
ABSTRACT
Stage 4S neuroblastoma (NB) is a special type of NB found in infants with metastases at diagnosis and is associated with an excellent outcome due to its remarkable capacity to undergo spontaneous regression. As genomics have not been able to explain this intriguing clinical presentation, we here aimed at profiling the DNA methylome of stage 4S NB to better understand this phenomenon. To this purpose, differential methylation analyses between International Neuroblastoma Staging System (INSS) stage 4S, stage 4 and stage 1/2 were performed, using methyl-CpG-binding domain (MBD) sequencing data of 14 stage 4S, 14 stage 4, and 13 stage 1/2 primary NB tumors (all MYCN non-amplified in order not to confound results). Stage 4S-specific hyper- and hypomethylated promoters were determined and further characterized for genomic localization and function by cytogenetic band enrichment, gene set enrichment, transcription factor target enrichment and differential RNA expression analyses. We show that specific chromosomal locations are enriched for stage 4S differentially methylated promoters and that stage 4S tumors show characteristic hypermethylation of specific subtelomeric promoters. Furthermore, genes involved in important oncogenic pathways, in neural crest development and differentiation, and in epigenetic processes are differentially methylated and expressed in stage 4S tumors. Based on these findings, we describe new biological mechanisms possibly contributing to the stage 4S-specific tumor biology and spontaneous regression. In conclusion, this study is the first to describe the highly characteristic stage 4S DNA methylome. These findings will open new avenues to further unravel the NB pathology in general and stage 4S disease specifically.
Abbreviations
| DMR | = | differentially methylated region |
| GSEA | = | gene set enrichment analysis |
| INRGSS | = | International Neuroblastoma Risk Group Staging System |
| INSS | = | International Neuroblastoma Staging System |
| MBD | = | methyl-CpG-binding domain |
| NB | = | neuroblastoma |
| NGF | = | nerve growth factor |
Introduction
Neuroblastoma (NB) is a childhood tumor that originates from precursor cells of the sympathetic nervous system and is a heterogeneous disease with prognosis ranging from long-term survival to fatal outcome. NB tumor staging systems have been established, based on clinical and biological parameters (age of the patient, tumor stage, MYCN amplification, DNA index, and histopathology), and are being used for therapeutic stratification. The special stage IV (IV-S) was first described in the staging system of Evans et al., introduced in 1971, and defined patients who would otherwise have been stage I or II (with localized primary tumors), but who had disseminated disease confined to liver, skin or bone marrow, or any combination of these, without involvement of the skeleton. Remarkably, these patients did not have the dismal prognosis of other metastatic NB, as most of these tumors are characterized by spontaneous regression without any therapeutic intervention.Citation1,2 Approximately 20 y later, the International Neuroblastoma Staging System (INSS) refined the criteria of this tumor stage, renamed as stage 4S, and limited it to children younger than 12 months of age at diagnosis.Citation3 In 2005, this age limit was extended to 18 months in the International Neuroblastoma Risk Group Staging System (INRGSS), as it was demonstrated that an age cut-off of 18 months resulted in a stratification that optimized the prognostic contribution of the age at diagnosis.Citation4 In the INRGSS, this special tumor stage is indicated as MS.Citation5
Several genomic alterations have been identified in NB, but so far no specific changes, apart from hyperdiploidy and near triploidy with numerical chromosome alterations common to localized tumors without MYCN amplification, have been associated with stage 4S NB. This indicates that other molecular mechanisms, such as epigenetic alterations, may play a role in stage 4S NB.Citation6 In this study, we therefore specifically focused on identifying patterns of DNA methylation that are characteristic of this particular NB tumor stage.
Results
Stage 4S tumors demonstrate a unique promoter methylation portrait
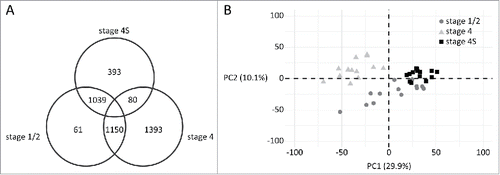
In order to profile the stage 4S NB methylome, we re-analyzed methyl-CpG-binding domain (MBD) sequencing data of 41 MYCN non-amplified primary NBs,Citation7,8 including INSS stage 1 (n = 7), stage 2 (n = 6), stage 4 (n = 14), and stage 4S (n = 14) tumors. As patients with stage 4S NB, in sharp contrast to stage 4 patients, show excellent outcome despite widespread metastases at diagnosis, we first performed a differential methylation analysis between these tumor stages. In total, 5,914 and 10,974 promoters were identified as being hyper- or hypomethylated in stage 4S tumors, respectively (Supplemental Table 1). Next, stage 4S tumors were also compared to stage 1/2 tumors, which are also associated with a favorable patient outcome, but patients with stage 1/2 tumors do not present with metastatic disease. Here, 1,288 and 3,854 promoters were hyper- and hypomethylated in stage 4S tumors, respectively (Supplemental Table 1). Subsequently, we determined the number of stage 4S-specific hyper- and hypomethylated promoters by determining the overlap between these 2 differential methylation analyses and by excluding promoters differentially methylated in the stage 1/2 vs. stage 4 comparison. As such, we identified 393 hypermethylated and 1,150 hypomethylated promoters that typify the stage 4S methylome (; Supplemental Table 1). Similarly, also for stage 4 and stage 1/2 NB, specific promoter methylation patterns were determined (; Supplemental Table 1). Principal component analysis using the normalized read counts of the promoters included in visualizes the NB stage-specific promoter methylation patterns (). A dense clustering is observed for stage 4S tumors and this tumor stage seems most similar to stage 1/2.
Figure 1. MBD sequencing analysis of MYCN non-amplified stage 4S, stage 4, and stage 1/2 tumors identifies stage-specific promoter methylation portraits. A. Diagram of the promoters identified as hypermethylated in the respective sample groups. For example, 393 and 1,150 promoters are specifically hyper- and hypomethylated in stage 4S, respectively. B. Principal component (PC) analysis using the normalized read counts of the promoters included in the diagram. Shown are the data for the first 2 PCs. PC1 explains 29.9% of the variability of the data and PC2 10.1%.
Specific chromosomal locations are enriched in stage 4S differentially methylated promoters
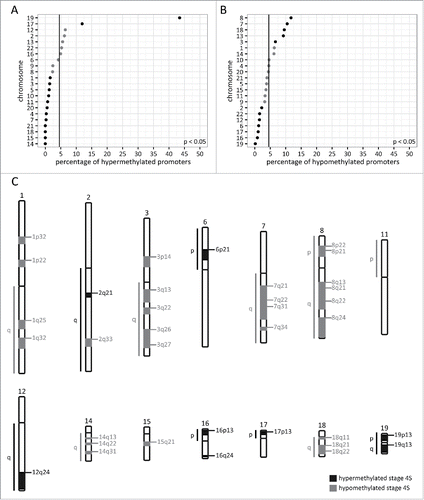
Next, we determined the chromosomal distribution of the promoters contributing to the specific stage 4S methylation portrait. The percentages of hyper- and hypomethylated promoters located on the different chromosomes is indicated in and , respectively. Importantly, as chromosomes differ in length and gene density, these chromosome percentages were calculated based on the number of identified promoters corrected for the total number of promoters located on the corresponding chromosome. Stage 4S hypermethylated promoters are overrepresented (P < 0.05) on chr17 and chr19, and underrepresented (P < 0.05) on chr1, chr3, chr4, chr5, chr7, chr10, chr11, chr14, chr15, chr18, chr20, and chr21 (). Stage 4S hypomethylated promoters are overrepresented (P < 0.05) on chr3, chr7, chr8, chr13, and chr18, and underrepresented (P < 0.05) on chr2, chr6, chr12, chr16, chr17, chr19, and chr22 (). To further pinpoint specific chromosomal regions enriched in stage 4S differentially methylated promoters, cytogenetic band enrichment analyses in WebGestaltCitation9,10 were performed ( and Supplemental Table 2). These analyses indicate that stage 4S hyper- and hypomethylated promoters are not randomly distributed across the genome, but that specific chromosomal cytogenetic bands are significantly enriched in stage 4S differentially methylated promoters ().
Figure 2. Stage 4S differentially methylated promoters are not randomly distributed across the genome. A. Percentage of stage 4S hypermethylated promoters per chromosome, based on promoter density corrected numbers. Significantly under- and overrepresented chromosomes are indicated with a dark dot. The vertical line represents the percentage if the stage 4S hypermethylated promoters would be randomly distributed across the genome (null hypothesis). B. Percentage of stage 4S hypomethylated promoters per chromosome, based on promoter density corrected numbers. Significantly under- and overrepresented chromosomes are indicated with a dark dot. The vertical line represents the percentage if the stage 4S hypomethylated promoters would be randomly distributed across the genome (null hypothesis). C. Significantly enriched cytogenetic locations of stage 4S hyper- and hypomethylated promoters. A list of genes located in the enriched regions can be found in Supplemental Table 2.
Specific subtelomeres are enriched in stage 4S hypermethylated promoters
Telomerase activity upregulation and alternative lengthening of telomeres are key mechanisms in counteracting telomere shortening in NB and contribute to tumor cell immortalization.Citation11-14 As DNA methylation of subtelomeres represents an additional way to suppress telomere elongationCitation15,16 and as tumor regression (and thus loss of immortality) is a hallmark of stage 4S NB, we specifically analyzed the subtelomeric methylation patterns of the different NB tumor stages. To this purpose, we first determined the proportions of promoters of the different stage-specific methylation portraits that are located in subtelomeres (1 Mb or 2 Mb regions proximal to the telomeres). and illustrate that stage 4S hypermethylated and stage 4 hypomethylated promoters have a significantly larger proportion of 1 Mb and 2 Mb subtelomeric promoters (P < 0.001), compared to stage 1/2 and stage 4 hypermethylated and stage 4S hypomethylated promoters. Stage 1/2 hypomethylated promoters also comprise a larger proportion of 2 Mb subtelomeric promoters (P < 0.001; ), compared to stage 1/2 and stage 4 hypermethylated and stage 4S hypomethylated promoters. Next, using the gene lists of the differential methylation analyses, gene set enrichment analyses (GSEA) were performed to evaluate whether specific subtelomeres are enriched in differentially methylated promoters (). These analyses firmly demonstrate that multiple subtelomeres are enriched in stage 4S hypermethylated promoters compared to stage 4 and stage 1/2 tumors. Remarkably, not a single subtelomere is enriched in stage 4 hypermethylated promoters. further shows that the chr5p 1 Mb and 2 Mb subtelomeres are enriched in promoters hypermethylated in stage 4S compared to both stage 1/2 and stage 4, and in stage 1/2 compared to stage 4. TERT, the gene encoding the catalytic subunit of telomerase, is located on the chr5p 2 Mb subtelomere, and its promoter is hypermethylated in stage 4S compared to stage 4 (Supplemental Table 1). Using publically available mRNA expression data,Citation17 we could further demonstrate that TERT is expressed at lower levels in stage 4S compared to stage 4 (P < 0.001; log2 fold change = 0.82).
Figure 3. Stage 4S hypermethylated, and stage 4 and 1/2 hypomethylated promoters are frequently located on subtelomeres. Depicted are the percentages of the stage-specific differentially methylated promoters located in the 1 Mb (A) and 2 Mb (B) subtelomeres. Stage 4S hypermethylated and stage 4 hypomethylated promoters have a significantly larger proportion of 1 Mb and 2 Mb subtelomeric promoters, compared to stage 1/2 and stage 4 hypermethylated and stage 4S hypomethylated promoters. Stage 1/2 hypomethylated promoters also comprise a larger proportion of 2 Mb subtelomeric promoters, compared to stage 1/2 and stage 4 hypermethylated and stage 4S hypomethylated promoters.
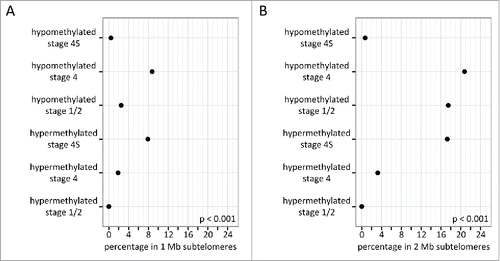
Table 1. One third of subtelomeres are enriched in stage 4S hypermethylated promoters.
Genes involved in important oncogenic pathways, neural crest development and differentiation, and epigenetic processes are differentially methylated and expressed in stage 4S tumors
To functionally characterize the stage 4S differentially methylated promoters, we first examined whether these promoters share common DNA motifs, by analyzing transcription factor target enrichment in WebGestalt.Citation9,10 In total, 24 and 320 motifs were significantly enriched for stage 4S hyper- and hypomethylated promoters, respectively (Supplemental Table 3), of which the top 10 (ranked based on the ratio of enrichment) are shown in . Motifs enriched in stage 4S differentially methylated promoters represent binding sites for transcription factors implicated in controlling cell cycle, apoptosis, neural crest development, and neural differentiation (). Further, we performed GSEA on the gene lists of the differential methylation analyses, using the oncogenic signatures database containing gene sets that represent signatures of cellular pathways dysregulated in cancer.Citation18 Enriched gene sets were only identified for promoters hypomethylated in stage 4S compared to stage 4, in stage 4S compared to stage 1/2, and in stage 1/2 compared to stage 4 (Supplemental Table 4). Of note, 19 gene sets () were specifically enriched in promoters hypomethylated in stage 4S compared to both stage 1/2 and stage 4, including gene sets involved in downstream signaling of important oncogenes and epigenetic actors. Transcription factor target gene sets () and the leading edge genes of the oncogenic signatures gene sets () significantly enriched in stage 4S differentially methylated promoters were further tested for differential expression, using the global testCitation19 and publically available mRNA expression data.Citation17 In total, 25 of the 39 (64.10%) gene sets were identified as differentially expressed in stage 4S compared to both stage 4 and stage 1/2 ( and ). Finally, using covariates plotsCitation19 and differential gene expression analyses on the individual gene level, we evaluated which genes contributed the most to the significant global test results (Supplemental Table 5 and ).
Table 2. Specific transcription factor targets are enriched in stage 4S differentially methylated promoters.
Table 3. Oncogenic signatures are enriched in stage 4S hypomethylated promoters.
Table 4. Multiple stage 4S differentially methylated genes are also differentially expressed.
Discussion
Genome-wide characterization studies in NB often combine patients with stage 4S or other low-risk tumors into one group for comparison to high-risk patients, as low-risk patients generally have an excellent outcome, which is in sharp contrast to high-risk patients who often show fatal progression. Patients with stage 4S tumors demonstrate a typical pattern of metastatic spread at diagnosis and have the capacity to undergo spontaneous regression, and these characteristics are not exhibited by stage 1/2 tumors. As genomics alone have not been able to explain this intriguing clinical presentation,Citation6 we aimed to characterize the DNA methylation pattern of stage 4S NB, in order to reveal new biological insights into this special type of NB.
By performing differential methylation analyses between stage 4S and stage 1/2 and stage 4, we could clearly define a unique stage 4S promoter methylation portrait. We further showed that stage 4S-specific hyper- and hypomethylated promoters are located on specific chromosomal locations. Non-random chromosomal distribution of regions differentially methylated between tumor and normal samples,Citation20,21 and between different tumor stages,Citation21,22 has been previously reported for other cancer types. In this study, we show that specific chromosomal cytogenetic bands are enriched in stage 4S differentially methylated promoters and, most importantly, that stage 4S tumors show characteristic hypermethylation of specific subtelomeric promoters. Telomere length and its regulatory mechanisms are well-studied in NB.Citation11-14,23-25 Most stage 4S tumors have low telomerase activity or short telomeres,Citation11,12,23 and it has been suggested that the association between stage 4S and spontaneous regression might be related to differences in telomere length regulation.Citation6 While subtelomeric DNA methylation has been shown to suppress telomere elongation in other cancer types,Citation15,16 such telomere length regulatory mechanism is unexplored in NB. In this study, we have shown that specific subtelomeric promoters are hypermethylated in stage 4S tumors compared to stage 1/2 and stage 4 tumors, which suggests that subtelomeric DNA methylation may represent an additional mechanism by which telomere length and spontaneous regression are controlled in NB. Therefore, the role of DNA methylation in telomere length regulation in stage 4S NB should definitely be further explored.
Further, we have shown that multiple transcription factor target gene sets are enriched in stage 4S differentially methylated promoters. Methylation of cytosines within transcription factor recognition motifs has been shown to cause structural changes influencing DNA-protein interactions (either in a positive or negative manner) and transcription factor activity.Citation26 As such, DNA methylation serves as a regulatory mechanism for transcription factor activity. Therefore, the transcription factor target gene sets enriched in stage 4S differentially methylated promoters may indicate differential activity of the respective transcription factors in stage 4S tumors compared to stage 1/2 and stage 4 tumors. Of note, this assumption is reinforced by the fact that several of the identified transcription factors of which multiple target genes are differentially methylated in stage 4S show proven dependency on the DNA methylation status of the binding site for transcription factor-DNA interaction. For example, DNA methylation inhibits the binding of E2F1 and CEBPG.Citation26-28 Additionally, the differentially methylated target gene sets are differentially expressed in stage 4S NB, further strengthening the possibility that DNA methylation affects the activity of these transcription factors. Of note, E2F1 also binds to the TERT promoter,Citation29 which is hypermethylated in stage 4S compared to stage 4, and TERT is expressed at lower levels in stage 4S compared to stage 4. This indicates that TERT DNA methylation might also regulate telomerase activity in stage 4S NB.
Furthermore, our transcription factor target enrichment analyses particularly demonstrate that the DNA methylation portrait of stage 4S NB is dominated by differential methylation of target genes of transcription factors involved in neural crest development and (sympathetic) neural differentiation (MSX1,Citation30,31 EVI1,Citation32,33 EGR3,Citation34 AHR,Citation35,36 MEF2A,Citation37,38 YY1,Citation39 PPARA,Citation40 POU2F1,Citation41 and GFI1Citation42,43). These findings suggest that differences in the pathogenic mechanisms between stage 4S and stage 1/2 and stage 4 tumors are at least in part related to differences in transcriptional activity during neural crest development and differentiation, which might indicate that stage 4S, stage 1/2 and stage 4 tumors arise during different phases of neural crest cell development. This is in line with the previous postulation that stage 4S NB originate from defective premigratory neural crest cells, which explains its clinical symptomology, as stage 4S tumor sites mirror developmental neural crest cell migration.Citation44
One of the genes contributing the most to the significant differential expression of the target genes of E2F1 and MEF2A is SLC9A5. The SLC9A5 promoter is hypermethylated in stage 4S and these tumors show low SLC9A5 expression levels compared to stage 4 and stage 1/2 tumors. SLC9A5 is a member of the Na(+)/H(+) exchanger family and is abundantly expressed in neurons. In rat pheochromocytoma, a neuroendocrine tumor of the adrenal medulla, Diering et al. have shown that SLC9A5 modulates trafficking of TrkA between recycling endosomes and the plasma membrane, neurotrophin signaling, and neural differentiation.Citation45 The authors demonstrate that upon knockdown of SLC9A5 in these tumor cells, there is no difference in the total abundance of TrkA compared to control cells, suggesting that reduced expression of SLC9A5 has little effect on TrkA degradation or synthesis. However, although the overall TrkA protein levels did not change, the cell surface abundance of TrkA was reduced upon knockdown of SLC9A5.Citation45 Stage 4S NB tumors generally show high levels of TrkA and, depending on the presence or absence of nerve growth factor (NGF) in the tumor microenvironment, the tumors undergo neural differentiation or spontaneous regression, respectively. Therefore, neurotrophin deprivation (TrkA without NGF) and activation of apoptosis have been proposed to explain the phenomenon of spontaneous regression in NB.Citation6 Given the crucial role of SLC9A5 in regulating the cellular distribution of TrkA and NGF signaling,Citation45 our findings suggest that, over time, stage 4S tumor cell surfaces might get depleted of TrkA (with an accompanying intracellular accumulation of TrkA) due to the low expression levels of SLC9A5, ultimately leading to apoptosis and regression.
Our gene set enrichment analyses also highlight differential methylation of genes involved in important oncogenic pathways, especially of genes implicated in EGFR and RAS signaling. Although the exact mechanisms by which DNA methylation contributes to differential regulation of these pathways in stage 4S specifically cannot be determined in our study, our findings indicate that it is crucial to separate stage 4S tumors from other low-stage (stage 1/2) tumors in studies focusing on NB biology, as this might reveal new insights into the stage 4S NB pathogenesis. The importance hereof is highlighted by the fact that all but one differentially methylated oncogenic signatures are also differentially expressed in stage 4S NB, confirming differential regulation of these oncogenic pathways in stage 4S on the one hand and stage 4 and stage 1/2 on the other hand. Finally, the enrichment analyses also illustrate that stage 4S tumors present differential activity of important epigenetic players, such as BMI1, PCGF2, EED, and EZH2. Potentially, differential regulation of epigenetic mechanisms might also contribute to the regression phenotype of stage 4S NB, as epigenetic alterations can more easily be reversed. Clearly, these epigenetic mechanisms need to be further studied in NB.
In conclusion, this study is the first to describe the stage 4S DNA methylome and demonstrates that stage 4S NB tumors show characteristic DNA methylation patterns compared to stage 1/2 and stage 4 tumors. Genomic localization and functional characterization analyses of this stage 4S DNA methylation portrait suggested new mechanisms that may contribute to the stage 4S-specific tumor biology and spontaneous regression. Future studies must confirm these observations and hypotheses.
Materials and methods
Neuroblastoma primary tumors
Sample collection for MBD sequencing (n = 41) and expression profiling (n = 180) is described in Decock et al.Citation7 and Oberthuer et al, Citation17 respectively. All tumor samples were assigned to one of the 3 following subgroups, based on the INSS tumor stageCitation3 and MYCN amplification status: (1) MYCN non-amplified stage 4S (MBD sequencing: n = 14; expression profiling: n = 28), (2) MYCN non-amplified stage 4 (MBD sequencing: n = 14; expression profiling: n = 46), or (3) MYCN non-amplified stage 1 or 2 (MBD sequencing: n = 13; expression profiling: n = 106). Detailed criteria for patient inclusion in the study are described in Decock et al.Citation8,46 MYCN amplified tumor samples were excluded. Detailed clinicobiological characteristics of the different sample cohorts are shown in Supplemental Table 6. The study was approved by the ethical committee of the Ghent University Hospital (approval number: B67020109912).
DNA methylome and transcriptome profiling
A detailed description of the generation and raw data analysis of the MBD sequencing data is provided in Decock et al.Citation7 Data can be accessed through the Gene Expression Omnibus numbers GSE69243 and GSE69268. For the mRNA expression analyses, microarray data of 180 NB tumors were used.Citation17
Bioinformatics and statistical analyses
Differential methylation analyses on the MBD sequencing data were performed as described in Decock et al.,Citation8 using DESeqCitation47 and promoter region count matrices with data on all Ensembl transcripts (release 68). Promoters located on X and Y chromosomes were excluded to avoid gender-related confounding. To estimate the background signal and to be able to exclude possible falsely identified differentially methylated regions (DMRs) in the DESeq analyses, a filtering procedure based on 2 correction steps was applied on the resulting DMR lists. A first correction step consisted of excluding DMRs, which were also significantly identified in the same comparison on the input (non-MBD-enriched) sample data (input correction).Citation8 Secondly, computational simulation analyses were performed, in which 100 random patient annotation swaps were executed to create 2 artificial sample groups on which the same statistics were applied as on the biologically relevant groups. Only biologically relevant DMRs with a π-value (= −ln pval * log2 fold change)Citation48 that equals or outperforms the π-value of the corresponding simulation DMR were considered for further evaluation (swap correction). DESeq normalized read counts of the stage-specific DNA methylation portraits were used for principal component analysis in R (package factoextra).
Cytogenetic band enrichment analysis and transcription factor target enrichment analysis were performed using WebGestaltCitation9,10 with gene symbols as input list. The entire human genome was used as reference list. Benjamini-Hochberg multiple testing correction was appliedCitation49 and only statistically enriched terms (q < 0.05) with at least 2 genes were considered.
Non-corrected transcript lists, including all transcripts included in the differential methylation analyses (preranked on the π-value) were used for GSEA.Citation18,50 To this purpose, Ensembl transcript IDs were converted to gene symbols. For genes with multiple transcripts, the data of the Ensembl transcript with the highest absolute π-value was retained. Tested gene set databases are the oncogenic signatures (version 5.1) of the Molecular Signatures DatabaseCitation18 and 2 in-house created subtelomere gene set lists. These two subtelomere gene set lists contain all genes in the 1 Mb or 2 Mb subtelomeres, which are defined as the 1 Mb or 2 Mb genomic regionsCitation24 proximal to the telomeres (10 kb chromosomal ends; hg19). GSEA was also performed on the input data to exclude falsely identified enriched gene sets.
The sets of differentially methylated transcription factor targets and the leading edge genes of the enriched oncogenic signature gene sets were further tested for differential expression using the globaltest R packageCitation19 and publically available mRNA expression data.Citation17 For significantly differentially expressed gene sets, covariates plotsCitation19 were generated to inspect the contributions of each gene to the test result. Differential expression analyses on the individual gene level were performed using the R package limma.Citation51 Benjamini-Hochberg multiple testing correction was applied.Citation49
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1226739_s07.zip
Download Zip (24 MB)Funding
This study is supported by the Fournier-Majoie Foundation (FFM), the Belgian National Lottery, the Flemish League Against Cancer (VLK), UGent GOA (grant number 01G01910) and the ISCIII Spain & FEDER (grant number PI14/01008). AD was a recipient of a grant of the Research Foundation Flanders (FWO) and an Emmanuel Van der Schueren research grant (VLK).
Related Research Data
References
- Evans AE, D'Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. Children's cancer study group A. Cancer 1971; 27:374-8; PMID:5100400; http://dx.doi.org/10.1002/1097-0142(197102)27:2%3c374::AID-CNCR2820270221%3e3.0.CO;2-G
- Evans AE, Albo V, D'Angio GJ, Finklestein JZ, Leiken S, Santulli T, Weiner J, Hammond GD. Factors influencing survival of children with nonmetastatic neuroblastoma. Cancer 1976; 38:661-6; PMID:788890; http://dx.doi.org/10.1002/1097-0142(197608)38:2%3c661::AID-CNCR2820380206%3e3.0.CO;2-M
- Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 1993; 11:1466-77; PMID:8336186; http://dx.doi.org/0732-183X/93/1108-0006$3.00/0
- London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol 2005; 23:6459-65; PMID:16116153; http://dx.doi.org/10.1200/JCO.2005.05.571
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 2009; 27:298-303; PMID:19047290; http://dx.doi.org/10.1200/JCO.2008.16.6876
- Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol 2014; 11:704-13; PMID:25331179; http://dx.doi.org/10.1038/nrclinonc.2014.168
- Decock A, Ongenaert M, Van Criekinge W, Speleman F, Vandesompele J. DNA methylation profiling of primary neuroblastoma tumors using methyl-CpG-binding domain sequencing. Sci Data 2016; 3:160004; PMID:26836295; http://dx.doi.org/10.1038/sdata.2016.4
- Decock A, Ongenaert M, Cannoodt R, Verniers K, De Wilde B, Laureys G, Van Roy N, Berbegall AP, Bienertova-Vasku J, Bown N, et al. Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget 2016; 7:1960-72; PMID:26646589; http://dx.doi.org/10.18632/oncotarget.6477
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005; 33:W741-8; PMID:15980575; http://dx.doi.org/10.1093/nar/gki475
- Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013; 41:W77-83; PMID:23703215; http://dx.doi.org/10.1093/nar/gkt439
- Hiyama E, Hiyama K, Yokoyama T, Matsuura Y, Piatyszek MA, Shay JW. Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1995; 1:249-55; PMID:7585042; http://dx.doi.org/10.1038/nm0395-249
- Onitake Y, Hiyama E, Kamei N, Yamaoka H, Sueda T, Hiyama K. Telomere biology in neuroblastoma: telomere binding proteins and alternative strengthening of telomeres. J Pediatr Surg 2009; 44:2258-66; PMID:20006006; http://dx.doi.org/10.1016/j.jpedsurg.2009.07.046
- Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE, Tytgat GAM, Molenaar JJ, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 2015; 47:1411-4; PMID:26523776; http://dx.doi.org/10.1038/ng.3438
- Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Krämer A, Roncaioli JL, Sand F, Heuckmann JM, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015; 526:700-4; PMID:26466568; http://dx.doi.org/10.1038/nature14980
- Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet 2007; 8:299-309; PMID:17363977; http://dx.doi.org/10.1038/nrg2047
- Vera E, Canela A, Fraga MF, Esteller M, Blasco MA. Epigenetic regulation of telomeres in human cancer. Oncogene 2008; 27:6817-33; PMID:18762811; http://dx.doi.org/10.1038/onc.2008.289
- Oberthuer A, Berthold F, Warnat P, Hero B, Kahlert Y, Spitz R, Ernestus K, König R, Haas S, Eils R, et al. Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J Clin Oncol 2006; 24:5070-8; PMID:17075126; http://dx.doi.org/10.1200/JCO.2006.06.1879
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/10.1073/pnas.0506580102
- Goeman JJ, Van de Geer SA, De Kort F, van Houwellingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics 2004; 20:93-9; PMID:14693814; http://dx.doi.org/10.1093/bioinformatics/btg382
- Gómez S, Castellano G, Mayol G, Suñol M, Queiros A, Bibikova M, Nazor KL, Loring JF, Lemos I, Rodríguez E, et al. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics 2015; 7:1137-53; PMID:26067621; http://dx.doi.org/10.2217/epi.15.49
- Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, Fernandez H, Tallman MS, Greally JM, Carraway H, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009; 114:3448-58; PMID:19652201; http://dx.doi.org/10.1182/blood-2009-01-200519
- Heuck CJ, Mehta J, Bhagat T, Gundabolu K, Yu Y, Khan S, Chrysofakis G, Schinke C, Tariman J, Vickrey E, et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J Immunol 2013; 190:2966-75; PMID:23408834; http://dx.doi.org/10.4049/jimmunol.1202493
- Krams M, Hero B, Berthold F, Parwaresch R, Harms D, Rudolph P. Full-length telomerase reverse transcriptase messenger RNA is an independent prognostic factor in neuroblastoma. Am J Pathol 2003; 162:1019-26; PMID:12598334; http://dx.doi.org/10.1016/S0002-9440(10)63896-5
- Buckley PG, Das S, Bryan K, Watters KM, Alcock L, Koster J, Versteeg R, Stallings RL. Genome-wide DNA methylation analysis of neuroblastic tumors reveals clinically relevant epigenetic events and large-scale epigenomic alterations localized to telomeric regions. Int J Cancer 2011; 128:2296-305; PMID:20669225; http://dx.doi.org/10.1002/ijc.25584
- Hertwig F, Peifer M, Fischer M. Telomere maintenance is pivotal for high-risk neuroblastoma. Cell Cycle 2016; 15:311-2; PMID:26653081; http://dx.doi.org/10.1080/15384101.2015.1125243
- Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem 2013; 288:34287-94; PMID:24151070; http://dx.doi.org/10.1074/jbc.R113.512517
- Campanero MR, Armstrong MI, Flemington EK. CpG methylation as a mechanism for the regulation of E2F activity. Proc Natl Acad Sci USA 2000; 97:6481-6; PMID:10823896; http://dx.doi.org/10.1073/pnas.100340697
- Mann IK, Chatterjee R, Zhao J, He X, Weirauch MT, Hughes TR, Vinson C. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res 2013; 23:988-97; PMID:23590861; http://dx.doi.org/10.1101/gr.146654.112
- Alonso MM, Fueyo J, Shay JW, Aldape KD, Jiang H, Lee OH, Johnson DG, Xu J, Kondo Y, Kanzawa T, et al. Expression of transcription factor E2F1 and telomerase in glioblastomas: mechanistic linkage and prognostic significance. J Natl Cancer Inst 2005; 97:1589-600; PMID:16264179; http://dx.doi.org/10.1093/jnci/dji340
- Tríbulo C, Aybar MJ, Sánchez SS, Mayor R. A balance between the anti-apoptotic activity of Slug and the apoptotic activity of msx1 is required for the proper development of the neural crest. Dev Biol 2004; 275:325-42; PMID:15501222; http://dx.doi.org/10.1016/j.ydbio.2004.07.041
- Monsoro-Burq AH, Wang E, Harland R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev Cell 2005; 8:167-78; PMID:15691759; http://dx.doi.org/10.1016/j.devcel.2004.12.017
- Hoyt PR, Bartholomew C, Davis AJ, Yutzey K, Gamer LW, Potter SS, Ihle JN, Mucenski ML. The Evi1 proto-oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech Dev 1997; 65:55-70; PMID:9256345; http://dx.doi.org/10.1016/S0925-4773(97)00057-9
- Kazama H, Kodera T, Shimizu S, Mizoguchi H, Morishita K. Ecotropic viral integration site-1 is activated during, and is sufficient for, neuroectodermal P19 cell differentiation. Cell Growth Differ 1999; 10:565-73; PMID:10470856; available at http://cgd.aacrjournals.org/cgi/content/full/10/8/565
- Quach DH, Oliveira-Fernandes M, Gruner KA, Tourtellotte WG. A sympathetic neuron autonomous role for Egr3-Mediated gene regulation in dendrite morphogenesis and target tissue innervation. J Neurosci 2013; 33:4570-83; PMID:23467373; http://dx.doi.org/10.1523/JNEUROSCI.5481-12.2013
- Akahoshi E, Yoshimura S, Ishihara-Sugano M. Over-expression of AhR (aryl hydrocarbon receptor) induces neural differentiation of Neuro2a cells: neurotoxicology study. Environ Health 2006; 5:24; PMID:16956419; http://dx.doi.org/10.1186/1476-069X-5-24
- Wu PY, Liao YF, Juan HF, Huang HC, Wang BJ, Lu YL, Yu IS, Shih YY, Jeng YM, Hsu WM, et al. Aryl hydrocarbon receptor downregulates MYCN expression and promotes cell differentiation of neuroblastoma. PLoS One 2014; 9:e88795; PMID:24586395; http://dx.doi.org/10.1371/journal.pone.0088795
- Ruffle RA, Mapley AC, Malik MK, Labruzzo SV, Chabla JM, Jose R, Hallas BH, Yu HG, Horowitz JM, Torres G. Distribution of constitutively expressed MEF2A in adult rat and human nervous systems. Synapse 2006; 59:513-20; PMID:16565967; http://dx.doi.org/10.1002/syn.20266
- Mao Z, Nadal-Ginard B. Functional and physical interactions between mammalian achaete-scute homolog 1 and myocyte enhancer factor 2A. J Biol Chem 1996; 271:14371-5; PMID:8662987; http://dx.doi.org/10.1074/jbc.271.24.14371
- He Y, Casaccia-Bonnefil P. The Yin and Yang of YY1 in the nervous system. J Neurochem 2008; 106:1493-502; PMID:18485096; http://dx.doi.org/10.1111/j.1471-4159.2008.05486.x
- Bento-Abreu A, Tabernero A, Medina JM. Peroxisome proliferator-activated receptor-α is required for the neurotrophic effect of oleic acid in neurons. J Neurochem 2007; 103:871-81; PMID:17683485; http://dx.doi.org/10.1111/j.1471-4159.2007.04807.x
- Dick T, Yang XH, Yeo SL, Chia W. Two closely linked Drosophila POU domain genes are expressed in neuroblasts and sensory elements. Proc Natl Acad Sci USA 1991; 88:7645-9; PMID:1881906; http://dx.doi.org/10.1073/pnas.88.17.7645
- Ossovskaya VS, Dolganov G, Basbaum AI. Loss of function genetic screens reveal MTGR1 as an intracellular repressor of beta1 integrin-dependent neurite outgrowth. J Neurosci Methods 2009; 177:322-33; PMID:19026687; http://dx.doi.org/10.1016/j.jneumeth.2008.10.031
- Cameron S, Clark SG, McDermott JB, Aamodt E, Horvitz HR. PAG-3, a Zn-finger transcription factor, determines neuroblast fate in C. elegans. Development 2002; 129:1763-74; PMID:11923211; available at http://dev.biologists.org/content/129/7/1763
- van Noesel MM. Neuroblastoma stage 4S: a multifocal stem-cell disease of the developing neural crest. Lancet Oncol 2012; 13:229-30; PMID:22381931; http://dx.doi.org/10.1016/S1470-2045(12)70012-8
- Diering GH, Numata Y, Fan S, Church J, Numata M. Endosomal acidification by Na+/H+ exchanger NHE5 regulates TrkA cell-surface targeting and NGF-induced PI3K signaling. Mol Biol Cell 2013; 24:3435-48; PMID:24006492; http://dx.doi.org/10.1091/mbc.E12-06-0445
- Decock A, Ongenaert M, Hoebeeck J, De Preter K, Van Peer G, Van Criekinge W, Ladenstein R, Schulte JH, Noguera R, Stallings RL, et al. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol 2012; 13:R95; PMID:23034519; http://dx.doi.org/10.1186/gb-2012-13-10-r95
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; PMID:20979621; http://dx.doi.org/10.1186/gb-2010-11-10-r106
- Xiao Y, Hsiao TH, Suresh U, Chen HI, Wu X, Wolf SE, Chen Y. A novel significance score for gene selection and ranking. Bioinformatics 2014; 30:801-7; PMID:22321699; http://dx.doi.org/10.1093/bioinformatics/btr671
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 1995; 57:289-300; available at http://www.jstor.org/stable/2346101
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34:267-73; PMID:12808457; http://dx.doi.org/10.1038/ng1180
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47; PMID:25605792; http://dx.doi.org/10.1093/nar/gkv007