT1 and DNMT3a by the combination compared to DAC alone may not be sufficient to induce a significant increase in DNA methylation reversal. Indeed, the difference between the nuclear protein level of DNMT3a upon treatment with DAC or the combination was almost null ().
Previous reports demonstrated induction of gene expression despite promoter methylation. For instance, the expression of the estrogen receptor α (ER-α) gene was shown to be silenced by DNA methylation in breast cancer. ER-α expression was induced by treatment with the HDAC inhibitor trichostatin A without reversing DNA methylation.Citation36 Class III HDAC SIRT1 was shown to localize to promoter-methylated silenced tumor suppressor genes. SIRT1 inhibition re-expressed the silenced genes in presence of promoter methylation.Citation37 Moreover, colorectal cancer cells treatment with different HDAC inhibitors induced the expression of promoter methylated genes without reversing DNA methylation, indicating that DNA methylation could not prevent gene reactivation by drug-induced resetting of the chromatin state.Citation38 In this study, DMC similarly induced the expression of both p15 and CDH-1 genes despite retention of promoter methylation. Furthermore, DMC combination with DAC enhanced the induction of both genes compared to single agents by increasing the H3K27Ac mark within the CpG island associated with exon 1. ChIP coupled with DNA microarray (ChIP-chip) analysis revealed that H3K27ac and the repressive epigenetic mark H3K27me3 are mutually exclusive.Citation39 The mechanism of H3K27Ac increase by either the single agents or the combination is not known; however, both DNMT inhibitors and curcumin were shown to increase histone acetylation previously.Citation23,40 Curcumin inhibited different HDAC classes and DMC is speculated to similarly inhibit these enzymes due to its structural similarity to curcumin, which needs to be confirmed. DAC was never reported to directly inhibit HDAC enzymes or stimulate HAT activity and the observed increase in H3K27Ac is probably an indirect consequential effect. Accordingly, the mechanism of the enhanced effect of the combination on H3K27Ac compared to the single agents appears to be complex and involves indirect mechanisms. Although the combination induced the expression of p15 and CDH-1 in both primary patient samples and cell lines, the induction was smaller in the primary cells. This may be explained by the difference in population doubling kinetics as the cell lines divide more rapidly than the primary cells and methylation reversal by DAC is dependent on cell division.
A drawback of epigenetic therapy is being non-specific. Chronic treatment with hypomethylating agents lead to global DNA hypomethylation and hypomethylation of proto-oncogenes and pro-metastatic genes that promote metastasis.Citation4,41 On the other hand, induction of immune-related genes was also observed and could be of benefit to improve clinical outcome of immune therapies.Citation42,43 Accordingly, we are not expecting the combination of DAC and DMC to specifically induce the expression of tumor suppressor genes only. Global analysis of gene expression after treatment with the combination is warranted to identify the complete list of induced genes. Nevertheless, treatment with DAC or 5-azacytidine in the clinic is well tolerated and was not associated with tumor development despite hypomethylation of proto-oncogenes.Citation44,45 Although no significant difference was observed between the DNA hypomethylating activity of DAC alone and in combination with DMC, it is possible that the chromatin enriched with the H3K27Ac epigenetic mark was demethylated to promote induction of gene expression. Sequential ChIP-DNA pyrosequencing demonstrated no significant difference in DNA methylation between the chromatin enriched H3K27Ac and other chromatin regions.
In summary, we provided mechanistic insight and evidence for the utility of combining DMC with the DNMT inhibitor DAC to enhance the re-expression of tumor suppressor genes silenced by promoter DNA methylation. Evaluation of this combination using in vivo leukemia animal models is warranted to evaluate its pharmacokinetics and pharmacodynamics properties against the single agents and to demonstrate the superiority of the combination on tumor growth and survival.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1226452_s02.pdf
Download PDF (592 KB)Acknowledgments
This work was supported by the Scholarship of Discovery Intramural Research Grant Program (SDIRGP) from Albany College of Pharmacy to TEF and University of Maryland intramural research grant to HH. The work was also supported by NIH Grant Numbers 5 P30 RR032135 from the COBRE Program of the National Center for Research Resources and 8 P30 GM 103498 from the National Institute of General Medical Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Related Research Data
References
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769; http://dx.doi.org/10.1038/nrg962
- Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002; 20:2429-40; PMID:12011120; http://dx.doi.org/10.1200/JCO.2002.04.117
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007; 12:1247-52; PMID:17962618; http://dx.doi.org/10.1634/theoncologist.12-10-1247
- Fandy TE. Development of DNA methyltransferase inhibitors for the treatment of neoplastic diseases. Curr Med Chem 2009; 16:2075-85; PMID:19519382; http://dx.doi.org/10.2174/092986709788612738
- Kotecha R, Takami A, Espinoza JL. Dietary phytochemicals and cancer chemoprevention: a review of the clinical evidence. Oncotarget; 7(32):52517-52529; PMID:27232756; http://dx.doi.org/10.18632/oncotarget.9593
- Aggarwal BB, Yuan W, Li S, Gupta SC. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities: Identification of novel components of turmeric. Mol Nutr Food Res 2013; 57:1529-42; PMID:23847105; http://dx.doi.org/10.1002/mnfr.201200838
- Aggarwal BB, Gupta SC, Sung B. Curcumin: an orally bioavailable blocker of TNF and other pro-inflammatory biomarkers. Br J Pharmacol 2013; 169:1672-92; PMID:23425071; http://dx.doi.org/10.1111/bph.12131
- Gupta SC, Patchva S, Aggarwal BB. Therapeutic roles of curcumin: lessons learned from clinical trials. Aaps J 2013; 15:195-218; PMID:23143785; http://dx.doi.org/10.1208/s12248-012-9432-8
- Tamvakopoulos C, Dimas K, Sofianos ZD, Hatziantoniou S, Han Z, Liu ZL, Wyche JH, Pantazis P. Metabolism and anticancer activity of the curcumin analogue, dimethoxycurcumin. Clin Cancer Res 2007; 13:1269-77; PMID:17317839; http://dx.doi.org/10.1158/1078-0432.CCR-06-1839
- Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, Li PK, Lin J, Fuchs JR, Marcucci G, et al. Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett 2009; 19:706-9; PMID:19112019; http://dx.doi.org/10.1016/j.bmcl.2008.12.041
- Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, Saw CL, Cheung KL, Kong AN. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. Aaps J 2011; 13:606-14; PMID:21938566; http://dx.doi.org/10.1208/s12248-011-9300-y
- Hassan HE, Carlson S, Abdallah I, Buttolph T, Glass KC, Fandy TE. Curcumin and dimethoxycurcumin induced epigenetic changes in leukemia cells. Pharm Res 2015; 32:863-75; PMID:25186441; http://dx.doi.org/10.1007/s11095-014-1502-4
- Parashar G, Capalash N. Promoter methylation-independent reactivation of PAX1 by curcumin and resveratrol is mediated by UHRF1. Clin Exp Med 2015; 16(3):471-478; PMID:26081871; http://dx.doi.org/10.1007/s10238-015-0366-1
- Jiemjit A, Fandy TE, Carraway H, Bailey KA, Baylin S, Herman JG, Gore SD. p21(WAF1/CIP1) induction by 5-azacytosine nucleosides requires DNA damage. Oncogene 2008; 27:3615-23; PMID:18223691; http://dx.doi.org/10.1038/sj.onc.1211018
- Brinkman AB, Gu H, Bartels SJ, Zhang Y, Matarese F, Simmer F, Marks H, Bock C, Gnirke A, Meissner A, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 2012; 22:1128-38; PMID:22466170; http://dx.doi.org/10.1101/gr.133728.111
- Statham AL, Robinson MD, Song JZ, Coolen MW, Stirzaker C, Clark SJ. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res 2012; 22:1120-7; PMID:22466171; http://dx.doi.org/10.1101/gr.132076.111
- Qin T, Youssef EM, Jelinek J, Chen R, Yang AS, Garcia-Manero G, Issa JP. Effect of cytarabine and decitabine in combination in human leukemic cell lines. Clin Cancer Res 2007; 13:4225-32; PMID:17634552; http://dx.doi.org/10.1158/1078-0432.CCR-06-2762
- Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res 2001; 61:1327-33; PMID:11245429
- Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm 2013; 10:337-52; PMID:23215027; http://dx.doi.org/10.1021/mp3004622
- Tsimberidou AM, Said R, Culotta K, Wistuba I, Jelinek J, Fu S, Falchook G, Naing A, Piha-Paul S, Zinner R, et al. Phase I study of azacitidine and oxaliplatin in patients with advanced cancers that have relapsed or are refractory to any platinum therapy. Clin Epigenetics 2015; 7:29; PMID:25806091; http://dx.doi.org/10.1186/s13148-015-0065-5
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440-6; PMID:20068163; http://dx.doi.org/10.1158/0008-5472.CAN-09-1947
- Yu J, Peng Y, Wu LC, Xie Z, Deng Y, Hughes T, He S, Mo X, Chiu M, Wang QE, et al. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS One 2013; 8:e55934; PMID:23457487; http://dx.doi.org/10.1371/journal.pone.0055934
- Chen CQ, Yu K, Yan QX, Xing CY, Chen Y, Yan Z, Shi YF, Zhao KW, Gao SM. Pure curcumin increases the expression of SOCS1 and SOCS3 in myeloproliferative neoplasms through suppressing class I histone deacetylases. Carcinogenesis 2013; 34:1442-9; PMID:23430957; http://dx.doi.org/10.1093/carcin/bgt070
- Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem 2004; 279:51163-71; PMID:15383533; http://dx.doi.org/10.1074/jbc.M409024200
- Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep 2015; 16:1467-81; PMID:26474904; http://dx.doi.org/10.15252/embr.201540945
- Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr 2011; 6:93-108; PMID:21516481; http://dx.doi.org/10.1007/s12263-011-0222-1
- Pesakhov S, Nachliely M, Barvish Z, Aqaqe N, Schwartzman B, Voronov E, Sharoni Y, Studzinski GP, Fishman D, Danilenko M. Cancer-selective cytotoxic Ca2+ overload in acute myeloid leukemia cells and attenuation of disease progression in mice by synergistically acting polyphenols curcumin and carnosic acid. Oncotarget 2016; 7(22):31847-31861; PMID:26870993; http://dx.doi.org/10.18632/oncotarget.7240
- Weng Q, Ren L, Guo L, Hu Y, Zhou H, Bao Y, Chen L, Liang G, Wang Y, Ruan J. Curcumin analogue, A13, exhibits anti-leukemia effect via inhibiting STAT3. Tumour Biol 2016; 37(7):9959-66; PMID:26815506; http://dx.doi.org/10.1007/s13277-016-4861-0
- Taverna S, Giallombardo M, Pucci M, Flugy A, Manno M, Raccosta S, Rolfo C, De Leo G, Alessandro R. Curcumin inhibits in vitro and in vivo chronic myelogenous leukemia cells growth: a possible role for exosomal disposal of miR-21. Oncotarget 2015; 6:21918-33; PMID:26116834; http://dx.doi.org/10.18632/oncotarget.4204
- Wu L, Yu J, Chen R, Liu Y, Lou L, Wu Y, Huang L, Fan Y, Gao P, Huang M, et al. Dual inhibition of Bcr-Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib-resistant CML cells. Clin Cancer Res 2015; 21:833-43; PMID:25501124; http://dx.doi.org/10.1158/1078-0432.CCR-13-3317
- Vendetti FP, Rudin CM. Epigenetic therapy in non-small-cell lung cancer: targeting DNA methyltransferases and histone deacetylases. Expert Opin Biol Ther 2013; 13:1273-85; PMID:23859704; http://dx.doi.org/10.1517/14712598.2013.819337
- Quintas-Cardama A, Santos FP, Garcia-Manero G. Therapy with azanucleosides for myelodysplastic syndromes. Nat Rev Clin Oncol 2010; 7:433-44; PMID:20551943; http://dx.doi.org/10.1038/nrclinonc.2010.87
- Schermelleh L, Spada F, Easwaran HP, Zolghadr K, Margot JB, Cardoso MC, Leonhardt H. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat Methods 2005; 2:751-6; PMID:16179921; http://dx.doi.org/10.1038/nmeth794
- Fu N, Drinnenberg I, Kelso J, Wu JR, Paabo S, Zeng R, Khaitovich P. Comparison of protein and mRNA expression evolution in humans and chimpanzees. PLoS One 2007; 2:e216; PMID:17299596; http://dx.doi.org/10.1371/journal.pone.0000216
- Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol 2003; 4:117; PMID:12952525; http://dx.doi.org/10.1186/gb-2003-4-9-117
- Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, Herman JG, Davidson NE. Transcriptional activation of estrogen receptor α in human breast cancer cells by histone deacetylase inhibition. Cancer Res 2000; 60:6890-4; PMID:11156387
- Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet 2006; 2:e40; PMID:16596166; http://dx.doi.org/10.1371/journal.pgen.0020040
- Raynal NJ, Si J, Taby RF, Gharibyan V, Ahmed S, Jelinek J, Estécio MR, Issa JP. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res 2012; 72:1170-81; PMID:22219169; http://dx.doi.org/10.1158/0008-5472.CAN-11-3248
- Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009; 136:3131-41; PMID:19700617; http://dx.doi.org/10.1242/dev.037127
- Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 2009; 114:2764-73; PMID:19546476; http://dx.doi.org/10.1182/blood-2009-02-203547
- Szyf M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry (Mosc) 2005; 70:533-49; PMID:15948707; http://dx.doi.org/10.1007/s10541-005-0147-7
- Dubovsky JA, McNeel DG. Inducible expression of a prostate cancer-testis antigen, SSX-2, following treatment with a DNA methylation inhibitor. Prostate 2007; 67:1781-90; PMID:17929270; http://dx.doi.org/10.1002/pros.20665
- Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, Treisman J, Rosenberg SA. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2′-deoxycytidine. Cancer Res 1994; 54:1766-71; PMID:7511051
- Kihslinger JE, Godley LA. The use of hypomethylating agents in the treatment of hematologic malignancies. Leuk Lymphoma 2007; 48:1676-95; PMID:17786703; http://dx.doi.org/10.1080/10428190701493910
- Al-Ali HK, Jaekel N, Niederwieser D. The role of hypomethylating agents in the treatment of elderly patients with AML. J Geriatr Oncol 2014; 5:89-105; PMID:24484723; http://dx.doi.org/10.1016/j.jgo.2013.08.004