
ABSTRACT
The cdk inhibitor p57kip2, encoded by the Cdkn1c gene, plays a critical role in mammalian development and in the differentiation of several tissues. Cdkn1c protein levels are carefully regulated via imprinting and other epigenetic mechanisms affecting both the promoter and distant regulatory elements, which restrict its expression to particular developmental phases or specific cell types. Inappropriate activation of these regulatory mechanisms leads to Cdkn1c silencing, causing growth disorders and cancer. We have previously reported that, in skeletal muscle cells, induction of Cdkn1c expression requires the binding of the bHLH myogenic factor MyoD to a long-distance regulatory element within the imprinting control region KvDMR1. Interestingly, MyoD binding to KvDMR1 is prevented in myogenic cell types refractory to the induction of Cdkn1c. In the present work, we took advantage of this model system to investigate the epigenetic determinants of the differential interaction of MyoD with KvDMR1. We show that treatment with the DNA demethylating agent 5-azacytidine restores the binding of MyoD to KvDMR1 in cells unresponsive to Cdkn1c induction. This, in turn, promotes the release of a repressive chromatin loop between KvDMR1 and Cdkn1c promoter and, thus, the upregulation of the gene. Analysis of the chromatin status of Cdkn1c promoter and KvDMR1 in unresponsive compared to responsive cell types showed that their differential responsiveness to the MyoD-dependent induction of the gene does not involve just their methylation status but, rather, the differential H3 lysine 9 dimethylation at KvDMR1. Finally, we report that the same histone modification also marks the KvDMR1 region of human cancer cells in which Cdkn1c is silenced. On the basis of these results, we suggest that the epigenetic status of KvDMR1 represents a critical determinant of the cell type-restricted expression of Cdkn1c and, possibly, of its aberrant silencing in some pathological conditions.
Abbreviations
| Cdkn1c | = | Cyclin-dependent kinase inhibitor 1C |
| CKI | = | Cyclin-dependent kinase inhibitor |
| KvDMR1 | = | Kv-differentially methylated region 1 |
| CTCF | = | CCCTC-binding factor |
| BWS | = | Beckwith-Wiedemann syndrome |
| bHLH | = | basic-helix-loop-helix |
| MeDIP | = | Methylated DNA immunoprecipitation |
| 5-AZA | = | 5-Azacytidine |
| H3K9me2 | = | Histone H3 lysine 9 dimethylation |
| H3K9me3 | = | Histone H3 lysine 9 trimethylation |
| ChIP | = | Chromatin immunoprecipitation |
| 3C | = | Chromosome conformation capture |
| DMEM | = | Dulbecco's modified Eagle's medium |
| FBS | = | Fetal bovine serum |
Introduction
p57kip2, encoded by the Cdkn1c gene, is a member of the Cip/Kip family of cyclin-dependent-kinase inhibitors (CKIs) and plays a key role in mammalian development by regulating cell proliferation and differentiation in a number of different tissues.Citation1 Compared to the other Cip/Kip CKIs p21cip1 and p27kip1, p57kip2 exerts unique functions not compensated by the other family members, as suggested by the lethal phenotype of knockout miceCitation2,3 and by the developmental anomalies associated with Cdkn1c loss or misexpression in humans.Citation4 The unique role of the Cdkn1c protein has been ascribed both to the presence of specific biochemical features of the protein, not shared by the other family members,Citation5,6 and to the specific expression pattern of the gene.Citation7 Cdkn1c is widely expressed in most tissues during embryogenesis but its expression drastically decreases at birth, becoming restricted to a subset of tissues and organs, such as heart, brain, lung, kidney, pancreas, skeletal muscle, testis, and placenta.Citation8
The levels of Cdkn1c protein are fine-tuned by multiple regulatory mechanisms, ranging from epigenetic to posttranslational control, in different cell types.Citation1 Moreover, Cdkn1c is a paternally imprinted gene, expressed exclusively from the maternal allele, in both human and mouse.Citation9,10 Paternal Cdkn1c is regulated by an imprinting control region, called Kv-differentially methylated region 1 (KvDMR1), which controls the imprinting of the Kcnq1 domain, a cluster of imprinted genes including Cdkn1c.Citation11 KvDMR1 is a 3.6 kb sequence located about 150 kb at the 3′ of Cdkn1c and bears differential epigenetic marks on the 2 parental chromosomes, such as DNA methylation and histone modifications.Citation12-14 The functional organization of KvDMR1 is highly conserved in human and mouse. Two mechanisms of action have been proposed to explain the cis-acting repressive effect of this imprinting control region. The first one would involve the enhancer-blocking activity of paternal KvDMR1, mediated by the insulator CCCTC-binding factor (CTCF).Citation15 The second one would involve chromatin silencing by paternally expressed Kcnq1ot1, a macro noncoding RNA, the promoter of which is comprised within the KvDMR1 CpG island,Citation16 and which has been suggested to regulate in cis the silencing of adjacent genes by recruiting epigenetic modifiers.Citation17 As found for many imprinted genes, especially those involved in growth control, alterations in the dosage of Cdkn1c expression cause important functional consequences. In particular, Cdkn1c defective expression is generally responsible for overgrowth disorders, such as the Beckwith-Wiedemann syndrome (BWS) and several cancer types, while Cdkn1c excessive expression is associated with growth retardation disorders, such as the Silver Russel syndrome.Citation4,18
Although some information has been accumulated on the regulatory strategies and molecular mechanisms involved in the Cdkn1c silencing in cancer and in the paternal allele-specific repression during imprinting, much less is known about the developmental regulation of this CKI, that is expected to affect its maternal allele.
We have previously investigated the differentiation-dependent regulation of Cdkn1c using the skeletal muscle system as a model. We found that Cdkn1c is a target of the muscle-specific basic helix-loop helix (bHLH) factor MyoD.Citation19 MyoD is the prototypical master regulator of differentiationCitation20 and induces Cdkn1c expression through a complex mechanism involving, on one hand, the mediation of MyoD-induced factors, which interact with Cdkn1c promoterCitation21,22 and, on the other hand, the direct interaction of MyoD with KvDMR1.Citation23,24 In particular, we found that, in undifferentiated myoblasts, KvDMR1 is involved in a repressive long-range chromatin interaction with Cdkn1c promoter, mediated by CTCF. Upon differentiation stimuli, MyoD binds to an E-box-like sequence adjacent to a CTCF binding site within KvDMR1, causing the disruption of the chromatin loop and the induction of maternal Cdkn1c expression. This finding indicates that KvDMR1, in addition to control the imprinting, is also involved in the differentiation-dependent regulation of Cdkn1c. Interestingly, we observed that MyoD also induces the expression of at least another gene of the domain, Kcnq1, again from the maternal allele. Moreover, other reports showed that Cdkn1c and Kcnq1 are co-induced in other cell types in response to other factors, such as p73Citation25 and E47,Citation26 suggesting a general role of KvDMR1, a common regulatory element for the 2 genes, in the differentiation-dependent regulation of the entire locus.
During our studies on Cdkn1c induction, we observed that some myogenic cell types are refractory to the induction of Cdkn1c. These cell types, that we termed unresponsive in comparison with the normally responsive cells, represented a very useful experimental tool to highlight the existence of cis-acting constraints preventing Cdkn1c expressionCitation21,22 and to reveal the functional importance of the MyoD binding to KvDMR1.Citation23,24 In particular, we found that, in unresponsive cells, MyoD is unable to interact with KvDMR1-chromatin and promote the release of the CTCF-mediated loop.
In the present work, we investigated the molecular mechanisms that prevent KvDMR1 accessibility to MyoD-dependent regulation. We suggest that the accumulation of H3 lysine 9 dimethylation (H3K9me2) at KvDMR1, by preventing the binding of MyoD and the consequent reorganization of chromatin architecture, plays a critical role in preventing Cdkn1c induction in muscle cells.
Results
5-AZA treatment restores the functional interaction of MyoD with KvDMR1 and the induction of Cdkn1c
To investigate the nature of the constraints underlying the inability to upregulate Cdkn1c, we took advantage of a myogenic cell system based on the muscle conversion of fibroblast cells by exogenously expressed MyoD. This system, in addition to be recognized as a reliable model of both in vitro and in vivo myogenesis,Citation27,28 is particularly suitable for the identification and analysis of MyoD targets. It was during the examination of a number of different fibroblast cell types, showing comparable abilities to undergo apparently normal differentiation in response to MyoD, that we found the occurrence of their differential responsiveness to the induction of Cdkn1c.Citation19,21,22 In the present work, we employed immortalized mouse C57BL/6 as responsive and C3H10T1/2 as unresponsive fibroblasts, the 2 cell types in which we had previously examined in most detail the role of MyoD in regulating the long-range interaction between KvDMR1 and Cdkn1c promoter.Citation23,24 Comparison of the promoterCitation21 and KvDMR1 (Supplementary ) sequences between the 2 cell types revealed a complete identity, thus suggesting the existence of some epigenetic restriction in unresponsive cells.
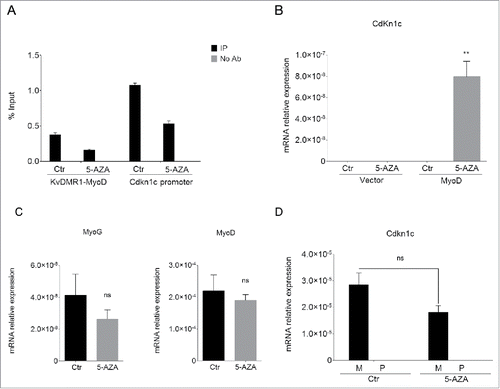
Figure 1. 5-AZA treatment restores Cdkn1c expression in unresponsive cells. (A) qPCR analysis of the MeDIP assay performed on the KvDMR1 sub-region bound by MyoD (KvDMR1-MyoD) and on the Cdkn1c promoter in untreated (Ctr) and 5-AZA-treated samples. Values are expressed as percentages of Input DNA. Error bars indicate standard error of the mean (SEM) for each sample analyzed in triplicate. (B) RT-qPCR in unresponsive cells (C3H10T1/2 fibroblasts) infected with either the empty vector retrovirus (Vector) or the MyoD-encoding retrovirus (MyoD) and treated as above. Statistical significance: P < 0.01 (**). (C) RT-qPCR analysis of myogenin and MyoD expression in unresponsive cells infected with MyoD and treated as above. Values, relative to those of 18S RNA, are the mean of 4 independent experiments ± SEM. Statistical significance: ns (not significant). (D) Allele-specific RT-qPCR analysis of Cdkn1c expression in hybrid fibroblasts (C57BL/6 female x SD7 male) infected with MyoD and treated as above; values, normalized on 18S expression, are the mean of 3 independent experiments ± SEM. Statistical significance: ns. M: maternal; P: paternal.
Cdkn1c is subject to a complex epigenetic regulation involving the cooperation of proximal, such as the promoter region, and distal regulatory elements, such as KvDMR1. DNA methylation is the most understood epigenetic modification, in regards to both the strategy of its propagation through cell divisions and the mechanisms by which it affects transcription.Citation29-31 CpG dinucleotides are the main target of DNA methylation, a modification consisting in the addition of a methyl group on the position 5 of cytosine. DNA methylation of regulatory elements is generally associated with altered gene expressionCitation32, due to direct interference with transcription factor binding and/or recruitment of chromatin modifying enzymes that promote chromatin compaction.Citation33-35 Both Cdkn1c promoter and KvDMR1 are within potentially methylatable CpG islands. In this regard, the use of pharmacological inhibitors of DNA methylation or genetic ablation of individual DNA methyltransferases suggested that the DNA methylation status of the 2 regulatory elements exert opposite effects on Cdkn1c expression. In fact, while promoter and gene body hypermethylation have been implicated in Cdkn1c silencing during imprinting,Citation36 prior to differentiation,Citation21 and in tumor cells.Citation37-40 KvDMR1 hypermethylation, present on the maternal allele, is believed to support maternal-specific Cdkn1c expression. The latter effect has been ascribed to the repression of Kcnq1ot1 promoter.Citation16
To explore whether and how DNA methylation was involved in the molecular mechanism that prevents Cdkn1c induction in unresponsive cells, we analyzed the effects of a pharmacological inhibitor of DNA methylation, 5-Azacytidine (5-AZA) on the MyoD-dependent induction of the gene. Unresponsive fibroblasts were infected with a MyoD-expressing retroviral vector in concomitance with 5-AZA or control treatment. Samples were collected 24 h after the shift to differentiation medium, which promotes MyoD activity, in order to carry out different kinds of analysis in parallel. To check the effect of 5-AZA we first performed a methylated DNA-immunoprecipitation (MeDIP) assay. Genomic DNA was prepared as described in the Materials and Methods section and subject to immunoprecipitation with an anti-5-methylcytidine antibody. Immunoprecipitated DNA was then analyzed by qPCR for the enrichment of the KvDMR1 sub-region bound by MyoD and of Cdkn1c promoter. As expected, 5-AZA treatment decreased the DNA methylation of both regions (). To ascertain whether DNA demethylation allows the restoration of Cdkn1c induction by MyoD, we examined its mRNA levels by RT-qPCR. As reported in , 5-AZA treatment causes an evident rescue of Cdkn1c expression in unresponsive cells. Importantly, the upregulation of Cdkn1c was only evident in the presence of MyoD and not in 5-AZA-treated control cells, supporting the hypothesis that 5-AZA was affecting the MyoD-dependent regulation of Cdkn1c rather than causing the direct de-repression of the gene. Unlike Cdkn1c, the expression of myogenin, that is not associated to a CpG island,Citation41 was not increased; rather, it appeared slightly decreased (). Interestingly, MyoD expression did not change after treatment with 5-AZA (). This was unexpected at first sight, since old seminal studies showed that 5-AZA treatment causes the myogenic conversion of C3H10T1/2 fibroblasts,Citation42,43 as a consequence of endogenous MyoD de-repression.Citation44,45 The apparent discrepancy of our result with previous studies could be easily explained if considering that we analyzed MyoD levels at earlier times after 5-AZA treatment, compared to those employed for detecting muscle-converted cell clones. Moreover, our cells overexpress exogenous MyoD, which constant levels probably mask minor variations caused by 5-AZA at the endogenous gene. Whatever the explanation, this finding further reinforces the claim that it is demethylation of Cdkn1c regulatory elements, and not an increased myogenic conversion, the major cause of the rescue of Cdkn1c expression. In light of the known contribution of promoter DNA methylation in the maintenance of imprinting, and of the previously reported ability of 5-AZA to cause loss of imprinting in uniparental mouse fibroblasts,Citation46 we wondered if the observed rescue of Cdkn1c expression following 5-AZA treatment of unresponsive cells reflected the reactivation of the paternal allele. We could not directly address this question in unresponsive cells due to the unavailability of allele-specific polymorphisms. However, in order to obtain at least indirect evidence of the parental origin of Cdkn1c during differentiation induced by MyoD in the presence of 5-AZA, we decided to take advantage of fibroblast cells derived from hybrid mice (C57BL/6 female X SD7 male), carrying allele-specific single nucleotide polymorphisms (SNPs) in the distal part of chromosome 7 and behaving as responsive.Citation24 Cells were treated as described above for unresponsive cells and Cdkn1c levels were measured by allele-specific RT-qPCR. As reported in , 5-AZA treatment not only did not increase the total Cdkn1c expression levels, consistently with the absence of the epigenetic restriction present in unresponsive cells, but also did not affect at all the paternal Cdkn1c allele. This renders unlikely the possibility that 5-AZA treatment restores the Cdkn1c responsiveness to MyoD by inducing loss of imprinting through promoter demethylation.
The possible effect of DNA demethylation on the functional interaction of MyoD with KvDMR1 was determined by chromatin immunoprecipitation (ChIP) assays. MyoD-bound chromatin was immunoprecipitated from unresponsive cells, either treated with 5-AZA or untreated, and analyzed by qPCR using primers specific for the amplicon indicated in (ChIP/MeDIP amplicon). As shown in , 5-AZA treatment restored the physical interaction of MyoD with KvDMR1. We have previously shown that this interaction, only enabled in responsive cells, accounts for the disruption of the higher-order chromatin structure that constrains Cdkn1c promoter.Citation24 Thus, we wanted to determine whether the rescue of MyoD binding was associated with the rescue of its ability to change the chromatin architecture of the locus. To this end, we performed chromosome conformation capture (3C) assays. Crosslinked chromatin was subject to digestion and ligation, after which hybrid fragments, deriving from the ligation of Cdkn1c promoter and KvDMR1 sequences, were amplified as previously described.Citation23,24 The results reported in show that 5-AZA treatment, in concomitance with decreased DNA methylation and increased MyoD binding, also caused a reduction of the interaction frequency between Cdkn1c promoter and KvDMR1, denoting the release of the chromatin loop. These findings, taken together, indicate that the 5-AZA effects on the MyoD-dependent induction of Cdkn1c are mediated by changes occurring at KvDMR1.
Figure 2. 5-AZA treatment restores the functional interaction of MyoD with KvDMR1. (A) Schematic diagram of the KvDMR1 region (as defined by Fitzpatrick and coworkersCitation11). The narrow vertical bars represent the position of CpG dinucleotides; the horizontal bars indicate the positions of the KvDMR1 sub-region spanning the transcriptional start site and the 3′-portion of Kcnq1ot1 promoter (KvDMR1-ot1) and of the KvDMR1 sub-region bound by MyoD (KvDMR1-MyoD); the small black rectangle indicate the MyoD-binding site; the lower enlargements represent the fragments sequenced after sodium bisulfite treatment (Bisulfite amplicons 1 and 2) or amplified in ChIP and MeDIP assays (ChIP/MeDIP amplicon), as well as the position of the CpG dinucleotides included. (B) ChIP-qPCR analysis of MyoD binding in unresponsive cells (C3H10T1/2 fibroblasts) expressing exogenous MyoD, and treated with 5-AZA or untreated (Ctr). Values obtained for the binding to KvDMR1 were normalized to those for the binding to myogenin promoter, which is a CpG poor region, and expressed as percentages of Input chromatin. The results, derived from 3 independent experiments, are reported as mean ± SEM. Statistical significance: P < 0.01 (**). (C) 3C-qPCR analysis of the KvDMR1-Cdkn1c promoter interaction in cells expressing MyoD and treated as in (A). Values are reported as fold change of the interaction frequencies in the presence of 5-AZA relative to untreated controls (Ctr). The 3C analysis of a constitutive and ubiquitous long-range interaction occurring at the ERCC3 locus was used as a positive control for digestion and ligation of the 3C samples. The results shown for both interactions correspond to the means of 2 independent experiments. Error bars represent SEM.

Responsive and unresponsive cells exhibit differential H3K9me2 at the KvDMR1 sub-region bound by MyoD
Previous studies addressing the genome-wide distribution of differentially methylated regions in muscle respect to other tissues revealed a significant association of these regions with the recognition sites for myogenic bHLH factors.Citation47,48 The MyoD binding site, which we found to be relevant for Cdkn1c induction, maps to a CpG-rich sub-region within KvDMR1 (indicated as KvDMR1-MyoD in ). In light of the observation that 5-AZA treatment restores MyoD binding in unresponsive cells, we asked whether the differential accessibility of MyoD to KvDMR1 in responsive vs. unresponsive cells could involve a differential DNA methylation status.
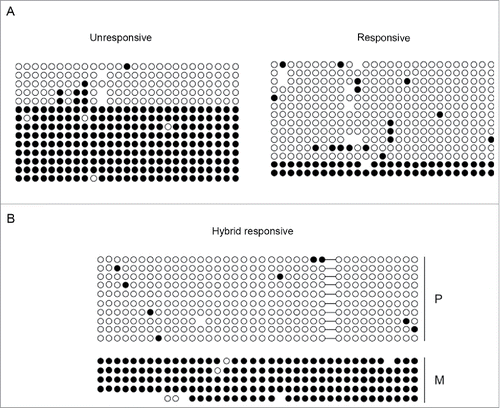
To address this issue, we performed a bisulfite sequence analysis. Genomic DNA from unresponsive and responsive cells, grown in the absence of exogenous MyoD expression, was treated with bisulfite and amplified with appropriated sets of primers for the fragments indicated in (Bisulfite amplicon). Cloned PCR products were then subjected to sequencing. As reported in , the percentages of methylated CpG sites, calculated in relation to the total CpGs of the region analyzed, appear significantly lower in responsive than in unresponsive cells. However, the presence of clones appearing either completely methylated or completely un-methylated, rather than partially methylated, suggested that the KvDMR1 sub-region bound by MyoD, like the adjacent sub-region that contains the Kcnq1ot1 promoter, considered critical for the imprinting of the cluster,Citation16 can be marked by allele-specific methylation. We could not determine the parental origin of the sequenced clones due to the lack of parental-specific polymorphisms. However, in order to address this question, we took advantage of a SNP present in the KvDMR1-MyoD region of C57BL/6 X SD7 fibroblasts. These cells not only allowed us to distinguish the parental alleles, but also represented an additional responsive cell line useful for verifying the results. As reported in , bisulfite sequencing of a larger region (Bisulfite amplicon 2) of KvDMR1-MyoD revealed the same DNA methylation pattern, with clones either completely methylated or completely un-methylated; even though in this case the percentage of methylated clones was higher respect to the other responsive cell line. More importantly, we found that the completely un-methylated clones are all of paternal origin, while the completely methylated clones are of maternal origin. It is likely that the same is true for the bisulfite-sequenced clones reported in and that the lower levels of DNA methylation observed in responsive cells reflected a bias toward the selection of paternal-specific clones, due to some technical reason.
Figure 3. Differential methylation at the KvDMR1 sub-region bound by MyoD correlates with the imprinting status rather than with the differential responsiveness. (A) Bisulfite sequence analysis showing the methylation status of the 27 CpG dinucleotides included in the Bisulfite amplicon 1 shown in A, in unresponsive (C3H10T1/2 fibroblasts) and responsive cells (C57BL/6 fibroblasts). Each circle indicates an individual CpG dinucleotide. Each row of circles corresponds to an individual clone of the bisulfite-PCR product. Empty and filled circles indicate unmethylated and methylated CpGs, respectively. Absent circles represent ambiguous data. (B) Allele-specific bisulfite sequence analysis in hybrid fibroblasts (C57BL/6 female x SD7 male) showing the methylation status of the 38 CpGs located in the Bisulfite amplicon 2 shown in . The SNP in this region causes the loss of a CpG on the paternal (P) respect to the maternal (M) allele. The gap is represented by a dash.
In light of the existence of a bidirectional cross-talk between DNA methylation and repressive histone modifications,Citation49,50 we reasoned that the epigenetic feature directly responsible for the differential MyoD binding could involve some histone modification, which would have been removed alongside DNA methylation after 5-AZA treatment. We focused our attention on H3K9me2, a modification catalyzed by G9a and G9a-like methyltransferasesCitation51, which have been recently recognized to play important roles in myoblast proliferation and differentiation.Citation52 In addition, H3K9me2 and DNA methylation are known to be coordinated by a complex and bidirectional relationship involving the recognition of DNA methylation by the histone methylation machinery, and vice versa.Citation53 Furthermore, treatment with DNA demethylating agents was reported to cause reduction of H3K9me2 (but not H3K9me3) levels at different regulatory regions.Citation54,55 These considerations prompted us to determine H3K9me2 levels at the KvDMR1-MyoD binding region in unresponsive compared to responsive cells and their possible reduction following 5-AZA treatment. As reported in the global levels of H3K9me2 are equivalent in the 2 cell types, as determined by Western blot analysis using an antibody specific for H3K9me2. In contrast, as shown in , the enrichment of the modification at the KvDMR1-MyoD binding region was significantly higher in unresponsive vs. responsive cells, as determined by ChIP assays performed with the primers for the ChIP/MeDIP amplicon indicated in . Unlike KvDMR1, the Cdkn1c promoter region did not appear significantly different in the 2 cell types, just as previously observed for DNA methylation;Citation21 rather, it showed slightly higher levels in responsive vs. unresponsive cells. This finding further highlights the role of KvDMR1, rather than the promoter, as the genomic region directing the epigenetic constraint that prevents Cdkn1c induction. Interestingly, a differential accumulation of H3K9me2 was also present on the adjacent KvDMR1 sub-region containing the Kcnq1ot1 promoter. However, the comparison of Kcnq1ot1 levels in responsive and unresponsive cells did not reveal any difference (Supplementary Fig. 2), thus excluding that the Cdkn1c unresponsiveness to MyoD involves the overexpression of the long noncoding RNA. Next, we determined whether the 5-AZA effects on the rescue of Cdkn1c expression were associated to a reduction of H3K9me2, in concomitance with DNA methylation levels, at the KvDMR1-MyoD region. Unresponsive cells, treated with 5-AZA as above, were collected for either genomic DNA or chromatin preparation. Bisulfite sequencing was performed both to confirm the MeDIP results and to determine the methylation status of the region at the single CpG level. As reported in , 5-AZA treatment caused a moderate but significant reduction of DNA methylation at the KvDMR1-MyoD region. Of course, DNA demethylation appeared to concern the completely hypermethylated clones that presumably represent, as suggested above, the maternal allele driving Cdkn1c expression. As expected, and according to the MeDIP assay, Cdkn1c promoter, which is equally hypermethylated in responsive and unresponsive cells,Citation21 also underwent moderate DNA demethylation throughout the region ().
Figure 4. Responsive and unresponsive cells display differential H3K9me2 at KvDMR1. (A) Western blot assay showing the total level of H3K9me2 in unresponsive (C3H10T1/2 fibroblasts) and responsive cells (C57BL/6 fibroblasts). Histone H3 was used as a loading control. (B) ChIP-qPCR analysis of H3K9me2 enrichment at the 2 KvDMR1 sub-regions and Cdkn1c promoter (Cdkn1c prom) in unresponsive and responsive cells. Actin promoter (Actin prom) was used as negative control. Values obtained for the enrichment were normalized to those on β-globin promoter, used as an invariant control, and expressed as percentages of Input chromatin. The results, derived from 2 independent experiments, are reported as mean ± SEM. Statistical significance: P < 0.05 (*). ns: not significant.
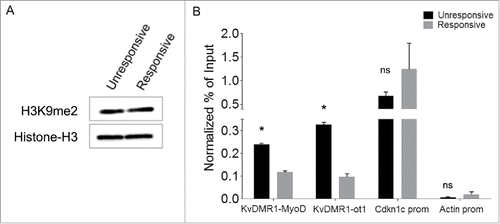
Figure 5. 5-AZA treatment reduces both DNA methylation and H3K9me2 at KvDMR1 in unresponsive cells. (A) The left panel shows the bisulfite sequence analysis of the same KvDMR1-MyoD sub-region analyzed in , performed in unresponsive cells (C3H10T1/2 fibroblasts) treated with 5-AZA. The results are reported as explained in . The right panel shows quantitative analysis of the DNA methylation percentages at the entire sub-region (KvDMR1-MyoD) in 5-AZA-treated cells respect to the control (Ctr). Values refer to the data shown in , for Ctr, and in for 5-AZA samples. Error bars represent SEM. Statistical significance: P < 0.001 (***). (B) The left panel shows the bisulfite sequence analysis of the Cdkn1c promoter, performed in unresponsive cells (C3H10T1/2 fibroblasts) treated with 5-AZA. The results are reported as explained in . The right panel shows the quantitative analysis of DNA methylation percentages in 5-AZA-reated respect to control sample; Values refer to our previously reported dataCitation21 for control and to those shown in for 5-AZA samples. Error bars represent SEM. Statistical significance: P < 0.001 (***). (C) ChIP-qPCR analysis of H3K9me2 enrichment at the 2 KvDMR1 sub-regions and Cdkn1c promoter (Cdkn1c prom) in untreated (Ctr) and 5-AZA treated unresponsive cells. β-globin promoter (β-globin prom) and actin promoter (Actin prom) were used as positive and negative controls respectively. Values obtained for the enrichment are the mean of 2 independent experiments and are expressed as percentages of Input chromatin. Error bars represent SEM. Statistical significance: P < 0.05 (*); ns: not significant.
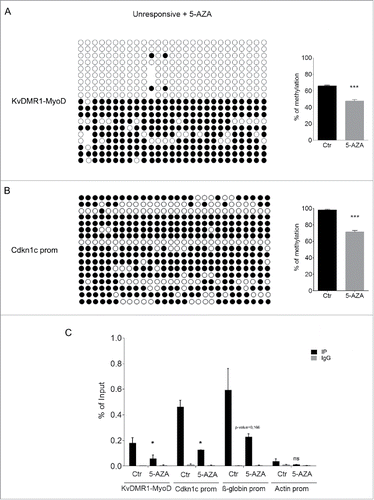
In parallel, 5-AZA-treated and control cells were analyzed by ChIP for H3K9me2. As reported in , and according to the previously reported influence of DNA methylation on this modification, we found that 5-AZA treatment caused a significant reduction of H3K9me2 at several regulatory regions and, most remarkably, at the same KvDMR1 region where it promotes the restoration of MyoD binding. These findings, taken together, support the conclusion that the epigenetic constraint that prevents Cdkn1c induction in unresponsive cells is related to the presence of increased levels of H3K9me2 at KvDMR1 and that DNA demethylation, by interfering with the maintenance of H3K9 methylation, leads to the removal of this constraint.
Differential H3K9me2 enrichment marks KvDMR1 in cancer cells differentially expressing Cdkn1c
Mouse and human KvDMR1 show a similar genomic organization, including the presence of E-box elements adjacent to CTCF binding sites.Citation23 Cdkn1c silencing has been observed in several cancer cell types and has been found to contribute to their malignant phenotype.Citation1 A number of studies, addressing the molecular mechanisms underlying Cdkn1c repression, focused on the gain of repressive epigenetic modifications at the promoter or on the loss of KvDMR1 function resulting in maternal expression of Kcnq1ot1 and biallelic silencing of Cdkn1c. In contrast, the possible role of the accumulation of repressive modifications at KvDMR1 in the cancer cell context has never been considered.
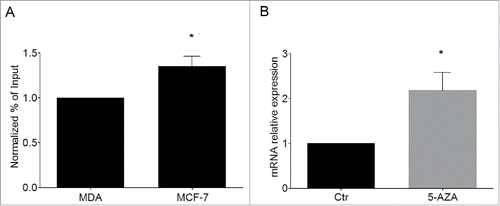
We started to explore this issue by analyzing the H3K9me2 status of KvDMR1 in 2 human breast cancer cell lines, MCF-7 and MDA-MB-231, expressing very low and high Cdkn1c levels, respectively.Citation56 ChIP assays were performed on the 2 cell types using the anti-H3K9me2 antibody and a set of primers encompassing a human KvDMR1 sub-region containing a putative bHLH binding site. As reported in , a small but significant enrichment of H3K9me2 is present in MCF-7 compared to MDA-MB-231 cells, reminiscent of the difference observed between unresponsive and responsive fibroblast cells. Interestingly, as reported in , the similarity also extends to the 5-AZA-response in MCF-7 cells, in which Cdkn1c is upregulated upon treatment.
Figure 6. Cdkn1c silencing in human cancer cells correlates with H3K9me2 enrichment at KvDMR1. (A) ChIP-qPCR analysis of H3K9me2 enrichment at KvDMR1 in MCF-7 and MDA-MB-231 (MDA). Values obtained for the enrichment were normalized to those of APOC-III promoter, used as a positive control, and expressed as percentages of Input chromatin. The results, derived from 2 independent experiments, are reported as mean ± SEM. Statistical significance: P < 0.05 (*). (B) RT-qPCR analysis of Cdkn1c expression in MCF-7 cells untreated (Ctr) or 5-AZA treated samples. Values, relative to those of GAPDH RNA, are the mean of 3 independent experiments ± SEM. Statistical significance: P < 0.05 (*).
These results support the hypothesis that similar mechanisms, involving repressive chromatin modifications at KvDMR1, may participate both in Cdkn1c control during muscle differentiation and Cdkn1c silencing in cancer cells.
Discussion
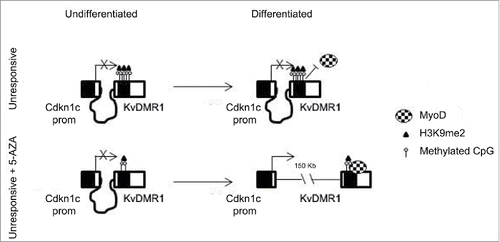
Using a model system previously characterized in our lab, a pair of responsive and unresponsive cell types, we obtained evidence that the presence of repressive chromatin modifications at the KvDMR1 sub-region bound by MyoD represents a critical determinant of the response of Cdkn1c to MyoD-dependent induction. In light of the observation that 5-AZA treatment of unresponsive cells restores the chromatin pathway involving the binding of MyoD to KvDMR1, the disruption of the repressive long-range interaction of KvDMR1 with Cdkn1c promoter and the upregulation of the gene, we first studied the methylation status of KvDMR1. Interestingly, however, we found that the key epigenetic feature underlying the differential responsiveness of the 2 cell types to the MyoD-dependent induction of Cdkn1c is the differential enrichment of H3K9me2, rather than DNA methylation, at KvDMR1 (see model depicted in )
Figure 7. Schematic model of the mechanism by which 5-AZA restores the functional interaction of MyoD with KvDMR1 and the rescue of Cdkn1c induction in unresponsive cells. In unresponsive cells MyoD cannot bind to KvDMR1, due to the presence of a repressive chromatin conformation involving H3K9me2. The failed MyoD binding does not allow the disruption of the repressive chromatin contact between KvDMR1 and Cdkn1c promoter and Cdkn1c is not induced. After 5-AZA treatment not only DNA methylation but also H3K9me2 levels decrease at KvDMR1, allowing MyoD binding to KvDMR1 and causing the release of the chromatin loop and thus induction of Cdkn1c.
The function of the dimethyl mark on H3K9 has been less investigated respect to the trimethyl mark on the same position. However, H3K9me2 appears generally associated with facultative heterochromatin while H3K9me3 is commonly associated with constitutive heterochromatin.Citation57 Moreover, genome-wide analysis of H3K9me2 distribution revealed that this modification is present at repressed genes during development and differentiation and seems important for gene silencing during lineage restriction.Citation58 The roles of H3K9me2 and G9a, the main enzyme catalyzing this modification, are currently extensively investigated in the muscle differentiation system. In this regard, it has been reported that, in undifferentiated myoblasts, H3K9me2 represses the promoters of some MyoD target genes, such as the muscle-specific gene myogeninCitation59 and the cell cycle exit genes Cdkn1a (coding for the cdk inhibitor p21cip1) and Rb1.Citation52 In the case of myogenin promoter, the molecular mechanism of repression would involve the interference with the activity of chromatin-bound MyoD through the cooperation with other histone modifications.Citation60 It has also been reported that G9a, in addition to methylate histone H3, also methylates the MyoD protein,Citation61 indicating that the relationship between G9a, H3K9me2, and MyoD functions is very complex. On the other hand, we found that the presence of high levels of H3K9me2 at KvDMR1 correlates with the failure of MyoD to access chromatin, thus suggesting an additional way by which this histone modification affects MyoD-dependent tran-scription.
H3K9me2 chromatin targeting can be mediated by several mechanisms involving sequence-specific transcription factors interacting with histone methyltransferases, DNA methylation, other chromatin-binding complexes, and noncoding RNAs,Citation57 suggesting that additional epigenetic features underpin the modification we have highlighted in the present work. It is worth mentioning that Kcnq1ot1 has been found to interact with G9a and other histone methyltransferases as well as with DNA methyltransferase 1.Citation17 As mentioned above, Kcnq1ot1 is expressed at comparable levels in unresponsive vs. responsive cells. Although this finding would indicate that Kcnq1ot1 does not play a primary role in determining the responsiveness to MyoD-dependent induction of Cdkn1c, we cannot exclude that the long noncoding RNA could differentially interact with the Cdkn1c locus in the 2 cell types.
The differential enrichment of H3K9me2 at KvDMR1 in responsive and unresponsive cells does not correspond to a pre-existing differential DNA methylation. In fact, the allele-specific analysis of bisulfite-sequenced clones from a cell line polymorphic for maternal and paternal KvDMR1 revealed that their DNA methylation status reflected their parental origin. The reduction of H3K9me2 levels in response to DNA demethylating agents has been already observed in previous studies concerning the epigenetic reactivation of tumor suppressor genes.Citation62,63 This effect was ascribed both the removal of a platform for the recruitment of histone methyltransferases (through methylated CpG binding proteins) and to a more indirect mechanism involving a DNA demethylation-independent decrease of G9a, the main enzyme promoting H3K9me2 deposition. Our finding that 5-AZA treatment concomitantly reduces both H3K9me2 and DNA methylation levels suggests that DNA demethylation is directly involved in the observed histone demethylation.
We recognize that the 5-AZA-induced rescue of Cdkn1c expression could involve modifications not only at KvDMR1, but also at the promoter region. However, we did not observe Cdkn1c expression after treatment with 5-AZA in the absence of MyoD, despite promoter demethylation. This suggests that the main determinant of the restored Cdkn1c expression is the restored MyoD binding to KvDMR1. It is worth to underline that the epigenetic changes compatible with MyoD binding do not influence the imprinting status of Cdkn1c, which is exclusively expressed from the maternal allele. This supports the conclusion that the KvDMR1 sub-region bound by MyoD also plays regulatory roles other than the imprinting control.
Remarkably, we found differential enrichment of H3K9me2 at KvDMR1 also in 2 human breast cancer cell lines, MCF-7 and MDA-MB-231, differentially expressing Cdkn1c. Moreover, MCF-7 cells, in which Cdkn1c is repressed, show upregulation of the gene following 5-AZA treatment. These observations not only support the results obtained in the muscle differentiation system, but also extend the importance of the proposed model. In light of previous findings showing that other bHLH proteins, such as Mash2, E47, and ID2, regulate Cdkn1c transcription in neural cells,Citation26,64 we hypothesize that a shared regulatory mechanism, involving a long-range interaction of KvDMR1 with Cdkn1c promoter and its reorganization upon binding of bHLH factors, may participate in Cdkn1c repression and de-repression in more than one cell type.
In summary, this work highlights a still uncharacterized mechanism of Cdkn1c silencing, pointing to the epigenetic status of KvDMR1 as a critical feature determining the availability of this CKI for induction by regulatory factors. Importantly, the KvDMR1 sub-region involved in the differentiation-dependent regulation of Cdkn1c appears functionally distinct from that involved in the imprinting control. These results could have an interesting follow up in the fields of cancer and BWS research. It will be compelling to investigate if H3K9me2 enrichment at the regulatory element identified in this work is also present in BWS patients and in other cancer types where Cdkn1c is not expressed. If this were the case, the epigenetic status of this region could be used as a prognostic or diagnostic marker.
A more detailed knowledge of the complex roles of DNA and histone methylation at KvDMR1 not only will clarify the molecular mechanisms underlying the restriction of Cdkn1c expression during development and its silencing in some pathologies, but also will provide a more integrated picture of the effects of using epigenetic drugs in attempts to restore Cdkn1c expression.
Materials and methods
Cell cultures
Mouse C3H10T1/2 (unresponsive), C57BL/6 (responsive), C57BL/6 x SD7 (responsive) fibroblasts, MCF-7, and MDA-MB-231 breast cancer cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). Production of MyoD-expressing retrovirus and retroviral infections were performed as previously described.Citation21 MyoD activity was stimulated by shifting cell cultures to low serum-containing differentiation medium (DMEM supplemented with 0.5% FBS). Five-AZA treatment of unresponsive fibroblasts and MCF-7 cells was performed by adding 4 µM 5-AZA (Sigma-Aldrich) to the culture medium. The 5-AZA-containing medium was changed every day for 4 d.
MeDIP assay
Genomic DNA was extracted and purified by standard procedures. DNA was sonicated to obtain fragments of an average length of 300–600 bp. Sonicated DNA (6 µg) was resuspended in 500 µL of Immunoprecipitation Buffer (Ip Buffer; 10 mM sodium phosphate pH = 7, 140 mM NaCl, 0.05% Triton X-100). DNA was denatured at 95°C and then incubated with 5 µg of 5-Methylcytidine antibody (Bi-Mecy 0100, Eurogentec) or without antibody (No Ab sample) overnight at 4°C. Protein G magnetic beads (40 µL) were added to each sample and incubated 4 h at 4°C. Before washing, the supernatant of the no antibody control was taken as Input sample. After 3 washes with Ip Buffer, samples were treated with proteinase K and incubated 2 h at 56°C and overnight at 37°C. DNA was extracted with phenol-chloroform, precipitated with ethanol, and re-suspended in 50 µl of distilled water. Quantitative PCR (qPCR) analyses of immunoprecipitated KvDMR1 and Cdkn1c promoter regions were performed each in triplicate using 5 ng of DNA, GoTaq qPCR Master Mix (Promega) and the primer pairs: KvDMR1, 5′-GCACAAGTCGCAAGTCCGCG-3′ and 5′-ATGGAGCCCAGCCGCGAAAG-3′, specific for the KvDMR1-MyoD sub-region indicated in ; Cdkn1c prom, 5′ACTGAGAGC-AAGCGAACAGG and 5′ACCTGGCTGATTGGTGATGG. The reactions were performed in the “CFX Connect Real Time system” termocycler (Bio-Rad).
Bisulfite modification and sequencing
Sodium bisulfite treatment was carried out as previously describedCitation21 with some modifications. DNA (5 μg), previously digested with BamHI, was denatured with 0.3 M NaOH at 42°C for 20 min. Bisulfite reactions were performed in 5.8 M urea, 3.7 M sodium bisulfite and 6.67 mM hydroquinone at 55°C for 4 h. The DNA was desalted using Wizard Clean-Up system (Promega). The reactions were terminated by further incubation in 0.3M NaOH at 37°C for 20 min. Samples were co-precipitated with 5 µg of molecular biology grade glycogen in sodium acetate/ethanol at −20°C for 16 h. Pellets were then resuspended in 50 µl of distilled water. Modified DNA was used as a template in PCR reactions using the following primers: For bisulfite amplicon 1: F, 5′-TTGGAGAGTTTT-TTTGTTTAGTTTG and R, 5′CAAAACCACCC-CTACTTCTATAAAC, which amplify the fragment indicated in . Amplification consisted of one cycle at 95°C for 5 min followed by 35 cycles at 95°C for 30 sec, 56°C for 60 sec and 72°C for 60 sec. For Bisulfite amplicon 2: F, 5′-GATTTTTATGGTGAGG-TTTTA-3′ and R, 5′-CAAAACCACCCCTACTTCTAT-3′ that amplify the fragment indicated in . Amplification consisted of one cycle at 95°C for 5 min followed by 35 cycles at 95°C for 30 sec, 57°C for 60 sec and 72°C for 60 sec. For Cdkn1c promoter: F, 5′-TGTTGAAATTGAAAATATTATATTATGTTA-3′ and R, 5′- TAAATAAAA-CCCCTTACACAAC-CC-3′. Amplification consisted of one cycle at 95°C for 5 min followed by 35 cycles at 95°C for 30 sec, 54°C for 60 sec and 72°C for 60 sec.
PCR products were cloned into pGEM-T Easy vector using TA cloning kit (Promega) and transformed into E. cloni 5-α chemically competent E. coli cells (Lucigen). A number of positive clones were randomly selected for automated sequencing.
ChIP assays
MyoD ChIP assays were carried out as described.Citation21 Chromatin was immunoprecipitated with anti-MyoD antibody (sc-760; Santa Cruz Biotechnology, Inc.) or with normal rabbit IgG (12–370; Merk Millipore). qPCR analysis was performed in triplicate for each of 3 independent experiments, using 5 ng of DNA, Go Taq qPCR Master Mix (Promega), and the following primer pairs: F, 5′-GCACAAGTCGCAAGTCCGCG-3′ and R, 5′-ATGGAGCCCAGCCGCGAAAG-3′ specific for the KvDMR1-MyoD sub-region indicated in ; 5′-TGGCAGGAACAAGCC-TT-TTGCGA-3′ and 5′-AGTCCGCTCATAGCCCG-GGG-3′ for myogenin promoter.
Histone-ChIP assays were performed using 50 µg of chromatin for sample and 5 µg of specific antibody anti dimethyl-histone H3K9 (Millipore, 05–1249) or with normal mouse IgG Antibody (Millipore, 12–371) . Each sample was diluted 10-fold with dilution buffer (5 mM EDTA, 50 mM Tris-HCl pH8.0, 0.5%NP40, 200 mM NaCl, and protease and phosphatase inhibitors). Samples were incubated overnight with gently rotation at 4°C. Before washing, the supernatant of the IgG sample was taken as Input. Each sample was washed twice with Low Salt buffer (0.1% SDS, Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl pH = 8.1, 150 mM NaCl) and with High Salt buffer (0.1% SDS, Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl pH = 8.1, 500 mM NaCl); one time with LiC Buffer (0.25 M LiCl, 1% NP40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH = 8,1) and TE Buffer (10 mM Tris-HCl, 1 mM EDTA pH = 8).
Samples were eluted with Elution Buffer (1x TE, 0.5% SDS), treated with RNAse A for 10 min at room temperature and with Proteinase K for 5 h at 62°C. DNA was extracted with phenol-chloroform, precipitated with ethanol, and re-suspended in 50 μl of distilled water. After immunoprecipitation, the DNA quantity of each sample was measured. The different regions were amplified using the same DNA amount with the following primers: F, 5′-GCACAAGTCGCAAGTCCGCG-3′ and R, 5′-ATGGAGCCCAGCCGCGAAAG-3′, specific for the KvDMR1-MyoD sub-region and F, 5′-GGCTGCCACGTCACCAA-3′ and R, 5′-CCTGACTGGACCAAAATGCA-3′, for KvDMR1-ot1 sub-region; F, 5′GAAGCCTGATTCCGTAGA-GC-3′ and R, 5′-CAACTGATCCTACCTCACCTTATATGC-3′ for β-globin promoter; F, 5′-GTGACATCCACACCCAGA-GG-3′ and R, 5′GAATAGCCTCCGCCCTTG-3′, for Actin promoter:; F, 5′-GGAGGAGGGGTCATTCTCTC-3′ and R, 5′-GACAGGGGTCACATCCATTC-3′ for Cdkn1c promoter; F, 5′-CCGTTCTGCCTGGAGACTG-3′ and R, 5′-AATGACAGGACACGGCACAT-3′, for hKvDMR1 E-box; F, 5′-CGGGACAGCAGCCTGGATT-3′ and R, 5′-GCTCCTCTTGCCCCT-CTTCAT-3′, for human APOC-III promoter.
Histone extraction and Western blot
Cells were harvested and washed twice with ice-cold PBS, then resuspended with Buffer A (20 mM Hepes pH = 7.9, 1 M KCl, 0.2 M EDTA pH = 7, 0.1 M EGTA pH = 8, 1 mM DTT and protease and phosphatase inhibitors). Cells were incubated in ice for 15 min, 0.6% NP40 was added and the solution was gently stirred. After centrifugation, the pellet was washed with Buffer A and resuspended with cold 0.4 M HCl. The extracted nuclei were incubated in ice for 1 h and then centrifuged. The supernatant was precipitated overnight with 5 volumes of cold acetone. After centrifugation, the pellet was washed with acetone, air dried, and resuspended with water. Proteins (15 µg) were loaded on a 15% polyacrylamide gel and transferred to nitrocellulose membranes by electroblotting. Membranes were blocked with 5% non-fat dry milk in PBS containing Tween 20 at room temperature and incubated with the primary antibody overnight at 4°C with anti-histone H3 (Millipore, 07–690) diluted 1:1000 and anti-dimethyl-histone H3K9 (Millipore, 05–1249) diluted 1:4000. After washing 3 times with TBST solution, membranes were incubated in a 1:10000 or 1:20000 dilution of peroxidase-conjugated anti-mouse or anti-rabbit antibodies for 1 h (Bio-Rad) at room temperature. Proteins were detected using the ECL chemiluminescence system (Cyanagen).
3C assays
Crosslinking, nuclei isolation, and DNA digestion and ligation were performed as previously described.Citation23,24 qPCR was performed in triplicate for each of 2 independent experiments with the following primers: 5′-CCTTCGACCATGGTGAGG-TC-3′, 5′GTGCTGAAACGATCCACACG-3′ for KvDMR1-Cdkn1c; 5′-ATGGCCTGAAGAAACCGC-3′, 5′-CTTAGGCA-ACACACTCAAGC-3′ for ERCC3 locus.
Gene expression analysis
Total cellular RNA was extracted using the High Pure RNA Isolation Kit according to the manufacturer's instructions (Roche Diagnostics) and 500 ng of total RNA was reverse transcribed with the iScript cDNA Synthesis Kit (Bio-Rad). Diluted cDNAs were subjected to qPCR with the following primer sets: 5′-AACTTCCAGCAGGATGTGCC-3′ and 5′-CATCCACTGCAGACGACCAG-3′ for Cdkn1c; 5′-GTCTCTTCCTGAAGCCAGTTGCG-3′ and 5′-TGCAAATGCTTGGCCCCCAGAG-3′ for myogenin; 5′-CCCGGCGGCAGAATGGCTAC-3′ and 5′-CTGCCAAAAGCAGCGCAGGC-3′ for MyoD; 5′-ACGA-CCCATTCGAACGTCTG-3′ and 5′-GCACGGCGACTACCA-TCG-3′ for 18S (used as a housekeeping gene); F, 5′-TTCTG-GAGGCGATTGAGGC-3′ and R, 5′-AGCAACCAGAACCA-GGTGAGAG-3′ for Kcnq1ot1; F, 5′-CAGATCTGACCTCA-GACCCA-3′ and R, 5′-CCTGTTCCTCGCCGTCCC-3′ for the maternal allele of Cdkn1c; F, 5′-CAGATCTGACCTCAGA-CCCG-3′ and R, 5′-GACCTGTTCCTCGCCATCCT-3′ for the paternal allele of Cdkn1c; F, 5′-GGACGAGACAGGCGAA-CCC-3′ and R, 5′-TCTGCGTGTGCGAGGGAC-3′ for human Cdkn1c; F, 5′-GACAGTCAGCCGCATCTTCT-3′ and R, 5′-GCGCCCAATACGACCAAATC3′ for human GAPDH.
Statistical analysis
For statistical analysis, comparisons were performed using parametric Student's t-test analysis or non-parametric Mann-Whitney U-tests, depending on the normality of the distribution, as assessed using the Kolmogorov-Smirnov statistics. Statistical significance is shown as *P < 0.05 or ** P < 0.01 or P < 0.001 (***)
For the quantitative analysis of bisulfite treated samples the percentage of methylation of each CpG was calculated and paired t-test was performed to compare control and 5-AZA samples.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1230576_s02.doc
Download MS Word (104 KB)Acknowledgements
We thank Prof. Paolo Amati for critical reading of the manuscript and Dr. Francesca Matteini for helpful discussions.
Funding
This work was supported by grants from Italian Ministry of University and Research - Basic research investment fund (MIUR/FIRB) - collaboration Istituto Pasteur-Fondazione Cenci Bolognetti/Institut Pasteur Paris. M.N. Rossi is holder of a fellowship from Fondazione Adriano Buzzati-Traverso.
Related Research Data
References
- Pateras IS, Apostolopoulou K, Niforou K, Kotsinas A, Gorgoulis VG. p57KIP2: “Kip”ing the cell under control. Mol Cancer Res 2009; 7:1902-19; PMID:19934273; http://dx.doi.org/10.1158/1541-7786.MCR-09-0317
- Yan Y, Frisen J, Lee MH, Massague J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 1997; 11:973-83; PMID:9136926; http://dx.doi.org/10.1101/gad.11.8.973
- Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997; 387:151-8; PMID:9144284; http://dx.doi.org/10.1038/387151a0
- Eggermann T, Binder G, Brioude F, Maher ER, Lapunzina P, Cubellis MV, Bergada I, Prawitt D, Begemann M. CDKN1C mutations: two sides of the same coin. Trends Mol Med 2014; 20:614-22; PMID:25262539; http://dx.doi.org/10.1016/j.molmed.2014.09.001
- Lee MH, Reynisdottir I, Massague J. Cloning or P57(Kip2), a Cyclin-Dependent Kinase Inhibitor with Unique Domain-Structure and Tissue Distribution. Genes Dev 1995; 9:639-49; PMID:7729683; http://dx.doi.org/10.1101/gad.9.6.639
- Matsuoka S, Edwards MC, Bai C, Parker S, Zhang PM, Baldini A, Harper JW, Elledge SJ. P57(Kip2), a Structurally Distinct Member of the P21(Cip1) Cdk Inhibitor Family, Is a Candidate Tumor-Suppressor Gene. Genes Dev 1995; 9:650-62; PMID:7729684; http://dx.doi.org/10.1101/gad.9.6.650
- Susaki E, Nakayama K, Yamasaki L, Nakayama KI. Common and specific roles of the related CDK inhibitors p27 and p57 revealed by a knock-in mouse model. Proc Nat Acad Sci U S A 2009; 106:5192-7; PMID:19276117; http://dx.doi.org/10.1073/pnas.0811712106
- Nagahama H, Hatakeyama S, Nakayama K, Nagata M, Tomita K, Nakayama K. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27(Kip1) and p57(Kip2) during mouse development. Anat Embryol 2001; 203:77-87; PMID:11218061; http://dx.doi.org/10.1007/s004290000146
- Hatada I, Mukai T. Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse. Nat Genet 1995; 11:204-6; PMID:7550351; http://dx.doi.org/10.1038/ng1095-204
- Matsuoka S, Thompson JS, Edwards MC, Bartletta JM, Grundy P, Kalikin LM, Harper JW, Elledge SJ, Feinberg AP. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Nat Acad Sci U S A 1996; 93:3026-30; PMID:8610162; http://dx.doi.org/10.1073/pnas.93.7.3026
- Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 2002; 32:426-31; PMID:12410230; http://dx.doi.org/10.1038/ng988
- Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 2004; 36:1291-5; PMID:15516931; http://dx.doi.org/10.1038/ng1468
- Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Nat Acad Sci U S A 1999; 96:8064-9; PMID:10393948; http://dx.doi.org/10.1073/pnas.96.14.8064
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 2004; 36:1296-300; PMID:15516932; http://dx.doi.org/10.1038/ng1467
- Fitzpatrick GV, Pugacheva EM, Shin JY, Abdullaev Z, Yang Y, Khatod K, Lobanenkov VV, Higgins MJ. Allele-specific binding of CTCF to the multipartite imprinting control region KvDMR1. Mol Cell Biol 2007; 27:2636-47; PMID:17242189; http://dx.doi.org/10.1128/MCB.02036-06
- Mancini-DiNardo D, Steele SJS, Ingram RS, Tilghman SM. A differentially methylated region within the gene Kcnq1 functions as an imprinted promoter and silencer. Hum Mol Genet 2003; 12:283-94; PMID:12554682; http://dx.doi.org/10.1093/hmg/ddg024
- Kanduri C. Kcnq1ot1: A chromatin regulatory RNA. Semin Cell Dev Biol 2011; 22:343-50; PMID:21345374; http://dx.doi.org/10.1016/j.semcdb.2011.02.020
- Demars J, Gicquel C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith-Wiedemann and Silver-Russell syndromes. Clin Genet 2012; 81:350-61; PMID:22150955; http://dx.doi.org/10.1111/j.1399-0004.2011.01822.x
- Figliola R, Maione R. MyoD induces the expression of p57Kip2 in cells lacking p21Cip1/Waf1: overlapping and distinct functions of the two cdk inhibitors. J Cell Physiol 2004; 200:468-75; PMID:15254975; http://dx.doi.org/10.1002/jcp.20044
- Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 2005; 16:585-95; PMID:16099183; http://dx.doi.org/10.1016/j.semcdb.2005.07.006
- Figliola R, Busanello A, Vaccarello G, Maione R. Regulation of p57(KIP2) during muscle differentiation: role of Egr1, Sp1 and DNA hypomethylation. J Mol Biol 2008; 380:265-77; PMID:18513743; http://dx.doi.org/10.1016/j.jmb.2008.05.004
- Vaccarello G, Figliola R, Cramerotti S, Novelli F, Maione R. p57Kip2 is induced by MyoD through a p73-dependent pathway. J Mol Biol 2006; 356:578-88; PMID:16405903; http://dx.doi.org/10.1016/j.jmb.2005.12.024
- Battistelli C, Busanello A, Maione R. Functional interplay between MyoD and CTCF in regulating long-range chromatin interactions during differentiation. J Cell Sci 2014; 127:3757-67; PMID:25002401; http://dx.doi.org/10.1242/jcs.149427
- Busanello A, Battistelli C, Carbone M, Mostocotto C, Maione R. MyoD regulates p57(kip2) expression by interacting with a distant cis-element and modifying a higher order chromatin structure. Nucleic Acids Res 2012; 40:8266-75; PMID:22740650; http://dx.doi.org/10.1093/nar/gks619
- Balint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57(KIP2) expression by p73 beta. Proc Nat Acad Sci U S A 2002; 99:3529-34; PMID:11891335; http://dx.doi.org/10.1073/pnas.0624-91899
- Rothschild G, Zhao X, Iavarone A, Lasorella A. E Proteins and Id2 converge on p57Kip2 to regulate cell cycle in neural cells. Mol Cell Biol 2006; 26:4351-61; PMID:16705184; http://dx.doi.org/10.1128/MCB.01743-05
- Bergstrom DA, Penn BH, Strand A, Perry RL, Rudnicki MA, Tapscott SJ. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell 2002; 9:587-600; PMID:11931766; http://dx.doi.org/10.1016/S1097-2765(02)00481-1
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 2010; 18:662-74; PMID:20412780; http://dx.doi.org/10.1016/j.devcel.2010.02.014
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25:1010-22; PMID:21576262; http://dx.doi.org/10.1101/gad.2037511
- Espada J, Esteller M. DNA methylation and the functional organization of the nuclear compartment. Semin Cell Dev Biol 2010; 21:238-46; PMID:19892028; http://dx.doi.org/10.1016/j.semcdb.2009.10.006
- Schubeler D. Function and information content of DNA methylation. Nature 2015; 517:321-6; PMID:25592537; http://dx.doi.org/10.1038/nature14192
- Miranda TB, Jones PA. DNA methylation: The nuts and bolts of repression. J Cell Physiol 2007; 213:384-90; PMID:17708532; http://dx.doi.org/10.1002/jcp.21224
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295-304; PMID:19308066; http://dx.doi.org/10.1038/nrg2540
- Clouaire T, Stancheva I. Methyl-CpG binding proteins: specialized transcriptional repressors or structural components of chromatin? Cell Mol Life Sci 2008; 65:1509-22; PMID:18322651; http://dx.doi.org/10.1007/s00018-008-7324-y
- Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev 1993; 3:226-31; PMID:8504247; http://dx.doi.org/10.1016/0959-437X(93)90027-M
- Bhogal B, Arnaudo A, Dymkowski A, Best A, Davis TL. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics 2004; 84:961-70; PMID:15533713; http://dx.doi.org/10.1016/j.ygeno.2004.08.004
- Kikuchi T, Toyota M, Itoh F, Suzuki H, Obata T, Yamamoto H, Kakiuchi H, Kusano M, Issa JP, Tokino T, et al. Inactivation of p57KIP2 by regional promoter hypermethylation and histone deacetylation in human tumors. Oncogene 2002; 21:2741-9; PMID:11965547; http://dx.doi.org/10.1038/sj.onc.1205376
- Kuang SQ, Ling X, Sanchez-Gonzalez B, Yang H, Andreeff M, Garcia-Manero G. Differential tumor suppressor properties and transforming growth factor-beta responsiveness of p57KIP2 in leukemia cells with aberrant p57KIP2 promoter DNA methylation. Oncogene 2007; 26:1439-48; PMID:16936778; http://dx.doi.org/10.1038/sj.onc.1209907
- Li Y, Nagai H, Ohno T, Yuge M, Hatano S, Ito E, Mori N, Saito H, Kinoshita T. Aberrant DNA methylation of p57(KIP2) gene in the promoter region in lymphoid malignancies of B-cell phenotype. Blood 2002; 100:2572-7; PMID:12239171; http://dx.doi.org/10.1182/blood-2001-11-0026
- Shin JY, Kim HS, Park J, Park JB, Lee JY. Mechanism for inactivation of the KIP family cyclin-dependent kinase inhibitor genes in gastric cancer cells. Cancer Res 2000; 60:262-5; PMID:10667572
- Fuso A, Ferraguti G, Grandoni F, Ruggeri R, Scarpa S, Strom R, Lucarelli M. Early demethylation of non-CpG, CpC-rich, elements in the myogenin 5′-flanking region: a priming effect on the spreading of active demethylation. Cell Cycle 2010; 9:3965-76; PMID:20935518; http://dx.doi.org/10.4161/cc.9.19.13193
- Konieczny SF, Emerson CP, Jr. 5-Azacytidine induction of stable mesodermal stem cell lineages from 10T1/2 cells: evidence for regulatory genes controlling determination. Cell 1984; 38:791-800; PMID:6207933; http://dx.doi.org/10.1016/0092-8674(84)90274-5
- Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell 1979; 17:771-9; PMID:90553; http://dx.doi.org/10.1016/0092-8674(79)90317-9
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987; 51:987-1000; PMID:3690668; http://dx.doi.org/10.1016/0092-8674(87)90585-X
- Lassar AB, Paterson BM, Weintraub H. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell 1986; 47:649-56; PMID:2430720; http://dx.doi.org/10.1016/0092-8674(86)90507-6
- El Kharroubi A, Piras G, Stewart CL. DNA demethylation reactivates a subset of imprinted genes in uniparental mouse embryonic fibroblasts. J Biol Chem 2001; 276:8674-80; PMID:11124954; http://dx.doi.org/10.1074/jbc.M009392200
- Chandra S, Baribault C, Lacey M, Ehrlich M. Myogenic differential methylation: diverse associations with chromatin structure. Biology 2014; 3:426-51; PMID:24949935; http://dx.doi.org/10.3390/biology30-20426
- Tsumagari K, Baribault C, Terragni J, Varley KE, Gertz J, Pradhan S, Badoo M, Crain CM, Song L, Crawford GE, et al. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics 2013; 8:317-32; PMID:23417056; http://dx.doi.org/10.4161/epi.23989
- Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2010; 2:657-69; PMID:21339843; http://dx.doi.org/10.2217/epi.10.44
- Torres IO, Fujimori DG. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr Opin Struct Biol 2015; 35:68-75; PMID:26496625; http://dx.doi.org/10.1016/j.sbi.2015.09.007
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 2003; 12:1591-8; PMID:14690610; http://dx.doi.org/10.1016/S1097-2765(03)00479-9
- Rao VK, Ow JR, Shankar SR, Bharathy N, Manikandan J, Wang Y, Taneja R. G9a promotes proliferation and inhibits cell cycle exit during myogenic differentiation. Nucleic Acids Res 2016.
- Cheng X. Structural and functional coordination of DNA and histone methylation. Cold Spring Harb Perspect Biol 2014; 6; PMID:25085914; http://dx.doi.org/10.1101/cshperspect.a018747
- Egger G, Aparicio AM, Escobar SG, Jones PA. Inhibition of histone deacetylation does not block resilencing of p16 after 5-aza-2′-deoxycytidine treatment. Cancer Res 2007; 67:346-53; PMID:17210717; http://dx.doi.org/10.1158/0008-5472.CAN-06-2845
- McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res 2006; 66:3541-9; PMID:16585178; http://dx.doi.org/10.1158/0008-5472.CAN-05-2481
- Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, Miller LD, Tan PB, Yu Q. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. Plos One 2009; 4:e5011; PMID:19340297; http://dx.doi.org/10.1371/journal.pone.0005011
- Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol 2015; 16:499-513; PMID:26204160; http://dx.doi.org/10.1038/nrm4029
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 2009; 41:246-50; PMID:19151716; http://dx.doi.org/10.1038/ng.297
- Ling BM, Gopinadhan S, Kok WK, Shankar SR, Gopal P, Bharathy N, Wang Y, Taneja R. G9a mediates Sharp-1-dependent inhibition of skeletal muscle differentiation. Mol Biol Cell 2012; 23:4778-85; PMID:23087213; http://dx.doi.org/10.1091/mbc.E12-04-0311
- Mal A, Harter ML. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc Nat Acad Sci U S A 2003; 100:1735-9; PMID:12578986; http://dx.doi.org/10.1073/pnas.0437843100
- Ling BM, Bharathy N, Chung TK, Kok WK, Li S, Tan YH, Rao VK, Gopinadhan S, Sartorelli V, Walsh MJ, et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Nat Acad Sci U S A 2012; 109:841-6; PMID:22215600; http://dx.doi.org/10.1073/pnas.1111628109
- Nguyen CT, Weisenberger DJ, Velicescu M, Gonzales FA, Lin JC, Liang G, Jones PA. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res 2002; 62:6456-61; PMID:12438235
- Wozniak RJ, Klimecki WT, Lau SS, Feinstein Y, Futscher BW. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene 2007; 26:77-90; PMID:16799634; http://dx.doi.org/10.1038/sj.onc.1209763
- Kury P, Greiner-Petter R, Cornely C, Jurgens T, Muller HW. Mammalian achaete scute homolog 2 is expressed in the adult sciatic nerve and regulates the expression of Krox24, Mob-1, CXCR4, and p57kip2 in Schwann cells. J Neurosci 2002; 22:7586-95; PMID:12196582