
ABSTRACT
Cholangiocarcinoma (CC) is a rare malignancy of the extrahepatic or intrahepatic biliary tract with an outstanding poor prognosis. Non-surgical therapeutic regimens result in minimally improved survival of CC patients. Global genomic analyses identified a few recurrently mutated genes, some of them in genes involved in epigenetic patterning. In a previous study, we demonstrated global DNA methylation changes in CC, indicating major contribution of epigenetic alterations to cholangiocarcinogenesis. Here, we aimed at the identification and characterization of CC-related, differentially methylated regions (DMRs) in potential microRNA promoters and of genes targeted by identified microRNAs. Twenty-seven hypermethylated and 13 hypomethylated potential promoter regions of microRNAs, known to be associated with cancer-related pathways like Wnt, ErbB, and PI3K-Akt signaling, were identified. Selected DMRs were confirmed in 2 independent patient cohorts. Inverse correlation between promoter methylation and expression suggested miR-129-2 and members of the miR-200 family (miR-200a, miR-200b, and miR-429) as novel tumor suppressors and oncomiRs, respectively, in CC. Tumor suppressor genes deleted in liver cancer 1 (DLC1), F-box/WD-repeat-containing protein 7 (FBXW7), and cadherin-6 (CDH6) were identified as presumed targets in CC. Tissue microarrays of a representative and well-characterized cohort of biliary tract cancers (n=212) displayed stepwise downregulation of CDH6 and association with poor patient outcome. Ectopic expression of CDH6 on the other hand, delayed growth in the CC cell lines EGI-1 and TFK-1, together suggesting a tumor suppressive function of CDH6. Our work represents a valuable repository for the study of epigenetically altered miRNAs in cholangiocarcinogenesis and novel putative, CC-related tumor suppressive miRNAs and oncomiRs.
Introduction
Cholangiocarcinomas (CCs) are heterogeneous hepatobiliary cancers with features of cholangiocyte differentiation.Citation1 From a clinical point of view, CC is an orphan cancer with poor patient outcome. Presently, therapeutic options for CC are limited, and surgical resection remains the only option with curative intent. Current clinical trials often employ mixed cohorts of biliary tract cancers (BTCs) including intrahepatic CC (ICC), extrahepatic CC (ECC), and gallbladder carcinoma (GBC) and, so far, promising chemotherapeutic treatment strategies have not been identified.Citation2 Recurrent mutations in CC have been found in TP53, KRAS, and SMAD4.Citation3 Profiling for additional mutations using exome sequencing identified mutations in genes associated with functions in epigenetic programming (e.g., BAP1, MLL3, ARID1A, IDH1, and IDH2) suggesting alterations in epigenetic patterns.Citation4,5
Aberrant gene promoter methylation is an early and driving event in carcinogenesis, often manifested in an inverse correlation between the levels of promoter methylation and gene expression.Citation6 In a recent global DNA methylation screen in ECC and ICC patients, we identified many aberrantly methylated candidate genes related to cancer-relevant signaling pathways, thereby demonstrating the substantial contribution of DNA methylation changes in the pathogenesis of CC.Citation7
Aberrant expression of oncogenic or tumor suppressive microRNAs (miRNAs) can also promote carcinogenesis and be attributed to epigenetic dysregulation, as exemplified by cancer-associated promoter hypermethylation of the tumor suppressive miRNAs miR-124aCitation8 and miR-34b/c.Citation9,10 When we screened for aberrant methylation of known or suspected miRNA promoters in patients with chronic lymphocytic leukemia, we also found correlations between promoter methylation alterations and miRNA dysregulation.Citation11 Dysregulation of miRNAs has been also described in CC,Citation12 but a systematic study of the relation between DNA methylation changes and miRNA expression in CC has not been presented so far.
Here, we correlated our data sets of known or suspected miRNA promoters and global DNA methylation to identify CC-related differentially methylated miRNA promoters. Our results represent a valuable repository enabling to study the role of epigenetically altered miRNAs in the onset and progression of CC. Novel putative oncomiRs and tumor suppressive miRNAs as well as their target genes were identified. For one of them, CDH6, encoding cadherin-6, we found a stepwise downregulation at the protein level in cholangiocarcinogenesis and association with patient survival, suggesting that CDH6 is a putative tumor suppressor in CC.
Results
Aberrantly methylated miRNA promoters in cholangiocarcinoma
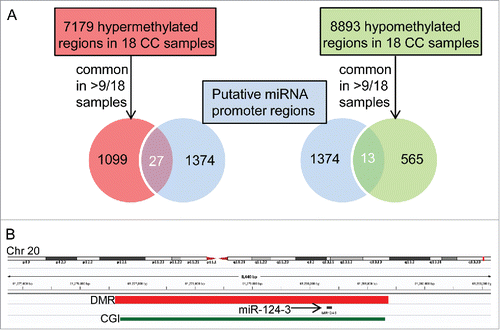
To identify aberrantly methylated miRNA promoters in CC, we searched for overlaps between previously published CC-related differentially methylated regions (DMRs)Citation7 and known or suspected miRNA promoters (for simplicity, henceforth designated miRNA promoters).Citation11 We considered 1099 hypermethylated and 565 hypomethylated regions, common to more than 50%, i.e., more than 9 of 18 CC cases examined in a previous study, and found overlaps with 27 and 13 miRNA promoters, respectively (; Supplementary Table S2 and S3). All overlapping DMRs were represented in both ECC and ICC. Screening miRCancer,Citation13 a literature-based database covering 454 cancer-related miRNAs (version December 2014), we found that 20 miRNAs or miRNA families with CC-related promoter DMR had been already described as dysregulated in cancer (Supplementary Table S4); yet, only one of them, miR-200b, in CC.Citation14
Figure 1. Hypermethylated and hypomethylated miRNA promoters in cholangiocarcinoma identified by overlapping genomic DMR and putative miRNA promoter coordinates. A) From 1374 putative miRNA promoter regions,Citation11 27 and 13 overlapped with 1099 hypermethylated and 565 hypomethylated regions, respectively, common to >9/18 CC samples.Citation7 B) Integrated Genomics Viewer (IGV 2.3)Citation41 browser view on an example of a hypermethylated DMR common to 17 of 18 CC patientsCitation7 and overlapping with the promoter of miR-124-3 (see Supplementary Table S2). CGI: CpG island.
To substantiate our previous array–based methylation results, we validated several of the miRNA-associated DMRs in CC cohort #1 (see CC cohort #1 in ) using MassARRAY. From the miRNAs with suspected hypermethylated promoters, we selected miR-129-2, miR-9-3, miR-124-2, and miR-9-2, since they have been described as dysregulated in various cancer types (see Supplementary Table S4). The respective DMRs were shared by 12 CC (6 ECC and 6 ICC), 14 CC (8 ECC and 6 ICC), 16 CC (each 8 ECC and ICC), and 14 CC (6 ECC and 8 ICC) samples (Supplementary Fig. S1A-D). MassARRAY analyses revealed significantly higher average methylation levels of the CC samples compared to normal tissue in all 4 selected regions (Supplementary Fig. S2A-D and Supplementary Fig. S3A-D) and confirmed our CpG island array data.Citation7 Among the miRNAs with hypomethylated promoters, we selected the frequently dysregulated cluster comprising miR-200a, miR-200b, and miR-429 for validation. The three miRNAs share a common promoter that overlaps with a single DMR observed in all 18 previously examined CC samples (Supplementary Fig. S1E). The average methylation levels in the CC samples of cohort #1 were significantly reduced compared to normal samples, confirming our earlier data (Supplementary Fig. S2E-F and Supplementary Fig. S3E-F). In summary, independent validation by MASSARRAY analysis confirmed our previous microarray results for DMRs that were shared by the majority of the CC samples.
Aberrantly methylated miRNA promoters in cholangiocarcinoma target cancer related signaling pathways
We tested, in silico, whether signaling pathways might be targeted by miRNAs aberrantly methylated at their promoters in the majority of the CC cohort #1 cases. Using miRPath v.2.0,Citation15 we identified 100 and 69 signaling pathways targeted by the hypermethylated and hypomethylated miRNA promoters, respectively; 63 of the pathways overlapped (Supplementary Fig. S4A-B and Supplementary Table S6). Pathways ranking high in both lists were cancer related, such as the neurotrophin, Wnt, ErbB, PI3K, MAPK, and TGF-β signaling pathway. Notably, the Wnt, PI3K/AKT, MAPK, and TGF-β pathway proved also affected in our earlier study, which focused on the global aberrant methylation of protein coding genes in CC.Citation7 Thus, additional data presented here not only complements our previous work but also confirms that cancer related signaling pathways are targeted by global epigenetic alterations in CC.
Correlation of miRNA expression and promoter methylation in an independent cholangiocarcinoma validation cohort
We used DNA and RNA preparations from microdissected formalin-fixed paraffin embedded (FFPE) material from 20 CCs (9 ICC and 11 ECC; see CC cohort #2 in ) and 10 control samples (normal bile duct) to correlate expression and promoter methylation of selected miRNAs. Promoter methylation of miR-129-2 was significantly higher in tumor samples compared to normal bile duct specimens ( and Supplementary Fig. S5A). Collectively, expression of miR-129 was lower in ECC and ICC sample groups as compared to the normal extra- and intra-hepatic bile duct samples, but the difference reached statistical significance only for the ECC group (). Promoter methylation of miR-9-2 and miR-9-3 in CC cohort #2 was less pronounced than in cohort #1, but still reached statistical significance (Supplementary Fig. S6A-B). No miR-9 expression differences, however, could be observed between normal and tumor samples (Supplementary Fig. S6C). For promoter methylation analysis of the clustered miR-200a, miR-200b, and miR-429 and for miR-429 expression analysis, we included additional patients samples (55 ECC, 37 ICC) and controls (92), which were also used in the tissue microarray (TMA) analyses. Methylation was significantly lower in ICC and lower in ECC with borderline significance ( and Supplementary Fig. S5B and S7A). Correspondingly, expression of miR-200a and miR-200b was significantly higher in ICC, but not in ECC, than in the corresponding controls (Supplementary Fig. S7B-C). Expression of miR-429, however, was significantly higher in both ECC and ICC samples, whether considering the samples of cohort #2 alone (data not shown) or in combination with those from the TMA cohort (). We also observed an inverse correlation between the promoter methylation and miR-429 expression, supporting the notion of promoter methylation-dependent regulation of this miRNA (Supplementary Fig. S8).
Figure 2. Correlation between miRNA promoter methylation and miRNA expression in CC. Methylation levels of the promoters of miR-129-2 (A) and the co-regulated miR-200a, miR-200b, and miR-429 (C) in cohort #2 and control samples. Corresponding relative expression is shown to the right (B and D). In C) and D), additional patient samples were included (see text). NEBD: normal extrahepatic bile duct; NIBD: normal intrahepatic bile duct.
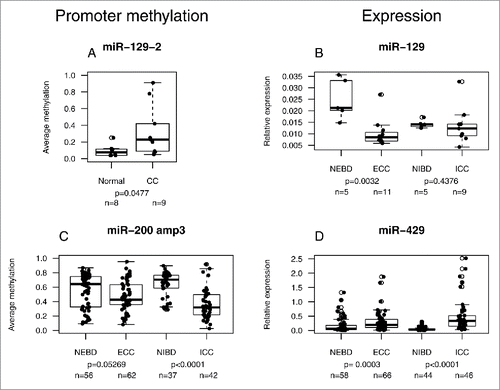
Table 1. Clinicopathological characteristics of cholangiocarcinoma cohorts #1 and #2 used for methylome and miRNA analyses. Cases with pNx had no lymph nodes resected. 16 cases (ID 1-16) were used in our methylome screen.Citation7
Tumor suppressor genes DLC1, FBXW7, and CDH6 are potential targets of miR-200a and miR-429
Upregulation by promoter demethylation of the clustered miR-200a, miR-200b, and miR-429 may contribute to CC by downregulation of target genes with tumor suppressive capability. We performed in silico prediction of genes potentially targeted by miR-200a and miR-429, the latter sharing the same seed sequence with miR-200b. Potential targets, 744 for miR-200a and 1057 for miR-429 (Supplementary Table S6), were screened for known downregulated genes in CC.Citation16,17 Targets with high scores and suspected tumor suppressive potential (Supplementary Table S7) were then tested in a luciferase reporter assay. We observed a reduction by about 40% in luciferase activity for the potential miR-200a target DLC1 (Supplementary Fig. S9A) and a reduction of about 30 and 20% for potential miR-429 targets FBXW7 and CDH6, respectively (Supplementary Fig. S9B), whereas the other targets remained unaffected (Supplementary Fig. S9A-B). The positive targets DLC1, FBXW7, and CDH6 each harbor 2 3′ UTR sequence motifs that perfectly match the seed sequences of either miR-200a or miR-429 (Supplementary Fig. S10A-C).
Protein expression of CDH6 is stepwise downregulated and associated with patient survival in biliary tract cancer
We studied the relevance of DLC1, FBXW7, and CDH6 expression in cholangiocarcinogenesis at the protein level by immunohistochemical analysis of conventional tissue slides and TMAs from normal and biliary tract cancer (BTC) samples. While DLC1 did not show expression in either sample type, FBXW7 displayed strong signals in both normal and tumor samples; thus, appropriate discrimination between expression levels was not possible (data not shown). Potential miR-429 target CDH6, however, showed strong cytoplasmic and membranous staining in normal cholangiocytes of gallbladder and extra- and intra-hepatic bile ducts (), but significantly reduced immunoreactivity in the vast majority of neoplastic tissues (). High CDH6 expression was visible also in some ICC cases (n=11; , ). Statistical analysis revealed a significantly higher immunoreactive score (IRS) of CDH6 in human normal biliary epithelium than in BTC (). Significant downregulation of CDH6 could be already observed in BTC precursor lesion BilIN 3 followed by an even more pronounced CDH6 reduction in invasive BTC (). These findings were consistent in all BTC-subgroups ().
Figure 3. CDH6 expression in CC. High immunoreactivity for CDH6 in (A) normal biliary epithelium (large intrahepatic bile ducts) and (D) exemplary in one intrahepatic cholangiocarcinoma. Low immunoreactivity for CDH6 in (B) one extrahepatic cholangiocarcinoma and (C) one intrahepatic cholangiocarcinoma. Original magnification: 50x, 200x (inset).
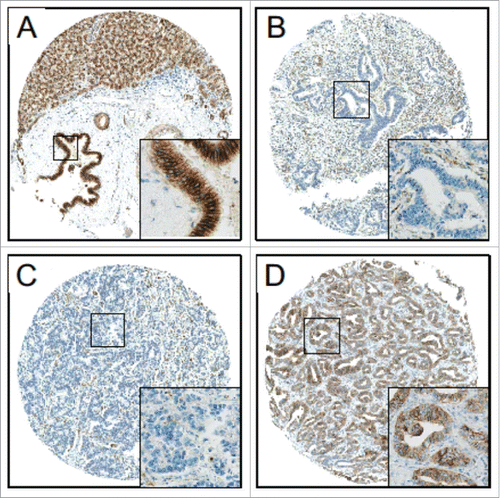
Figure 4. Statistical analyses of CDH6 expression in BTC and correlation with patient survival. (A) CDH6 immunoreactivity is already significantly reduced in pre-invasive BillN 3 lesions and decreases even more in invasive BTC compared to normal biliary epithelium. (B) Subgroup analysis shows a significant stronger decrease of CDH6 expression for ECC and GBAC compared to ICC (Unpaired non-parametric Mann-Whitney-U test: ** P < 0.005; ns: not significant; Kruskal-Wallis test P < 0.001). (C) Kaplan-Meier curves show that BTC patients with very high CDH6 expression levels (IRS = 12, n = 11 ICCs; see also and Fig. 6D) show significantly prolonged overall survival (red Kaplan-Meier curve) compared to patients with lower CDH6 IRS (blue Kaplan-Meier curve). (D) This also holds true in ICC subtype-specific survival analysis.
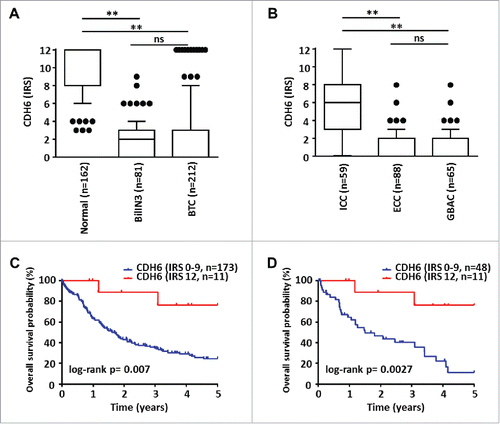
Table 2. Clinicopathological characteristics of the biliary tract cancer cohort used for TMA analysis. Cases with pNx had no lymph nodes resected; therefore, UICC status could not be assessed precisely in these cases. ‡Mann Whitney U-test; ‡‡Fisher's exact test; ‡‡‡Chi-square test.
We correlated CDH6 expression with miR-429 promoter methylation and expression and compared the data of tumor with that of normal samples for which all 3 data sets were available. Generally, lower promoter methylation correlated with higher miR-429 expression, which, in turn, was inversely correlated with lower CDH6 expression (Supplementary Fig. S11A-D). Comparing ECC (n = 55) with normal EBD (n = 34), CDH6-IRS and miRNA promoter methylation were lower [P < 0.0001 and P = 0.1859 (not significant), respectively] and miR-429 expression was higher (P = 0.0006) in ECC (Supplementary Fig. S11A-B). Similarly, in ICC (n = 36), CDH6-IRS and miRNA promoter methylation were lower (P = 0.0006 and P < 0.0001, respectively), while miR-429 expression was higher (P < 0.0001) than in normal IBD (n = 31) (Supplementary Fig. S11C-D).
Relating CDH6 expression with clinical data, we found a significant positive correlation between expression and tumor-associated patient survival (; P = 0.007). The vast majority of BTC cases showed absent to low (n = 178, IRS 0-4) CDH6 expression. A smaller proportion of tumors showed intermediate/high (n = 23, IRS 6-9) CDH6 expression. These two groups displayed very similar survival curves (data not shown). A minority of cases (n = 11) showed very high (IRS = 12) CDH 6 expression. This group consisted only of ICCs (; P < 0.0001) and was associated with prolonged overall survival both in the complete BTC and in the ICC subgroup (; P = 0.007 and P = 0.0027, respectively). We also observed a significant positive correlation between very high CDH6 expression (IRS = 12) and lower pT-status of tumors (i.e., pT1 and pT2; ; P = 0.008); none of the very high CDH6 expressing ICCs showed perineural tumor invasion (; P = 0.008). For other clinicopathological features, no significant correlation with CDH6 expression was observed (). Moreover, there was no significant correlation between overall survival and the promoter methylation status of the clustered miR-200a, miR-200b, and miR-429 (Supplementary Fig. S12).
CDH6 delays growth of CC cell lines
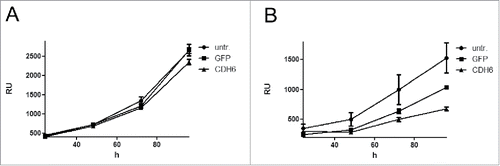
To corroborate the potential tumor suppressive function of CDH6, we stably transfected the 2 CC cell lines EGI-1 and TFK-1 with a lentiviral construct harboring CDH6 under the control of the CMV promoter. While CDH6-transfected cells displayed expression of the CDH6 protein, untransfected and GFP transfected cells did not (Supplementary Fig. S13). We monitored growth of serial dilutions of CDH6 transfectants and observed delayed growth of both cell lines as compared to the untransfected lines or the GFP transfectants ( and Supplementary Fig. S14). Growth delay by CDH6 was more pronounced for the faster growing TFK-1 than for EGI-1 and could be confirmed in TFK-1 by colony formation assay (Supplementary Fig. S15).
Figure 5. Growth curves of the CC cell lines EGI-1 (A) and TFK-1 (B) after lentiviral transfection with CDH6 cDNA or GFP. Initial cell number of EGI-1 was 2,500, that of TFK-1 was 313. Relative fluorescence units (RU) were determined after 24, 48, 72, and 96 h. Values are means of 3 technical replicates; error bars indicate standard deviation. untr.: untransfected.
Discussion
Exome and whole-genome sequencing studies of CC patients revealed a number of recurrent somatic mutations, including FGFR2-associated translocationsCitation18 and point mutations affecting epigenetically relevant genes, such as ARID1, IDH1, and IDH2.Citation5 Furthermore, expression profiling demonstrated recurrent dysregulation of miRNAs in CC (reviewed inCitation19). In the present study, we aimed to globally identify and exemplarily characterize epigenetic alterations of miRNA promoters in ECC and ICC to advance the epigenetic characterization of CC. Overlapping the genomic coordinates of CC-related DMRs identified in our previous studyCitation7 and of known or suspected miRNA promoters,Citation11 we identified 28 and 16 miRNAs associated with hypermethylated and hypomethylated promoters, respectively, in more than 50% of the analyzed CC cases. All DMRs were found in both ECC and ICC, and many of the identified miRNAs targeted signaling pathways that were already known to be affected in cholangiocarcinogenesis, such as Wnt, ErbB, PI3K, MAPK, and TGF-β, but had not been described in the context of altered miRNA promoter methylation in CC.
In cancer, downregulation of tumor suppressor genes is frequently caused by promoter hypermethylation. We observed hypermethylation of promoters from several miRNAs with suspected tumor suppressive ability in the initial CC patient cohort and confirmed promoter hypermethylation of miR-129-2, miR-9-2, and miR-9-3 in an independent cohort. miR-129 was also downregulated in ECC. MiR-129 was reported to be epigenetically downregulated in various cancers (e.g.,Citation20,21) and to exert tumor suppressive functions by inhibiting cell proliferation and inducing cell death.Citation22,23 Thus, its downregulation in ECC might promote proliferation and survival of tumor cells. Promoter hypermethylation and downregulation of miR-9 have been described in several tumor types.Citation24,25 Though confirmed in CC cohort #2, promoter hypermethylation of miR-9-2 and miR-9-3 was less pronounced and was not associated with dysregulation of expression. The downregulation of these miRNAs might be compensated by concomitant upregulation of miR-9-1. Notably, the miR-9 family was reported to be both up- and down-regulated depending on the cancer type (see Supplementary Table S4).
The miRNA cluster encompassing miR-200a, miR-200b, and miR-429 did not only show promoter hypomethylation, but also all 3 miRNAs were found to be upregulated in CC. Both up- and down-regulation of the miR-200 family have been observed in cancer (see Supplementary Table S4), possibly depending on type and stage of the malignancy. Supporting our data, miR-200b was already reported as upregulated in CC.Citation26
DLC1, FBXW7, and CDH6 were found to be potential targets for downregulation by miR-200a and miR-429, respectively, and, thus, to be potential tumor suppressor genes in cholangiocarcinogenesis. Notably, the promoters of DLC1, FBXW7, and CDH6 did not show CC-related hypermethylation in our previous study,Citation7 supporting the role of miRNA-mediated downregulation of these tumor suppressor genes in CC. Knockdown of DLC1, encoding an activator of Rho family GTPases and involved in the regulation of the cytoskeleton and cell motility,Citation27 contributes to carcinogenesis of HCC in an animal model;Citation28 DLC1 downregulation might also contribute to ICC, since both malignancies share common molecular characteristics.Citation29 FBXW7 was shown to be affected by loss-of-function mutationsCitation30 and to suppress epithelial-mesenchymal transition, stemness, and metastasis in CC.Citation31 CDH6 is a cell adhesion molecule that contributes to the induction of apoptosis by dephosphorylation of ERK.Citation32 CDH6 was already shown to be transcriptionally downregulated in CC (see Supplementary Table S7), and we also observed downregulation of the CDH6 protein in 2 CC cell lines, EGI-1 and TFK-1. On the other hand, CDH6 was reported to be upregulated in other cancer types as well.Citation33,34 In line with its potential tumor suppressive function, we observed stepwise downregulation of CDH6 in a comprehensive and well characterized cohort, including normal tissue, precursor lesions, and invasive tumors of BTC patients. Additionally, we detected an ICC subgroup with very high CDH6 expression, better patient survival, and lower local tumor extent. When we expressed CDH6 in the CC cell lines EGI-1 and TFK-1, both cell lines displayed delayed growth. Together, these features suggest that CDH6 is a potential tumor suppressor in cholangiocarcinogenesis. CDH6 could also serve as a biomarker due to its stepwise downregulation that can already be detected in pre-invasive BilIN 3 lesions.
In summary, our systematic approach revealed a considerable number of aberrantly methylated miRNA promoters in CC. We presented examples of inverse correlation between miRNA promoter methylation and miRNA expression, suggesting miR-129 as a novel tumor suppressor and members of the miR-200 family as novel oncomiRs in CC. CDH6 was identified as a potential tumor suppressor and biomarker in cholangiocarcinogenesis. The presented compilation of CC-related miRNAs constitutes a profound repository for future studies to dissect the role of epigenetically dysregulated miRNAs in the onset and progression of CC.
Material and methods
Clinicopathological characteristics of cholangiocarcinoma cohorts #1 and #2 used for methylome and miRNA analyses
Tissue samples from 42 CC patients (20 perihilar ECCs and 22 ICCs) who underwent bile duct and/or liver surgery in the University Hospital Heidelberg between 2004 and 2010 were included in these 2 cohorts (). Sixteen cases (ID 1-16) were already part of our previous methylome screen.Citation7 Patient cohort #1, initially used here for quantitative DNA methylation validation by mass spectrometry (MassARRAY) consisted of these 16 CC cases and additional 2 ECC and 4 ICC patients. As validation controls, fresh-frozen tissue specimens of 9 matched normal samples and one non-matched normal sample were used. Cohort #2, used for correlating miRNA expression with miRNA promoter methylation, consisted of 11 ECC and 9 ICC cases (FFPE samples). The normal controls used for comparison with cohort #2 consisted of extra- and intra-hepatic bile duct samples from non-neoplastic cases. Microdissection was performed as previously described.Citation35 Only patients with primary adenocarcinomas of the biliary tract and without other known malignancies at the time of diagnosis were included. Tumors were classified, graded, and staged according to the current World Health Organization (WHO) tumor classification system (4th ed., 2010) and the 7th TNM Classification of Malignant Tumors, respectively. Patients who received radiochemotherapy prior to surgery were excluded. The study was approved by the institutional ethics committee (206/05).
Clinicopathological characteristics of the biliary tract cancer cohort used for tissue microarray analysis
Tissue samples from 212 patients with a median age of 64.7 y who underwent bile duct and/or liver surgery in the University Hospital Heidelberg between 1995 and 2010 were included in this cohort (). Only patients with primary adenocarcinomas of the biliary tract and without other known malignancies at the time of diagnosis were included. Patients who received radiochemotherapy prior to surgery were excluded. BTCs of this study consisted of 88 ECCs, 59 ICCs, and 65 adenocarcinomas of the gallbladder (GBACs). For 162 cases, corresponding histologically normal tissue, and for 81 cases corresponding biliary intraepithelial neoplasia grade 3 (BilIN 3) lesions could be included. For most patients, complete clinicopathological data, including sex, age, tumor grade, TNM/UICC status, lymph- and haemangiosis carcinomatosa, perineural tumor invasion, as well as overall survival data were available.
Cell lines, cloning, and transfection
Cell line HEK293T and the human CC cell lines EGI-1 and TFK-1 were obtained from the German Collection of Microorganims and Cell Cultures (DSMZ) and stored as low-passage aliquots in liquid nitrogen. For virus production and functional assays, freshly thawed aliquots are regularly authenticated and tested for absence of mycoplasmas using the Venor® GeM Classic kit (Minerva Biolabs, cat. no. 11-1025). HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, cat. no. 41965-039) supplemented with 10% fetal calf serum (FCS). EGI-1 and TFK-1 were grown in high glucose Dulbecco's modified Eagle's medium (DMEM; GE Healthcare Life Sciences, cat. no. SH30243.02) and RPMI 1640 (Life Technologies, cat. no. 21875-034), respectively, supplemented with 10% FCS. A human CDH6 full ORF cDNA clone (Refseq no. BC000019)Citation36 was obtained from the DKFZ Genomics and Proteomics Core Facility. The CDH6 ORF was re-cloned behind the CMV promoter into lentiviral vector rwpLX305 (R. W., unpublished), a puromycin resistance-conferring derivative of pLX304.Citation37 The GFP gene instead of CDH6 was used in the same lentiviral vector setting as a control. Lentiviral particles were generated in HEK293T cells using a second-generation packaging system and transfected in diverse dilutions into both EGI-1 and TFK-1 cells under puromycin selection.
Cell proliferation and colony formation assay
Triplicates of 10,000 cells and twofold serial dilutions were seeded into 96-well plates. Cell proliferation was evaluated after 24, 48, 72 and 96 h with the Cell Titer-Blue Cell Viability Assay (Promega, cat. no. G8081), according to manufacturer's instructions using Spectramax M5e (Molecular Devices). To determine relative units, mean values of fluorescence units of medium only were subtracted from the fluorescence units obtained with cells. Growth curves were generated with GraphPad Prism 6 software. To monitor colony formation, 1,000 cells were seeded into 10 cm dishes, grown for 13 d washed with PBS, fixed with methanol, and stained with Giemsa (Sigma-Aldrich, cat. no. T862.1).
Immunoblotting
Proteins were isolated from roughly 2 × 106 cells using lysis buffer composed of 0.0625 M Tris/HCL pH 6.8, 2% SDS, 10% glycerol, 1 mM dithiothreitol, 1 mM NaVO4, 5 mM NaF and 1x protease inhibitors (Complete; Roche Diagnostics, cat.no. 11 697 498 001). Proteins (25 µg) were separated by 4-20% SDS-PAGE (Sigma Aldrich, PCG2012-10EA) and transferred to PVDF membrane (Immobilon, Milipore, cat. no. IPVH00010). Either anti-CDH6 monoclonal antibody (dilution 1:300; Thermo Fischer Scientific, cat.no. MA1-06305) or monoclonal anti-ACTB (dilution 1:5000; Santa Cruz, cat. no. sc47778) were used, followed by anti-mouse IgG horseradish peroxidase-linked antibody (dilution 1:5000; Cell Signaling, cat. no. 7076P2). For signal detection, we used Western Lightning Plus ECL (Perkin Elmer; cat. no. NEL 104001EA) and Amersham Hyperfilm ECL (GE Healthcare; cat. no. 28906836).
Tissue microarray preparation, immunohistochemistry, and analysis
Representative areas from vital tissue of invasive adenocarcinoma, BilIN 3, and normal tissue (NT) of biliary epithelium were marked by 2 pathologists experienced in BTC diagnostics (BG and WW). Tissue cores (1 mm diameter) from the selected representative areas were punched out of the sample tissue blocks and embedded into a new paraffin array block using a manual tissue microarrayer (Beecher Instruments, Woodland, CA, USA). Immunohistochemistry was performed according to standard protocols using the avidin-biotin complex-method and diaminobenzidine as chromogen. Immunohistochemistry was performed using an anti-CDH6 monoclonal antibody (dilution 1:100; Thermo Fischer Scientific, cat. no. MA1-06305), an anti-FBXW7 monoclonal antibody (dilutions 1:100 down to 1:2000; Abcam, cat. no. ab55036), and 2 anti-DLC1 antibodies (both polyclonal; dilution 1:50, each; Sigma Hamburg, cat. no. HPA017753 and Santa Cruz Biotechnology, cat. no. sc-271915). All sections were counterstained with hemalum. The specificity of the reaction was controlled by omitting the primary antibody. For semi-quantitative immunohistochemical assessment of cytoplasmic CDH6 expression, the product of the scores of staining intensity and percentage of immunoreactive cells was calculated based on the following scoring system: the intensity ranged from 0 = negative, 1 = low, 2 = medium to 3 = high; the quantity comprised 0 = no expression, 1 = positivity in less than 10 %, 2 = positivity in 10 % to 50 %, 3 = positivity in 51 % to 80 %, and 4 = positivity in more than 80 % of biliary cells. The final immunoreactive score (IRS) was obtained by multiplication of the intensity score and the quantity score according to IRS (ranging from 0 to 12). Only biliary cells were counted. Evaluations were performed independently by 2 experienced pathologists (BG and WW).
Statistical analyses of IRS in normal biliary tissue, BilIN3, and BTC samples were performed by unpaired non-parametric Mann-Whitney-U and Kruskal-Wallis test with GraphPad Prism 6. The same statistical test was also applied to the comparison of ICC, ECC, and GBAC groups. Kaplan-Meier survival analysis and determination of corresponding log-rank P-values were performed with GraphPad Prism 6 software. Survival data of 28 cases was not available. All P-values were 2-tailed and a P-value of less than 0.05 was considered significant.
Screening differentially methylated regions and miRNA promoters for overlaps of genomic coordinates
In a previous study, we identified 7179 hypermethylated and 8893 hypomethylated regions (collectively designated DMRs) in 10 ICC and 8 ECC samples using human CpG island tiling arrays (Agilent; genome assembly hg18).Citation7 In an independent analysis, we identified 1374 putative miRNA promoter regions by integration of H3K4me3-enriched genomic regions with sequences upstream of pre-miRNA (miRbase v15).Citation11 The genomic coordinates of DMRs and putative miRNA promoter regions (for simplicity, designated miRNA promoters) were harmonized to hg18 using the LiftOver tool from the UCSC genome browser.Citation38 We screened for overlaps between coordinates using a self-designed Perl script (kindly provided by Lei Gu, DKFZ Heidelberg) considering only DMRs represented by >9/18 (>50 %) CC samples.
Isolation of nucleic acids, cDNA synthesis, and expression analysis
DNA preparations from fresh-frozen tissue specimens from cohort #1 and its controls were performed as described previously.Citation7 FFPE samples from cohort #2 and corresponding control samples were used for simultaneous extraction of DNA and RNA using the AllPrepR DNA/RNA FFPE Kit as recommended (Qiagen, cat.no. 80234). Due to limited material, microdissected healthy bile duct samples were processed using the RNeasy FFPE Kit (Qiagen, cat. no. 73504) following the manufacturer's protocol to ensure high RNA yield. RNA isolation was done as recommended; the pellet from step 6 was used to follow the DNA isolation protocol of the AllPrepR DNA/RNA FFPE Kit starting at step 25 (Qiagen, version 2010a). In contrast to the miRNA quality, DNA quality proved appropriate for only a fraction of the CC cohort #2 and corresponding control samples for MassARRAY analysis, most likely owing to the FFPE derivation of the DNAs and their harsh bisulfite treatment.
Isolation of miRNA was performed using the RNeasy Mini Kit (Qiagen, cat. no. 74104) largely according to the manufacturer's instructions. The Qiagen protocol was modified at 2 steps: for precipitation of RNA, 1.5 volumes of 100% ethanol were used instead of 1 volume of 70% ethanol (step 4), and the RW1 wash buffer from the first wash step was replaced with RWT wash buffer (step 6). On-column DNase digestion was performed as indicated in the RNeasy Mini Handbook (D1-D4). For cDNA synthesis from 500 ng total RNA, the miScript PCR system (Qiagen, cat.no. 218073) was used as recommended (miScript II RT protocol).
For expression analysis, 2.1 μl of 1:20 diluted cDNA were used in a 7 μl PCR reaction with 3.5 μl 2x QuantiTect SYBR Green PCR Master Mix and 0.7 μl each of 10x miScript Universal Primer and respective 10x miScript Primer Assay (Supplementary Table S1C). Cycling conditions on the Lightcycler 480 (Roche, Mannheim, Germany) were used as described in the manufacturer's protocol.
For FFPE tissue-derived RNA, miR-574 was used as a reference gene, which was identified as the best-suited reference gene for this sample cohort using the web-based tool RefFinder (http://www.leonxie.com/referencegene.php).
Relative expression was calculated using the ΔCt method.Citation39 Statistical evaluation of expression differences was performed by Mann-Whitney-U test, and accompanying boxplots were generated using the R statistical environment, version 3.0.1 (http://www.R-project.org). A P-value <0.05 was considered significant.
Quantitative determination of methylation levels by mass spectrometry
Quantitative methylation levels of CpGs or CpG units were determined by mass spectrometric analysis using the Epityper MassARRAY approach as described previouslyCitation7 and listed primers (Supplementary Table S1A). Use of FFPE samples for cohort #2 analyses partially required smaller amplicons than the analyses performed with cohort #1. Statistical evaluation of differences between average methylation values was performed by Mann-Whitney-U test, and accompanying boxplots and heat maps were generated using the R statistical and graphical environment, version 3.0.1. A P-value < 0.05 was considered significant. Kaplan-Meier survival analysis was done as described above.
Prediction of miRNA target genes and dual luciferase/renilla reporter assays
We used “TargetScanHuman” (release 6.2; http://www.targetscan.org/vert_61/) of the miRGator online tool for in silico prediction of miRNA target genes.Citation40 TargetScan predictions are ranked based on the predicted efficacy of targeting and calculated as “context scores” as described (http://www.targetscan.org/vert_61/docs/context_score.html). From the 3′ UTRs of selected target genes, amplicons containing the seed sequences were generated and cloned into the 3′ UTR of the firefly luciferase reporter gene of vector pMIR-REPORT (Ambion, cat. no. AM5795). Positive controls cloned into the reporter vector were short double-stranded sequence stretches formed by complementary phosphorylated oligonucleotides covering 4 times the seed sequence of the respective miRNA with a 6 bp spacer after the first and another 6 bp spacer after the third seed sequence. Reporter constructs were transfected into HEK293T cells together with synthetic microRNA Mimics (Qiagen, Supplementary Table S1D) or a non-target small RNA control (AllStar, Qiagen, cat. no. SI03650318) to test for physical interactions between the microRNA Mimics and the cloned 3′ UTR. HEK293T cells were transfected with 1 pmol of the miRNA mimic or AllStar non-target control using the DharmaFECT 1 transfection reagent (Thermo Scientific, cat. no. T-2001-01), according to the manufacturer's instructions. After overnight incubation, cells were transfected using the TransIT LT1 transfection reagent (Mirus, cat.no. MIR 2304) with 300 pg of the reporter construct or empty vector pMIR-Report together with 10 ng of the pRL-TK Renilla luciferase reporter vector (Promega, cat.no. E2241). The dual luciferase readout was performed with nearly confluent cells using the SpectraMax M5 (Berthold, Bad Wildbad, Germany); the luciferase signals were normalized to the renilla signals. Mean values were calculated from 6 or 8 parallel measurements per transfection, and 4 independent transfections were performed per construct. Barplots were generated with the mean values using the R statistical environment, version 3.0.1. Error bars indicate standard deviations. Statistical evaluation of differences between mean values of target 3′ UTRs and empty vector was performed by one-sample t-test.
Pathway enrichment analysis
Lists of 28 hypermethylated and 16 hypomethylated miRNAs present in >9/18 CC cases from patient cohort #1 (see ) were loaded separately into the DIANA miRPath v.2.0 online tool to search with default settings for possibly affected signaling pathways.Citation15 In those cases, where the miRPath tool indicates ambiguity, for example miR-200a, both options, miR-200a-3p and miR-200a-5p, were loaded.
Oligonucleotides
The oligonucleotides used in this study are listed in Supplementary Table S1.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1227899_s02.doc
Download MS Word (2.6 MB)KEPI_A_1227899_s02.doc
Download MS Word (2.6 MB)Acknowledgments
The excellent technical assistance of Veronika Geissler, John Moyers (Institute of Pathology, University of Heidelberg), Monika Helf and Oliver Mücke as well as helpful discussions with Daniel Lipka, Christopher Oakes, Yassen Assenov and Daniela Mancarella (Division of Epigenomics and Cancer Risk Factor, German Cancer Research Center) are gratefully acknowledged. We would also like to thank the Genomics and Proteomics Core Facility of the German Cancer Research Center for providing the CDH6 ORFeome clone.
The work was supported by the Biobank of the National Center of Tumor Diseases (NCT) Heidelberg and, in part, by the Helmholtz Association.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) to P. Schirmacher (SFB/TRR77) and by the German Cancer Consortium to C. Plass and P. Schirmacher.
Related Research Data
References
- Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013; 145:1215-29; PMID:24140396; http://dx.doi.org/10.1053/j.gastro.2013.10.013
- Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011; 8:512-22; PMID:21808282; http://dx.doi.org/10.1038/nrgastro.2011.131
- Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet 2012; 44:690-3; PMID:22561520; http://dx.doi.org/10.1038/ng.2273
- Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 2013; 45:1474-8; PMID:24185513; http://dx.doi.org/10.1038/ng.2806
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013; 45:1470-3; PMID:24185509; http://dx.doi.org/10.1038/ng.2813
- Costello JF, Plass C. Methylation matters. J Med Genet 2001; 38:285-303; PMID:11333864; http://dx.doi.org/10.1136/jmg.38.5.285
- Goeppert B, Konermann C, Schmidt CR, Bogatyrova O, Geiselhart L, Ernst C, Gu L, Becker N, Zucknick M, Mehrabi A, et al. Global alterations of DNA methylation in cholangiocarcinoma targets the Wnt signaling pathway. Hepatology 2014; 59:544-54; PMID:24002901; http://dx.doi.org/10.1002/hep.26721
- Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Gitt A, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 2007; 67:1424-9; PMID:17308079; http://dx.doi.org/10.1158/0008-5472.CAN-06-4218
- Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 2008; 105:13556-61; PMID:18768788; http://dx.doi.org/10.1073/pnas.0803055105
- Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68:4123-32; PMID:18519671; http://dx.doi.org/10.1158/0008-5472.CAN-08-0325
- Baer C, Claus R, Frenzel LP, Zucknick M, Park YJ, Gu L, Weichenhan D, Fischer M, Pallasch CP, Herpel E, et al. Extensive promoter DNA hypermethylation and hypomethylation is associated with aberrant microRNA expression in chronic lymphocytic leukemia. Cancer Res 2012; 72:3775-85; PMID:22710432; http://dx.doi.org/10.1158/0008-5472.CAN-12-0803
- Karakatsanis A, Papaconstantinou I, Gazouli M, Lyberopoulou A, Polymeneas G, Voros D. Expression of microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c, miR-221, miR-222, and miR-223 in patients with hepatocellular carcinoma or intrahepatic cholangiocarcinoma and its prognostic significance. Mol Carcinogenesis 2013; 52:297-303; PMID:22213236; http://dx.doi.org/10.1002/mc.21864
- Xie B, Ding Q, Han H, Wu D. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013; 29:638-44; PMID:23325619; http://dx.doi.org/10.1093/bioinformatics/btt014
- Peng F, Jiang J, Yu Y, Tian R, Guo X, Li X, Shen M, Xu M, Zhu F, Shi C, et al. Direct targeting of SUZ12/ROCK2 by miR-200b/c inhibits cholangiocarcinoma tumourigenesis and metastasis. Br J Cancer 2013; 109:3092-104; PMID:24169343; http://dx.doi.org/10.1038/bjc.2013.655
- Vlachos IS, Kostoulas N, Vergoulis T, Georgakilas G, Reczko M, Maragkakis M, Paraskevopoulou MD, Prionidis K, Dalamagas T, Hatzigeorgiou AG. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res 2012; 40:W498-504; PMID:22649059; http://dx.doi.org/10.1093/nar/gks494
- Jinawath N, Chamgramol Y, Furukawa Y, Obama K, Tsunoda T, Sripa B, Pairojkul C, Nakamura Y. Comparison of gene expression profiles between Opisthorchis viverrini and non-Opisthorchis viverrini associated human intrahepatic cholangiocarcinoma. Hepatology 2006; 44:1025-38; PMID:17006947; http://dx.doi.org/10.1002/hep.21330
- Obama K, Ura K, Li M, Katagiri T, Tsunoda T, Nomura A, Satoh S, Nakamura Y, Furukawa Y. Genome-wide analysis of gene expression in human intrahepatic cholangiocarcinoma. Hepatology 2005; 41:1339-48; PMID:15880566; http://dx.doi.org/10.1002/hep.20718
- Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, McCullough AE, Barrett MT, Hunt K, Patel MD, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 2014; 10:e1004135; PMID:24550739; http://dx.doi.org/10.1371/journal.pgen.1004135
- Haga H, Yan I, Takahashi K, Wood J, Patel T. Emerging insights into the role of microRNAs in the pathogenesis of cholangiocarcinoma. Gene Expression 2014; 16:93-9; PMID:24801170; http://dx.doi.org/10.3727/105221614X13919976902174
- Chen X, Zhang L, Zhang T, Hao M, Zhang X, Zhang J, Xie Q, Wang Y, Guo M, Zhuang H, et al. Methylation-mediated repression of microRNA 129-2 enhances oncogenic SOX4 expression in HCC. Liver Int: Official J Int Association For Study Liver 2013; 33:476-86; PMID:23402613; http://dx.doi.org/10.1111/liv.12097
- Huang YW, Liu JC, Deatherage DE, Luo J, Mutch DG, Goodfellow PJ, Miller DS, Huang TH. Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res 2009; 69:9038-46; PMID:19887623; http://dx.doi.org/10.1158/0008-5472.CAN-09-1499
- Brest P, Lassalle S, Hofman V, Bordone O, Gavric Tanga V, Bonnetaud C, Moreilhon C, Rios G, Santini J, Barbry P, et al. MiR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocrine-Related Cancer 2011; 18:711-9; PMID:21946411; http://dx.doi.org/10.1530/ERC-10-0257
- Kang M, Li Y, Liu W, Wang R, Tang A, Hao H, Liu Z, Ou H. miR-129-2 suppresses proliferation and migration of esophageal carcinoma cells through downregulation of SOX4 expression. Internat J Mol Med 2013; 32:51-8; PMID:23677061; http://dx.doi.org/10.3892/ijmm.2013.1384
- Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, Kreipe H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol 2008; 214:17-24; PMID:17948228; http://dx.doi.org/10.1002/path.2251
- Tsai KW, Liao YL, Wu CW, Hu LY, Li SC, Chan WC, Ho MR, Lai CH, Kao HW, Fang WL, et al. Aberrant hypermethylation of miR-9 genes in gastric cancer. Epigenetics 2011; 6:1189-97; PMID:21931274; http://dx.doi.org/10.4161/epi.6.10.16535
- Meng F, Wehbe-Janek H, Henson R, Smith H, Patel T. Epigenetic regulation of microRNA-370 by interleukin-6 in malignant human cholangiocytes. Oncogene 2008; 27:378-86; PMID:17621267; http://dx.doi.org/10.1038/sj.onc.1210648
- Kim TY, Vigil D, Der CJ, Juliano RL. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev 2009; 28:77-83; PMID:19221866; http://dx.doi.org/10.1007/s10555-008-9167-2
- Zimonjic DB, Popescu NC. Role of DLC1 tumor suppressor gene and MYC oncogene in pathogenesis of human hepatocellular carcinoma: potential prospects for combined targeted therapeutics (review). Internat J Oncol 2012; 41:393-406; PMID:22580498; http://dx.doi.org/10.3892/ijo.2012.1474
- Oishi N, Kumar MR, Roessler S, Ji J, Forgues M, Budhu A, Zhao X, Andersen JB, Ye QH, Jia HL, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012; 56:1792-803; PMID:22707408; http://dx.doi.org/10.1002/hep.25890
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007; 67:9006-12; PMID:17909001; http://dx.doi.org/10.1158/0008-5472.CAN-07-1320
- Yang H, Lu X, Liu Z, Chen L, Xu Y, Wang Y, Wie G, Chen Y. FBXW7 suppresses epithelial-mesenchymal transition, stemness and metastatic potential of cholangiocarcinoma cells. Oncotarget 2015; 6:6310-25; PMID:25749036; http://dx.doi.org/10.18632/oncotarget.3355
- Park MC, Kang T, Jin D, Han JM, Kim SB, Park YJ, Cho K, Park YW, Guo M, He W, et al. Secreted human glycyl-tRNA synthetase implicated in defense against ERK-activated tumorigenesis. Proc Natl Acad Sci USA 2012; 109:E640-7; PMID:22345558; http://dx.doi.org/10.1073/pnas.1200194109
- Paul R, Ewing CM, Robinson JC, Marshall FF, Johnson KR, Wheelock MJ, Isaacs WB. Cadherin-6, a cell adhesion molecule specifically expressed in the proximal renal tubule and renal cell carcinoma. Cancer Res 1997; 57:2741-8; PMID:9205085
- Sancisi V, Gandolfi G, Ragazzi M, Nicoli D, Tamagnini I, Piana S, Ciarrocchi A. Cadherin 6 is a new RUNX2 target in TGF-β signalling pathway. PloS One 2013; 8:e75489; PMID:24069422; http://dx.doi.org/10.1371/journal.pone.0075489
- Goeppert B, Schmezer P, Dutruel C, Oakes C, Renner M, Breinig M, Warth A, Vogel MN, Mittelbronn M, Mehrabi A, et al. Downregulation of tumor suppressor A kinase anchor protein 12 in human hepatocarcinogenesis by epigenetic mechanisms. Hepatology 2010; 52:2023-33; PMID:20979053; http://dx.doi.org/10.1002/hep.23939
- Collaboration OR. The ORFeome Collaboration: a genome-scale human ORF-clone resource. Nat Methods 2016; 13:191-2; PMID:26914201; http://dx.doi.org/10.1038/nmeth.3776
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas, SR, Alkan O, Bhimdi T, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 2011; 8:659-61; PMID:21706014; http://dx.doi.org/10.1038/nmeth.1638
- Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res 2014; 42:D764-70; PMID:24270787; http://dx.doi.org/10.1093/nar/gkt1168
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262
- Cho S, Jang I, Jun Y, Yoon S, Ko M, Kwon Y, Choi I, Chang H, Ryu D, Lee B, et al. MiRGator v3.0: a microRNA portal for deep sequencing, expression profiling and mRNA targeting. Nucleic Acids Res 2013; 41:D252-7; PMID:23193297; http://dx.doi.org/10.1093/nar/gks1168
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol 2011; 29:24-6; PMID:21221095; http://dx.doi.org/10.1038/nbt.1754