
ABSTRACT
Neuroinflammation plays a critical role in the development of reward-related behavior in cocaine self-administration rodents. Cocaine, one of most commonly abused drugs, has been shown to activate microglia both in vitro and in vivo. Detailed molecular mechanisms underlying cocaine-mediated microglial activation remain poorly understood. microRNAs (miRs) belonging to a class of small noncoding RNA superfamily have been shown to modulate the activation status of microglia. miR-124, one of the microglia-enriched miRs, functions as an anti-inflammatory regulator that maintains microglia in a quiescent state. To date, the possible effects of cocaine on microglial miR-124 levels and the associated underlying mechanisms have not been explored. In the current study, we demonstrated that cocaine exposure decreased miR-124 levels in both BV-2 cells and rat primary microglia. These findings were further validated in vivo, wherein we demonstrated decreased abundance of miR-124 in purified microglia isolated from cocaine-administered mice brains compared with cells from saline administered animals. Molecular mechanisms underlying these effects involved cocaine-mediated increased mRNA and protein expression of DNMTs in microglia. Consistently, cocaine substantially increased promoter DNA methylation levels of miR-124 precursors (pri-miR-124-1 and −2), but not that of pri-miR-124-3, both in vitro and in vivo. In summary, our findings demonstrated that cocaine exposure increased DNA methylation of miR-124 promoter resulting into its downregulation, which, in turn, led to microglial activation. Our results thus implicate that epigenetic modulation of miR-124 could be considered as a potential therapeutic approach to ameliorate microglial activation and, possibly, the development of cocaine addiction.
Introduction
In recent times, the role of microglia in drug addiction has drawn much attention. Emerging evidence also points to the fact that abused drugs including cocaine are capable of activating microglia, exemplified by the release of a plethora of cytokines and chemokines, leading ultimately to impaired cross-talk between glia and neurons, thereby contributing to the drug addiction.Citation1,2 In contrast, pharmacological inhibition and/or genetic silencing of microglial activation blocked cocaine-mediated reward-related behavioral changes in experimental animal models.Citation2,3 To date several signaling pathways have been suggested to be associated with cocaine-mediated neuroinflammation, including upregulation of reactive oxygen species (ROS), ER stress, and ensuing autophagy.Citation4,5 Cocaine has also been reported to directly interact with microglial toll-like receptor 4 (TLR4) leading, in turn, to increased secretion of IL-1β, which ultimately results in enhanced synaptic dopamine concentration in the striatum.Citation2 The detailed mechanism(s) underlying cocaine-mediated microglial activation, however, remain less known.
microRNAs (miR), members of the small noncoding RNA superfamily, are an important class of biological molecules with critical roles in many fundamental cellular processes, such as proliferation, differentiation, and apoptosis.Citation6-8 Mature miRs (∼22 nucleotides) downregulate the levels of their specific target(s) by binding to the 3′-UTR of the mRNA leading, in turn, to mRNA destabilization or translational repression. The brain is a particularly prominent organ wherein miRNAs play important roles in controlling gene expression, microglial quiescence, and neuronal activities.Citation9,10 Dysregulation of the expression levels of miRs, such as miR-155, has been involved in microglial activation.Citation11,12 Cocaine is known to modulate the levels of miRs in the brain to contribute to drug addiction.Citation13,14 Whether cocaine can regulate miRs specifically in microglia and lead to alteration of microglial activation status remains understudied.
miR-124 is a brain-enriched miR that plays key roles in neurogenesis, synaptic signal transmission, and glial-neuronal interactions to maintain brain homeostasis.Citation15 At the cellular level, miR-124 is highly abundant in neurons as well as in microglia. miR-124 is critical for the maintenance of microglia quiescence,Citation16,17 with reports suggesting that overexpression of miR-124 blocked lipopolysaccharide-induced microglial activation.Citation18 Decreased levels of miR-124 have been associated with multiple neuroinflammatory disorders, such as Parkinson disease, dementia, and multiple sclerosis.Citation17,19-21
Cocaine is capable of modulating the levels of targeted genes through epigenetic mechanisms.Citation22,23 Promoter DNA methylation is recognized as one of the important epigenetic regulatory mechanisms.Citation24 DNA methyltransferases (DNMTs), such as DNMT-1, 3a, and 3b, are key proteins responsible for modulating and maintaining promoter DNA methylation levels.Citation25 Alteration of DNA methylation levels at the promoter region has a negative correlation with the level of gene expression. For example, increased promoter DNA methylation leads to decreased gene expression. Alterations in miR promoter DNA methylation levels have been demonstrated to modulate the abundance of miRs.Citation26,27 Interestingly, in multiple tumors, including hepatocellular carcinoma,Citation28 stomach adenocarcinoma,Citation29 and prostate cancer,Citation30 it has been shown that miR-124 levels rely strongly on the DNA methylation of its promoter. Downregulation of miR-124 in these tumors promoted their metastasis and/or development.
In the current study, we explored the role of cocaine in regulating the expression of miR-124, specifically in microglia, and also uncovered the underlying mechanism(s) responsible for this modulation. We provide evidence that cocaine decreased the expression of microglial miR-124 both in vitro and in vivo, through increased promoter DNA methylation of pri-miR-124-1 and -2. These findings suggest a hitherto unexplored mechanism underlying cocaine-mediated microglial activation and provide a potential target for the development of therapeutics aimed at ameliorating neuroinflammation and possibly cocaine addiction.
Results
Cocaine exposure decreased miR-124 and increased DNMTs levels in BV-2 microglial cells
miR-124 is abundantly expressed in microglia, maintaining these cells in a resting state.Citation16,17 To investigate the possible effects of cocaine on microglial miR-124, we exposed BV-2 cells to varying doses of cocaine (1, 10, and 100 µM) for 24 h. Following the treatment, total cellular RNAs were isolated to determine miR-124 expression levels by qRT-PCR. Our findings () showed that cocaine significantly decreased miR-124 levels at both 10 and 100 µM (48.28 ± 5.34% and 53.87 ± 3.05% of control, respectively; P < 0.05) with no effects at lower concentration (1 µM). Next, we performed time course experiments, in which BV-2 cells were exposed to 10 µM cocaine for different time periods (6–48 h). Following that, total RNAs were isolated for miR-124 expression analysis. As shown in , exposure of BV-2 cells to 10 µM cocaine significantly decreased miR-124 levels at 24 and 48 h post-treatment (48.54 ± 12.57% and 47.02 ± 17.18% of control, respectively; P < 0.05), while negligible effects at the earlier time point (6 h) were observed. To ensure that the decrease observed in mature miR-124 was not due to alterations in the mechanisms underlying the processing of the corresponding pri-mRNAs, time course expression of pri-miR-124-1, -2, and -3 levels was also assessed. As shown in supplemental Fig. 1, cocaine exposure time-dependently decreased the expression of the pri-miR-124-1 and -2 levels, but not that of the pri-miR-124-3. Further validation of miR-124 downregulation was obtained using in situ hybridization (ISH) of miR-124 in BV-2 cells exposed to cocaine. Cells were seeded onto coverslips placed in 24-well plates, exposed to cocaine for 24 h, fixed with 4% paraformaldehyde (PFA) and subject to ISH using DIG-labeled miR-124 sense probes. miR-124 levels were observed by immunofluorescence microscopy. Lipopolysaccharide (LPS)-treated cells were used as positive controls. Similar to the findings with qPCR, ISH analyses also validated that cocaine exposure resulted in significant decrease in miR-124 levels compared to unexposed cells ().
Figure 1. Cocaine decreased microglial miR-124 levels in BV-2 cells. BV-2 cells were seeded into 6-well plates and exposed to various doses of cocaine (1–100 µM) for 24 h or with 10 µM of cocaine for the indicated time periods. After various treatments, cells were collected for RNA extraction and subjected to assessment of miR-124 by qPCR. (A and B) cocaine significantly decreased miR-124 levels in BV-2 cells. BV-2 cells were seeded into 6-well plates with cocaine exposure for 24 h. Then, cells were fixed with 4% PFA and miR-124 levels were determined by in situ immunofluorescence. LPS-treated cells served as positive controls. (C) miR-124 immunofluorescence signal intensity significantly decreased in cocaine-treated BV-2 cells. BV-2 cells were seeded into 6-well plates and exposed to various doses of cocaine (1–100 µM) for 24 h or with 10 µM of cocaine for the indicated time periods. After various treatments for 24 h, cells were collected for protein extraction and subject to assessment by Western blot. Cocaine exposure substantially increased DNMT1 and DNMT3a levels in all 3 tested doses in BV-2 cells (D, E, and F). Cocaine also significantly increased DNMT1 and DNMT3a in BV-2 cells in a time-dependent manner (G, H, and I). All experiments were done 4 times, independently. For all Western blots, β-actin served as protein loading control. Data are expressed as mean ± SEM and were analyzed using ANOVA (* P < 0.05 vs. control).
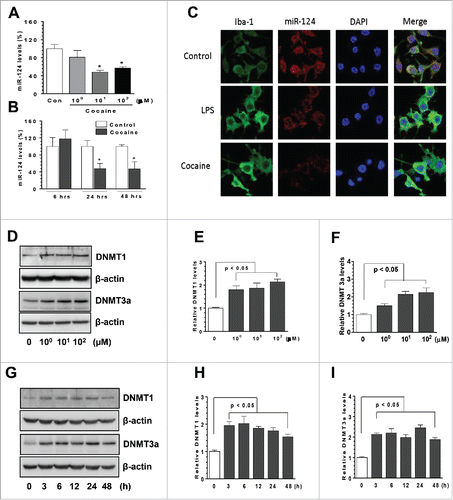
Previous reports have demonstrated that cocaine can modulate the levels of targeted genes by epigenetic mechanisms (promoter DNA methylation).Citation31,32 DNMTs including, DNMT1, 3a, and 3b, are the proteins responsible for regulating and maintaining promoter DNA methylation levels.Citation33 To investigate whether DNMTs are associated with cocaine-mediated miR-124 downregulation, we examined the protein levels of DNMT1 and 3a in those cocaine-treated BV-2 cells. Interestingly, we found that cocaine exposure significantly upregulated both DNMT1 and 3a levels at all 3 tested doses of cocaine (1, 10, 100 µM), with maximal effects at 100 µM cocaine (). Also, cocaine exposure at the dose of 10 µM significantly increased microglial DNMT1 and 3a levels at 3 h, and this upregulation persisted up to 48 h post-treatment ( < 0.05). Taken together, our results demonstrated that cocaine decreased miR-124 levels in BV-2 cells and that the upregulation of DNMTs levels was associated with miR-124 downregulation.
Cocaine exposure decreased miR-124 and increased DNMTs levels in rat primary microglia
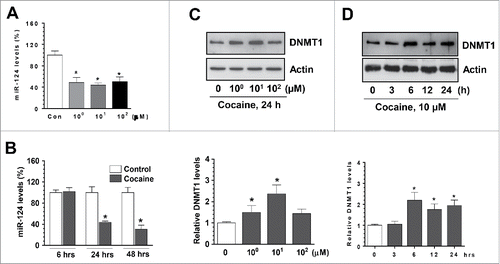
Having determined the functional link between cocaine and miR-124 in BV-2 cells, we next explored whether cocaine exposure could decrease miR-124 levels in rat primary microglia (rPM) cells that closely resemble the in vivo characteristics of microglia.Citation34 rPM were isolated from newborn pups and cultured in vitro for 7–10 d. Similarly, we exposed rPMs with cocaine at different doses (1, 10, 100 µM); then, total RNAs were extracted for detection of miR-124 levels. Consistent with the results obtained from BV-2 cells, exposure of cocaine to rPM also resulted in significant downregulation of miR-124 levels at all 3 doses of cocaine (). Furthermore, cocaine also significantly downregulated expression of miR-124 in a time-dependent manner (). We investigated the effects of cocaine on DNMT levels and found that DNMT1 protein levels were significantly upregulated by cocaine exposure (). Also, we revealed that cocaine significantly increased mRNA levels of all 3 DNMTs (data not shown). In summary, we showed evidence that cocaine decreased miR-124 levels and, in parallel, increased DNMTs levels in rPM.
Figure 2. Cocaine decreased microglial miR-124 levels in rPM. rPM were seeded into 6-well plates and exposed to various doses of cocaine (1–100 µM) for 24 h or with 10 µM of cocaine for the indicated time periods. After various treatments, cells were collected for RNA extraction and subjected to assessment of miR-124 by qPCR. (A and B) cocaine significantly decreased miR-124 levels in rPM cells. (C and D) Cocaine also increased DNMT1 levels in rPM. All experiments were done 4 times, independently. For all Western blots, β-actin served as protein loading control. Data are expressed as mean ± SEM and were analyzed using ANOVA (* P < 0.05 vs. control).
Cocaine exposure decreased microglial miR-124 and increased DNMTs levels in vivo
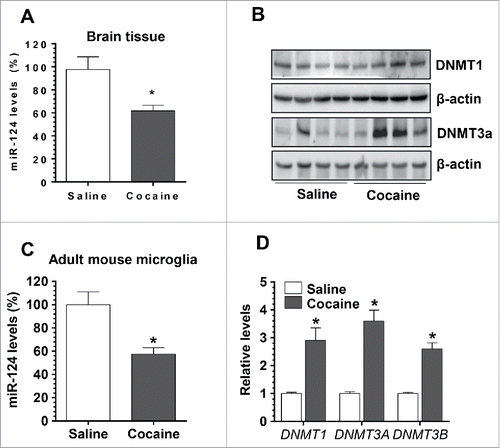
We next sought to examine whether cocaine could exert similar effects on miR-124 and DNMTs in vivo. For this, Balb/C57 mice were injected with either cocaine [20 mg/kg, intraperitoneal (i.p.) injection] or saline for 7 consecutive days. One hour after the last injection, mice were sacrificed and brains were removed. Total RNA was extracted from whole brain homogenates for assessment of miR-124 levels by qPCR, and protein lysates were used for detection of DNMT1 and DNMT3a levels by Western blotting. To specifically explore the effects of cocaine on microglia in vivo, adult microglia were also isolated and purified using 2 markers: CD11b and CD45. CD11bhighCD45low microglia was purified by fluorescence-activated cell sorting and assessed for expression of miR-124. In parallel, mRNA levels of the 3 DNMTs were also determined in CD11bhighCD45low microglia. Our findings clearly demonstrated that cocaine exposure in mice resulted in significant downregulation of miR-124 levels (67.72 ± 4.90% of controls, P < 0.05, ) with concomitant upregulation of DNMT1 and 3a proteins in brain homogenates (). Interestingly, and as expected, adult microglia isolated from cocaine-treated mice brains also exhibited decreased levels of miR-124 (57.40 ± 5.52% of controls, P < 0.05, ). In parallel, mRNA levels of all 3 DNMTs were substantially increased in adult mouse microglia (DNMT-1, 2.91 ± 0.45-fold; DNMT3a, 3.62 ± 0.39-fold; DNMT3b, 2.61 ± 0.22-fold; P < 0.05). In keeping with the cell culture findings, cocaine exposure in vivo can also downregulate levels of microglial miR-124.
Figure 3. Cocaine exposure decreased microglial miR-124 and increased DNMTs levels in vivo. Wild type Balb/C57 mice were injected with cocaine (20 mg/kg) for 7 consecutive days. One hour after the last injection, mice were sacrificed for brain removal. The striatal region was separated for RNA isolation. (A) Cocaine significantly decreased miR-124 levels in the brains of mice with cocaine administration. (B) DNMT1 and DNMT3a levels were also significantly increased in cocaine-treated mouse brains. (C) Adult purified mouse microglia isolated from cocaine-treated mice showed decreased miR-124 levels compared to the controls. (D) The mRNA levels of DNMT1, 3a, and 3b were significantly increased in adult mouse primary microglia isolated from cocaine-treated mouse brains. All experiments were done 4 times, independently. For all Western blots, β-actin served as protein loading control. Data are expressed as mean ± SEM and were analyzed using ANOVA (* P < 0.05 vs. control).
DNMT1 inhibitor 5-Azacytidine (5-AZA) reversed cocaine-mediated effects on miR-124 and DNMTs
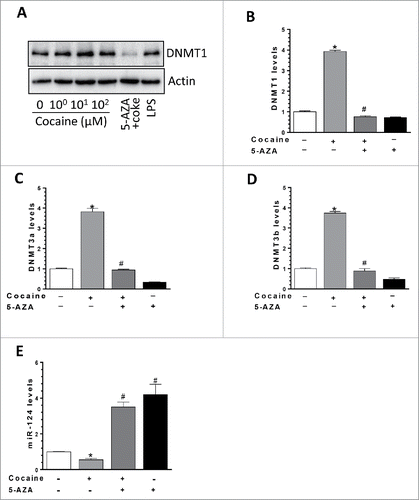
We have demonstrated a negative correlation between the levels of miR-124 and DNMTs in the context of cocaine both in vitro and in vivo. Next, we sought to explore the direct functional link between these 2 molecules. For this purpose, 5-Azacytidine (5-AZA), a DNMT1 inhibitor, was used here. BV-2 cells were first pre-treated with 5-AZA (5 µM) for an hour and then exposed to cocaine (10 µM) for 24 h. Total RNA and proteins were extracted for detection of DNMTs levels. Our results demonstrated that while cocaine increased DNMT1 protein levels, pre-treatment with 5-AZA could completely block this effect (). Similarly, inhibitory effects of 5-AZA were also observed in mRNA levels of all 3 DNMTs; cocaine exposure led to increased expression of DNMT1 (3.93 ± 0.16-fold, P < 0.05), DNMT3a (3.82 ± 0.17-fold; P < 0.05), and DNMT3b (3.76 ± 0.08-fold; P < 0.05) and these effects were significantly blocked by pre-treatment of BV-2 cells with 5-AZA (). More importantly, we observed that cocaine-mediated downregulation of miR-124 levels was abrogated in cells pretreated with 5-AZA (a group of cocaine vs. a group of 5-AZA + cocaine, P < 0.05; ). Taken together, these findings demonstrate a direct link between the expression levels of DNMTs and miR-124 in the context of cocaine, implying thereby that epigenetic changes (promoter DNA methylation) in the miR-124 promoter could likely be playing a role in cocaine-mediated downregulation of miR-124.
Figure 4. DNMT1 inhibitor 5-AZA reversed cocaine-mediated effects on DNMTs and miR-124. (A) BV-2 cells were seeded into 6-well plates and pre-treated with DNMTs inhibitor 5-AZA followed by cocaine exposure for another 24 h. DNMT1 mRNA and protein levels were determined by qPCR and Western blot, respectively. (A) DNMTs inhibitor 5-AZA significantly blocked cocaine-mediated upregulation on DNMT1 protein levels. (B, C, and D) DNMTs inhibitor 5-AZA significantly blocked cocaine-mediated upregulation on DNMT1, 3a, and 3b mRNA levels. (E) DNMTs inhibitor 5-AZA significantly blocked cocaine-mediated miR-124 downregulation in BV-2 cells. All experiments were done 4 times, independently. For all western blots, β-actin served as protein loading control. Data are expressed as mean ± SEM and were analyzed using ANOVA (* P < 0.05 vs. control; # P < 0.05 vs. cocaine-treated samples).
Cocaine increased DNA methylation levels in pri-miR-124-1 promoter regions both in vitro and in vivo
The above findings implied that cocaine-mediated downregulation of miR-124 involved alterations in the DNA methylation status of the pri-miR-124 promoter. To validate this, BV-2 cells were exposed to cocaine, followed by extraction of genomic DNA (gDNA) for bisulfite-mediated nucleotide conversion (unmethylated cytosine nucleotide will be converted into thymine while methylated cytosine remains unchanged). PCR products were then sequenced to identify specific methylation of cytosine. Mature miR-124 comprises of 3 pri-miRNAs: pri-miR-124-1, -2, and -3, which are located on chromosomes 14, 3, and 2, respectively; pri-miR-124-1 is the primary resource for mature miR-124 production.Citation35 We first investigated the effects of cocaine on DNA methylation levels of the pri-miR-124-1 promoter in BV-2 cells as well as in adult isolated mouse microglia. Bioinformatics analyses indicated there were dense CpG islands in the promoter region of pri-miR-124-1 (). We thus designed 2 sets of primers so that their PCR products (fragments 1 and 2) encompassed ∼1 kb of the promoter region (primer sequences are shown in ). As shown in , cocaine exposure of BV-2 cells resulted in substantial increase in promoter DNA methylation levels. In fragment-1, encompassing the region from −905 to −498 nucleotides (nt) upstream of the transcription site, CpG island methylation rate increased from ∼45% (control) to ∼78% (cocaine); in fragment-2 (−531 to −179 nt), methylation rate increased from ∼43 to ∼64%. Validation of these findings was also done in purified adult primary mouse microglia isolated from cocaine/saline-treated mice brains. The methylation rate for pri-miR-124-1 promoter increased from ∼5 to ∼30% and from ∼0.1 to ∼24% in fragment-1 and fragment-2, respectively, in the presence of cocaine (). Our results clearly demonstrated that cocaine significantly increased the methylation levels of pri-miR-124-1 promoter region both in vitro and in vivo.
Figure 5. Cocaine increased methylation levels in pri-miR-124-1 promoter regions in BV-2 and adult isolated mouse microglia. BV-2 cells were exposed to cocaine (10 µM) for 24 h. Adult mouse microglia were purified from mice on a 7-day cocaine administration regimen. BV-2 cells and isolated adult mouse microglia were subjected to genomic DNA extraction. Genomic DNA was then treated with bisulfite for nucleotide conversion followed by bisulfite genomic DNA PCR and DNA sequencing. (A) Schematic of CpG islands located in the pri-miR-124-1 promoter region. (B and C) Cocaine significantly increased pri-miR-124-1 promoter methylation rates in BV-2 cells as well as in adult mouse microglia.
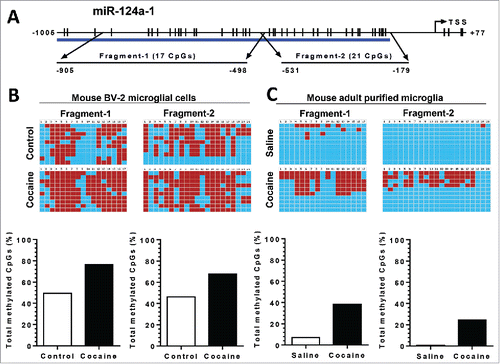
Table 1. List of primer sequences used for bisulfite genomic DNA sequencing.
Cocaine increased DNA methylation levels in pri-miR-124-2 but not in pri-miR-124-3 promoter regions both in vitro and in vivo
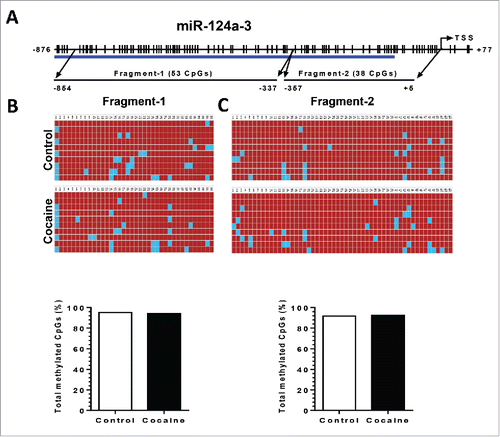
Since the other 2 precursors, pri-miR-124-2 and -3, also contribute to mature miR-124 production, we next investigated the DNA methylation status of the pri-miR-124-2 and -3 promoter regions in both BV-2 cells and in purified adult primary microglia. For pri-miR-124-2, cocaine significantly increased the CpG methylation rate from ∼62 to ∼70% and from ∼10 to ∼25% in fragment-1 and 2, respectively, in BV-2 cells (). A similar upregulation of methylation was also observed in isolated adult mouse microglia: increased methylation rates from ∼32 to ∼58% in fragment-1 and from ∼20 to ∼42% in fragment-2 (). For pri-miR-124-3, however, we did not detect significant changes in the methylation status of its promoter following cocaine exposure in BV-2 cells ().
Figure 6. Cocaine increased methylation levels in pri-miR-124-2 promoter regions in BV-2 and adult isolated mouse microglia. BV-2 cells were exposed to cocaine (10 µM) for 24 h. Adult mouse microglia were purified from mice on a 7-day cocaine administration regimen. BV-2 cells and isolated adult mouse microglia were subjected to genomic DNA extraction. Genomic DNA was then treated with bisulfite for nucleotide conversion followed by bisulfite genomic DNA PCR and DNA sequencing. (A) Schematic of CpG islands located in the pri-miR-124-2 promoter region. (B and C) Cocaine significantly increased pri-miR-124-2 promoter methylation rates in BV-2 cells as well as in adult mouse microglia.
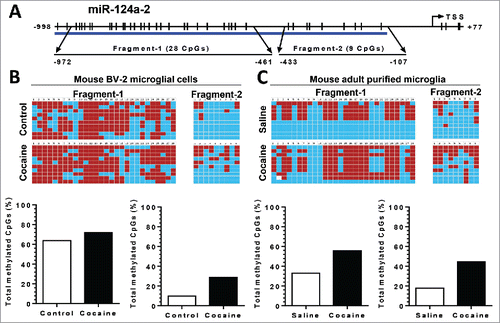
Figure 7. Cocaine did not increase methylation levels in pri-miR-124-3 promoter regions in BV-2 and adult isolated mouse microglia. BV-2 cells were exposed to cocaine (10 µM) for 24 h. Adult mouse microglia were purified from mice on a 7-day cocaine administration regimen. BV-2 cells and isolated adult mouse microglia were subjected to genomic DNA extraction. Genomic DNA was then treated with bisulfite for nucleotide conversion followed by bisulfite genomic DNA PCR and DNA sequencing. (A) Schematic of CpG islands located in the pri-miR-124-3 promoter region. (B and C) Cocaine did not alter pri-miR-124-2 promoter methylation rates neither in BV-2 cells nor in adult mouse microglia.
Discussion
In this study, we demonstrated that cocaine decreased microglial miR-124 levels as well as upregulated the expression levels of DNMTs both in vitro and in vivo. Increased promoter DNA methylation levels of pri-miR-124-1 and -2 were found responsible for the downregulation of mature miR-124. These findings uncover a novel molecular pathway responsible for cocaine-mediated microglial activation.
In the brain, psychostimulants, including cocaine and amphetamine, have long been known to block dopamine transporter to increase synaptic dopamine concentration, which, in turn, leads to elevated neuronal excitement in the striatum. In recent times, microglial activation and glia-neuron cross-talk have gradually been recognized as playing important roles in the development of drug addiction. Clinical evidence showed increased microglial/astrocytic activation markers in the brains of human addicts.Citation36 There is a statistically significant increase in activated macrophages/microglia in chronic cocaine abusers.Citation37 In another study comparing cognitive function with peripheral markers of immune activation, significant correlations were observed in methamphetamine users, but not in matched controls.Citation38 Findings from positron emission tomography have suggested a diffuse persistent increase in activated microglia in all brain regions of human methamphetamine abusers compared to control subjects.Citation36 Consistently, both in vitro and in vivo laboratory findings have also demonstrated that cocaine and amphetamine-mediated activation of microglia underlie neuroinflammation. Findings from our lab have demonstrated the ability of cocaine to activate microglia (BV-2 and rPM), leading to the production and release of pro-inflammatory factors, including TNFα, IL-6, and CCL2.Citation4,5,39 Other reports have also demonstrated increased neuroinflammation following exposure of rodents to commonly abused drugs in animal models. Amphetamine administration (4 mg/kg at 2-h intervals, 4 times) was shown to activate microglia in mice.Citation40 Similarly, acute high dose (30 mg/kg) and even lower dose of methamphetamine increased the mRNA levels of pro-inflammatory cytokines IL-6 and TNFα in the striatum and hippocampus of mice.Citation41,42 The relationship between psychostimulant exposure and ensuing neuroinflammation has also been confirmed by analyzing annotated datasets, which are available in the GEO public database.Citation43-45 Adding further validity to the psychostimulant-mediated neuroinflammation theory is the recent study by Watkin's group that provided direct evidence of interaction of cocaine with TLR4 in enhancing the microglial innate immune response, thereby contributing to cocaine addiction. In this study, TLR4 knockout mice showed decrease in cocaine-induced behavioral changes in both conditioned place preference (CPP) and self-administration assays, implying thereby that microglial activation via TLR4 was critical for cocaine addiction.Citation2 Beside the genetic approach, inhibitors of glial activation (pharmacological approach) have also been suggested as potential treatment strategies for psychostimulant abuse.Citation3 Taken together, these findings suggest that microglial activation induced by addictive drugs plays an important role in drug addiction. Detailed molecular mechanisms underlying this phenomenon, however, remain less well understood. Several intracellular pathways, including elevated ROS, ER stress, and ensuing autophagy, have been implicated in this process.Citation4,5
Expression levels of various genes are dysregulated during drug addiction through multiple mechanisms, such as epigenetic modifications, that primarily include histone modification, DNA methylation, and noncoding RNAs.Citation22 Promoter DNA methylation is well known to negatively regulate downstream gene transcription activity leading, in turn, to decreased expression levels. Cocaine has been demonstrated to modulate global DNA methylation levels with dynamic epigenetic changes suggested to play critical roles in cocaine addiction-related effects in memory and behavioral changes.Citation23 Besides chronic cocaine administration, acute cocaine treatment has been also shown to cause upregulation of DNMTs (3a and 3b) in the nucleus accumbens (NAc), a critical component of brain reward-related circuitry related to behavior sensitization.Citation46 Cocaine is capable of activating microglia and is known to induce global changes in the expression profiles of proteins and noncoding RNAs. It is not clear, however, whether alterations in promoter methylation of miRs also constitute a mechanism of cocaine-mediated microglial activation.
microRNAs have been demonstrated to be associated with many fundamental cellular processes, such as proliferation, differentiation, and apoptosis.Citation6-8 Dysregulation of miR levels has been reported in various neuroinflammatory disorders and also in many types of cancers. In the brain, many miRs have been shown to modulate the brain immune responses, being characterized as “NeurimmiRs.” Belonging to this category is the brain-enriched miR124 that is highly expressed in both neurons and microglia. miR-124 was first identified as a neuroprotective factor promoting neurogenesis during brain development and maintenance of synaptic homeostasis in the adult brain.Citation47,48 Later on, miR-124 was identified as one of the miRs whose expression levels closely correlated with microglial quiescence.Citation16,17 Decreased miR-124 levels have been reported in various neuroinflammatory related disorders, including Parkinson disease, dementia, and multiple sclerosis.Citation17,19-21
Modulation of miR-124 levels was also implicated in drug addiction, which is now recognized as a neuroinflammatory disease. Several abused drugs have been found to regulate the expression levels of miR-124. Chronic administration of cocaine was shown to decrease miR-124 levels in the NAc and, furthermore, downregulation of miR-124 was shown to be critical for cocaine-induced synaptic plasticity and behavioral changes.Citation49 Consistently, overexpression of miR-124 by lentivirus transduction in the NAc resulted in attenuation of cocaine-induced CPP.Citation50 Interestingly, reduced levels of miR-124 have also been found in the ventral tegmental area of methamphetamine self-administering rats by microarray analysis.Citation51 There are also reports demonstrating decreased miR-124 levels in ethanol-withdrawn rats, which likely involved histone acetylation.Citation52 All these studies allude to the fact that miR-124 downregulation is critical for reward-related behavior changes induced by abused drugs. The findings from our study are in agreement with previous reports and are novel in that: (1) cocaine-mediated downregulation of microglial miR-124 both in vitro and in vivo was linked with microglial activation (and thereby drug addiction); (2) increased promoter methylation was responsible for cocaine-mediated downregulation of miR-124, thereby implicating epigenetic mechanisms underlying cocaine-mediated microglial activation.
In summary, we have demonstrated that epigenetic downregulation of miR-124 was induced by cocaine in both primary and BV-2 microglia and that this effect was critical for microglial activation, with a possible impact on the ensuing cross-talk between glia and neurons. Since miR-124 is also expressed by neurons, the effect of cocaine exposure on the expression of miR-124 in primary rat hippocampal as well as striatal neurons was also assessed. Unlike what we observed in microglia, however, we failed to detect significant downregulation of miR-124 in these cells following cocaine exposure (data not shown). Taken together, in microglial cells, miR-124 can thus be envisioned as a potential therapeutic target for modulating drug abuse-mediated neuroinflammation and, likely, for ameliorating drug addiction. Adding credence to this is the fact that overexpression of miR-124 has already been shown to prevent microglial activation through inhibition of reactive oxygen species production.Citation53
Materials and methods
Reagents
Cocaine hydrochloride (C5776) and 5-Azacytidine (5-AZA; A3656) were purchased from Sigma-Aldrich. TaqMan® microRNA assays for miR-124 (001182) quantification were purchased from Applied Biosystems. miR-124 probe was purchased from Exiqon (88066-5). Antibodies against DNMT1 (sc-20701) and DNMT3a (sc-20703) were purchased from Santa Cruz Biotechnology. Goat anti-rabbit (sc-2004) and goat anti-mouse (sc-2005) secondary antibodies were also from Santa Cruz Biotechnology. DNMT1 (NM_001199431.1) primers: forward 5′-cagagactcccgaggacaga-3′ and reverse 5′-tttacgtgtcgtttttcgtctc-3′. DNMT3a (NM_007872.4) primers: forward 5′-aaacggaaacgggatgagt-3′ and reverse 5′-actgcaattaccttggctttct-3′. DNMT3b (NM_001003961.3) primers: forward 5′-ccctcccccatccatagt-3′ and reverse 5′-tctgctgtctcccttcattgt-3′. Primers for bisulfite genomic DNA sequencing of mouse pri-miR-124 were shown in .
BV-2 cell culture
BV-2 immortalized cell line was obtained from Dr. Sanjay Maggirwar (University of Rochester Medical Center, Rochester, NY, USA) and was grown and routinely maintained in DMEM with 10% fetal bovine serum (FBS) at 37°C and 5% CO2 and used up to 20 passages.
Primary rat microglial cell isolation
Primary microglia cells were obtained from Sprague-Dawley newborn (1–3 days) pups. After digestion and dissociation of the dissected brain cortices in Hanks buffered salt solution (Invitrogen, 14025076) supplemented with 0.25% trypsin (Invitrogen, 25300–054), mixed glial cultures were prepared by resuspending the cell suspension in DMEM (Invitrogen, 11995–065) supplemented with 10% heat-inactivated FBS (Invitrogen, 16000–044) with 100 U/ml penicillin and 0.1 mg/ml streptomycin. Cells were plated at 20 × 106 cells/flask density onto 75 cm2 cell culture flasks. The cell medium was replaced every 5 d and, after the first medium change, macrophage colony-stimulating factor (0.25 ng/ml; Invitrogen, PHC9504) was added to the flasks to promote microglial proliferation. When confluent (7 to 10 days), mixed glial cultures were subjected to shaking at 37°C at 220 g for 2 h to promote microglia detachment from the flasks. The cell medium, containing the released microglia cells was collected from each flask and centrifuged at 1000 g for 5 min to collect cells and plated onto cell culture plates for all subsequent experiments. The purity of microglial cultures was evaluated by immunocytochemical staining using the antibody specific for Iba-1 (Wako Pure Chemical Industries, 019-19741) and used only if >95% pure.
Western blotting
Treated cells were lysed using the Mammalian Cell Lysis kit (Sigma, MCL1-1KT). Equal amounts of proteins were electrophoresed on a sodium dodecyl sulfate-polyacrylamide gel (12%) under reducing conditions, followed by transfer to PVDF membranes (Millipore, IPVH00010). Blots were blocked with 5% nonfat dry milk in phosphate-buffered saline (PBS; 137 mM NaCl; 2.7 mM KCl; 10 mM Na2HPO4; 2 mM KH2PO4). Western blots were then probed with antibodies recognizing the indicated proteins. Protein amounts loaded were normalized according to the β-actin signal, using an anti-β-actin antibody (Sigma, A5441). The secondary antibodies were HRP conjugated to goat anti-mouse/rabbit IgG.
RNA extraction, reverse transcription, and qPCR
Total RNA was extracted using Trizol reagent (Invitrogen, 15596–018). Briefly, monolayer cells in 6-well plates were washed with PBS and lysed directly adding 1 ml Trizol. The cell lysate was aspirated into new 1.5 ml microcentrifuge tubes and 0.2 ml of chloroform was added. After extensive vortexing, the samples were centrifuged at 10,000 g for 15 min at 4°C. The upper aqueous phase was transferred to a new tube and 500 µl isopropyl alcohol was added. Samples were incubated for 10 min and centrifuged again to precipitate total RNA. Total RNA was dissolved in DEPC-treated water and quantified. Reverse transcription reactions were performed using a Verso cDNA kit (Invitrogen, AB-1453/B). The reaction system (20 µl) included 4 µl 5X cDNA synthesis buffer, 2 µl dNTP mix, 1 µl RNA primer, 1 µl RT enhancer, 1 µl Verso Enzyme Mix (Invitrogen, AB-1453/B), total RNA template (1 µg) and a variable volume of water. Reaction conditions were set at 42°C for 30 min. qPCRs were performed by using SYBR Green ROX qPCR Mastermix (Qiagen, 330510). Reaction systems were set up as follows: 10 µl SYBR Green Mastermix, 0.5 µl forward primers, 0.5 µl reverse primers, and 9 µl distilled water. Plates (96-well) were placed into a 7500 fast real-time PCR system (Applied Biosystems, Grand Island, NY) for a program running. Actin levels were set as an internal control for calculation.
miR-124 qPCR
TaqMan® microRNA assays for miR-124 were commercially available from Applied Biosystems. TaqMan® MicroRNA Reverse Transcription Kit (PN 4366596) was utilized according to indicated protocol. The reaction system (15 µl) was set as follows: 10 mM dNTPs (with dTTP), 1.5 µl MultiScribe™ Reverse Transcriptase (50 U/μL), 1.0 µl 10× Reverse Transcription Buffer, 1.5 µl RNase Inhibitor (20U/μL), 0.2 µl Nuclease-free water to 7 µl, 5 µl (∼50 ng) total RNA, and 3 μL of RT (5X) primer. The tube containing the reaction mixture was centrifuged and loaded onto the thermal cycler: 16°C for 30 min; 42°C for 30 min; 85°C for 5 min. The reverse transcription product was then diluted 1:10 for the following PCR reaction: 1 µl TaqMan® PCR primer (20×), 1.5 µl RT reaction product, TaqMan® 2× Universal PCR Master Mix, 10 µl No AmpErase UNGa (PN 4324018), and distilled water up to 20 µl. PCR conditions were as follows: hold 95°C for 10 min, then 40 cycles for 95°C for 15 sec and 60°C for 1 min. All reactions were run in triplicate. The expression level of miR-124 was calculated by normalizing to U6 snRNA.
In situ hybridization
BV-2 cells or mouse brain sections were fixed and pre-hybridized in hybridization buffer (50% formamide, 10 mM Tris-HCl pH 8.0, 200 µg/mg yeast tRNA, 1X Denhardt's solution, 600 mM NaCl, 0.25% SDS, 1 mM EDTA, 10% Dextran sulfate) at a concentration of 9 pM for the commercially available digoxigenin-labeled miR-124 probe (Exiqon). LNA-modified miR-124, labeled at both the 5′ and 3′ ends with digoxigenin (Exiqon), was diluted to a final concentration of 2 pM in hybridization buffer, heated to 65°C for 5 min, and separately hybridized to the sections at 37°C overnight. The slides were then washed twice in hybridization buffer (without probe) at 37°C, followed by washing 3 times in 2X SSC and twice in 0.2X SSC at 42°C. This was followed by 2 PBS washes and signal amplification using TSA Cy5 kit (PerkinElmer, Waltham, MA) according to the manufacturer's protocol. The slides were cover-slipped using Prolong gold antifade reagent with DAPI (Invitrogen). For BV-2 cells we also co-stained Iba-1 antibody. The specificity of the miR-124 signal in FISH experiments was confirmed as the scrambled probe showed no signal in microglia.
Bisulfite genomic DNA sequencing
Genomic DNA extracted from BV-2 cells or mouse adult microglia were subjected to bisulfite conversion by EZ DNA Methylation-Direct™ kit (Zymo Research). The bisulfite-modified DNA was amplified by bisulfite sequencing PCR using Platinum PCR SuperMix High Fidelity (Invitrogen) with primers specific to mouse pri-miR-124-1, pri-miR-124-2, and pri-miR-124-3 promoter regions. Subsequently, the amplified PCR products were cleaned by gel extraction with Zymoclean™ Gel DNA recovery kit (Zymo Research), then cloned into pCR®4-TOPO vectors using TOPO TA Cloning® kit (Invitrogen). The recombinant plasmids were transformed into One Shot® TOP10 chemically competent E. coli (Invitrogen) using the regular chemical transformation method. Plasmid DNA was isolated from about 10 independent clones of each amplicon with PureLink® Quick Plasmid Miniprep kit (Invitrogen) and then sequenced (High-Throughput DNA Sequencing and Genotyping Core Facility, University of Nebraska Medical Center, Omaha, NE) to determine the status of CpG methylation. Only the clones with insert containing >99.5% bisulfite conversion, i.e., non-methylated cytosine residues to thymine, were included in this study. The sequence data of each clone was analyzed for methylation in the miR-124 promoter by BISMA software (http://biochem.jacobs-university.de/BDPC/BISMA/) using default filtering threshold settings.
Animals
Sprague-Dawley rats and C57BL/6N mice were purchased from Charles River Laboratories (Wilmington, MA, USA). All the animals were housed under conditions of constant temperature and humidity on a 12-h light, 12-h dark cycle, with lights on at 0700. Food and water were available ad libitum. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center and the National Institutes of Health.
Chronic cocaine administration
Male C57BL/6N mice (8–10 weeks old) were selected for chronic cocaine administration. Mice received cocaine injections once a day (20 mg/kg, i.p.) for 7 consecutive days and sacrificed 1 h following the last cocaine injection for harvesting brains, which were divided into 2 parts. One part of the brains was subjected to the isolation of total RNA and striatal protein homogenates for assessment of DNMTs expression. Another part of mice brain was used for isolation of primary microglia.
Isolation of adult mice microglia
Groups of mice were injected with cocaine or saline for 7 d. One hour after the last injection, microglia was isolated from whole brain homogenates by percoll gradient centrifugation. Briefly, the brains were homogenized in HBSS with 10% serum (pH 7.4) by passing through a 70-mm nylon cell strainer. Resulting homogenates were centrifuged at 200 g for 10 min at 4°C. Supernatants were removed and cell pellets were re-suspended in digestion media (HBSS without serum, 28 U/ml DNase I, 0.2 mg/ml Collagenase I) and incubated for 30 min with shaking. One volume of HBSS and 20% serum (final concentration) was added to quench the digestion, following one-time washing by HBSS. For 5 mice, cells were resuspended in a final volume of 15.8 ml HBSS without serum, and mixed with 4.2 ml 1.22 g/ml percoll (GE-healthcare, Uppsala, Sweden). Percoll (7 ml at 1.088 g/ml) was then added to the percoll/cell suspension to form a discontinuous percoll density gradient. The gradient was centrifuged for 20 min at 250 g and microglia were collected from the interphase. Cells were washed and then re-suspended in sterile PBS followed by flow cytometry analysis by gating the myeloid cells for the CD11b+/CD45dim population.
Statistical analysis
Statistical tests were run and graphs created in GraphPad Prism Version 5 (San Diego, CA, USA). Data are presented as mean ± standard error of the mean (SEM). Appropriate statistical analyses were chosen based on experimental design. The specific statistical analysis used is indicated in the text and in each figure caption for all studies. Bonferroni post hoc tests were used for one-way analysis of variance (ANOVA). The significance threshold was set to P < 0.05 for all analyses.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1232233_s02.pptx
Download MS Power Point (46.6 KB)Funding
This work was supported by NIH grants: DA033614, DA035203, DA036157, and DA033150.
ORCID
Ming-Lei Guo http://orcid.org/0000-0003-2969-0802
Palsamy Periyasamy http://orcid.org/0000-0002-0386-5611
Shilpa Buch http://orcid.org/0000-0002-3103-6685
Related Research Data
References
- Clark KH, Wiley CA, Bradberry CW. Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotox Res 2013; 23:174-88; PMID:22714667; http://dx.doi.org/10.1007/s12640-012-9334-7
- Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, et al. DAT isn't all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol Psychiatry 2015; 20:1525-37; PMID:25644383; http://dx.doi.org/10.1038/mp.2014.177
- Beardsley PM, Hauser KF. Glial modulators as potential treatments of psychostimulant abuse. Adv Pharmacol 2014; 69:1-69; PMID:24484974; http://dx.doi.org/10.1016/B978-0-12-420118-7.00001-9
- Liao K, Guo M, Niu F, Yang L, Callen SE, Buch S. Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J Neuroinflammation 2016; 13:33; PMID:26860188; http://dx.doi.org/10.1186/s12974-016-0501-2
- Guo ML, Liao K, Periyasamy P, Yang L, Cai Y, Callen SE, Buch S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015; 11:995-1009; PMID:26043790; http://dx.doi.org/10.1080/15548627.2015.1052205
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009; 136:642-55; PMID:19239886; http://dx.doi.org/10.1016/j.cell.2009.01.035
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/10.1016/j.cell.2009.01.002
- Hobert O. Gene regulation by transcription factors and microRNAs. Science 2008; 319:1785-6; PMID:18369135; http://dx.doi.org/10.1126/science.1151651
- Davis GM, Haas MA, Pocock R. MicroRNAs: Not “Fine-Tuners” but Key Regulators of Neuronal Development and Function. Front Neurol 2015; 6:245; PMID:26635721; http://dx.doi.org/10.3389/fneur.2015.00245
- Yu B, Zhou S, Yi S, Gu X. The regulatory roles of non-coding RNAs in nerve injury and regeneration. Prog Neurobiol 2015; 134:122-39; PMID:26432164; http://dx.doi.org/10.1016/j.pneurobio.2015.09.006
- Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG. microRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J Neurosci 2016; 36:2383-90; PMID:26911687; http://dx.doi.org/10.1523/JNEUROSCI.3900-15.2016
- Parisi C, Napoli G, Amadio S, Spalloni A, Apolloni S, Longone P, Volonte C. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ 2016; 23:531-41; PMID:26794445; http://dx.doi.org/10.1038/cdd.2015.153
- Sadakierska-Chudy A, Frankowska M, Miszkiel J, Wydra K, Jastrzebska J, Filip M. Prolonged Induction of miR-212/132 and REST Expression in Rat Striatum Following Cocaine Self-Administration. Mol Neurobiol 2016; PMID:26944283; http://dx.doi.org/10.1007/s12035-016-9817-2
- Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, Willoughby D, Wahlestedt C, Conkright MD, Kenny PJ. Striatal microRNA controls cocaine intake through CREB signalling. Nature 2010; 466:197-202; PMID:20613834; http://dx.doi.org/10.1038/nature09202
- Sun Y, Luo ZM, Guo XM, Su DF, Liu X. An updated role of microRNA-124 in central nervous system disorders: a review. Front Cell Neurosci 2015; 9:193; PMID:26041995; http://dx.doi.org/10.3389/fncel.2015.00193
- Svahn AJ, Giacomotto J, Graeber MB, Rinkwitz S, Becker TS. miR-124 Contributes to the functional maturity of microglia. Dev Neurobiol 2016; 76:507-18; PMID:26184457; http://dx.doi.org/10.1002/dneu.22328
- Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med 2011; 17:64-70; PMID:21131957; http://dx.doi.org/10.1038/nm.2266
- Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED. IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS One 2013; 8:e81774; PMID:24358127; http://dx.doi.org/10.1371/journal.pone.0081774
- Gascon E, Lynch K, Ruan H, Almeida S, Verheyden JM, Seeley WW, Dickson DW, Petrucelli L, Sun D, Jiao J, et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat Med 2014; 20:1444-51; PMID:25401692; http://dx.doi.org/10.1038/nm.3717
- Kanagaraj N, Beiping H, Dheen ST, Tay SS. Downregulation of miR-124 in MPTP-treated mouse model of Parkinson's disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 2014; 272:167-79; PMID:24792712; http://dx.doi.org/10.1016/j.neuroscience.2014.04.039
- Wang H, Ye Y, Zhu Z, Mo L, Lin C, Wang Q, Wang H, Gong X, He X, Lu G, et al. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of Parkinson's Disease by Targeting to Bim. Brain Pathol 2016; 26:167-76; PMID:25976060; http://dx.doi.org/10.1111/bpa.12267
- Kalda A, Zharkovsky A. Epigenetic Mechanisms of Psychostimulant-Induced Addiction. Int Rev Neurobiol 2015; 120:85-105; PMID:26070754; http://dx.doi.org/10.1016/bs.irn.2015.02.010
- Sadri-Vakili G. Cocaine triggers epigenetic alterations in the corticostriatal circuit. Brain Res 2015; 1628:50-9; PMID:25301690; http://dx.doi.org/10.1016/j.brainres.2014.09.069
- Clermont PL, Parolia A, Liu HH, Helgason CD. DNA methylation at enhancer regions: Novel avenues for epigenetic biomarker development. Front Biosci (Landmark Ed) 2016; 21:430-46; PMID:26709784; http://dx.doi.org/10.2741/4399
- Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol 2014; 4:80; PMID:24822169; http://dx.doi.org/10.3389/fonc.2014.00080
- Wong KY, Chim CS. DNA methylation of tumor suppressor protein-coding and non-coding genes in multiple myeloma. Epigenomics 2015; 7:985-1001; PMID:26417914; http://dx.doi.org/10.2217/epi.15.57
- Loginov VI, Rykov SV, Fridman MV, Braga EA. Methylation of miRNA genes and oncogenesis. Biochemistry (Mosc) 2015; 80:145-62; PMID:25756530; http://dx.doi.org/10.1134/S0006297915020029
- Xu L, Dai W, Li J, He L, Wang F, Xia Y, Chen K, Li S, Liu T, Lu J, et al. Methylation-regulated miR-124-1 suppresses tumorigenesis in hepatocellular carcinoma by targeting CASC3. Oncotarget 2016; 7:26027-41; PMID:27029030; http://dx.doi.org/10.18632/oncotarget.8266
- Kim BC, Jeong HO, Park D, Kim CH, Lee EK, Kim DH, Im E, Kim ND, Lee S, Yu BP, et al. Profiling age-related epigenetic markers of stomach adenocarcinoma in young and old subjects. Cancer Inform 2015; 14:47-54; PMID:25983541; http://dx.doi.org/10.4137/CIN.S16912
- Chu M, Chang Y, Guo Y, Wang N, Cui J, Gao WQ. Regulation and methylation of tumor suppressor miR-124 by androgen receptor in prostate cancer cells. PLoS One 2015; 10:e0116197; PMID:25860954; http://dx.doi.org/10.1371/journal.pone.0116197
- Shi HS, Luo YX, Yin X, Wu HH, Xue G, Geng XH, Hou YN. Reconsolidation of a cocaine associated memory requires DNA methyltransferase activity in the basolateral amygdala. Sci Rep 2015; 5:13327; PMID:26289919; http://dx.doi.org/10.1038/srep13327
- Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology 2014; 76(Pt B):259-68; PMID:23643695; http://dx.doi.org/10.1016/j.neuropharm.2013.04.004
- Leppert S, Matarazzo MR. De novo DNMTs and DNA methylation: novel insights into disease pathogenesis and therapy from epigenomics. Curr Pharm Des 2014; 20:1812-8; PMID:23888951; http://dx.doi.org/10.2174/13816128113199990534
- Stansley B, Post J, Hensley K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer's disease. J Neuroinflammation 2012; 9:115; PMID:22651808; http://dx.doi.org/10.1186/1742-2094-9-115
- Sanuki R, Onishi A, Koike C, Muramatsu R, Watanabe S, Muranishi Y, Irie S, Uneo S, Koyasu T, Matsui R, et al. miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat Neurosci 2011; 14:1125-34; PMID:21857657; http://dx.doi.org/10.1038/nn.2897
- Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, et al. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci 2008; 28:5756-61; PMID:18509037; http://dx.doi.org/10.1523/JNEUROSCI.1179-08.2008
- Little KY, Ramssen E, Welchko R, Volberg V, Roland CJ, Cassin B. Decreased brain dopamine cell numbers in human cocaine users. Psychiatry Res 2009; 168:173-80; PMID:19233481; http://dx.doi.org/10.1016/j.psychres.2008.10.034
- Loftis JM, Choi D, Hoffman W, Huckans MS. Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox Res 2011; 20:59-68; PMID:20953917; http://dx.doi.org/10.1007/s12640-010-9223-x
- Yao H, Yang Y, Kim KJ, Bethel-Brown C, Gong N, Funa K, Gendelman HE, Su TP, Wang JQ, Buch S. Molecular mechanisms involving sigma receptor-mediated induction of MCP-1: implication for increased monocyte transmigration. Blood 2010; 115:4951-62; PMID:20354174; http://dx.doi.org/10.1182/blood-2010-01-266221
- Asanuma M, Miyazaki I, Higashi Y, Tsuji T, Ogawa N. Specific gene expression and possible involvement of inflammation in methamphetamine-induced neurotoxicity. Ann N Y Acad Sci 2004; 1025:69-75; PMID:15542702; http://dx.doi.org/10.1196/annals.1316.009
- Goncalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF, Malva JO, Macedo TR, Silva AP. Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Ann N Y Acad Sci 2008; 1139:103-11; PMID:18991854; http://dx.doi.org/10.1196/annals.1432.043
- Wisor JP, Schmidt MA, Clegern WC. Cerebral microglia mediate sleep/wake and neuroinflammatory effects of methamphetamine. Brain Behav Immun 2011; 25:767-76; PMID:21333736; http://dx.doi.org/10.1016/j.bbi.2011.02.002
- Piechota M, Korostynski M, Solecki W, Gieryk A, Slezak M, Bilecki W, Ziolkowska B, Kostrzewa E, Cymerman I, Swiech L, et al. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol 2010; 11:R48; PMID:20459597; http://dx.doi.org/10.1186/gb-2010-11-5-r48
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 2007; 56:517-29; PMID:17988634; http://dx.doi.org/10.1016/j.neuron.2007.09.032
- Ahmed SH, Lutjens R, van der Stap LD, Lekic D, Romano-Spica V, Morales M, Koob GF, Repunte-Canonigo V, Sanna PP. Gene expression evidence for remodeling of lateral hypothalamic circuitry in cocaine addiction. Proc Natl Acad Sci U S A 2005; 102:11533-8; PMID:16076954; http://dx.doi.org/10.1073/pnas.0504438102
- Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology 2010; 35:2450-61; PMID:20720536; http://dx.doi.org/10.1038/npp.2010.128
- Deo M, Yu JY, Chung KH, Tippens M, Turner DL. Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn 2006; 235:2538-48; PMID:16736490; http://dx.doi.org/10.1002/dvdy.20847
- Krichevsky AM, Sonntag KC, Isacson O, Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 2006; 24:857-64; PMID:16357340; http://dx.doi.org/10.1634/stemcells.2005-0441
- Chandrasekar V, Dreyer JL. microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol Cell Neurosci 2009; 42:350-62; PMID:19703567; http://dx.doi.org/10.1016/j.mcn.2009.08.009
- Chandrasekar V, Dreyer JL. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology 2011; 36:1149-64; PMID:21307844; http://dx.doi.org/10.1038/npp.2010.250
- Bosch PJ, Benton MC, Macartney-Coxson D, Kivell BM. mRNA and microRNA analysis reveals modulation of biochemical pathways related to addiction in the ventral tegmental area of methamphetamine self-administering rats. BMC Neurosci 2015; 16:43; PMID:26188473; http://dx.doi.org/10.1186/s12868-015-0186-y
- Mizuo K, Katada R, Okazaki S, Tateda K, Watanabe S, Matsumoto H. Epigenetic regulation of MIR-124 under ethanol dependence and withdrawal. Nihon Arukoru Yakubutsu Igakkai Zasshi 2012; 47:155-63; PMID:22894056.
- Louw AM, Kolar MK, Novikova LN, Kingham PJ, Wiberg M, Kjems J, Novikov LN. Chitosan polyplex mediated delivery of miRNA-124 reduces activation of microglial cells in vitro and in rat models of spinal cord injury. Nanomedicine 2016; 12:643-53; PMID:26582736; http://dx.doi.org/10.1016/j.nano.2015.10.011