
ABSTRACT
Epigenetic factors such as DNA methylation are DNA alterations affecting gene expression that can convey environmental information through generations. Only a few studies have demonstrated epigenetic inheritance in humans. Our objective is to quantify genetic and common environmental determinants of familial resemblances in DNA methylation levels, using a family based sample. DNA methylation was measured in 48 French Canadians from 16 families as part of the GENERATION Study. We used the Illumina HumanMethylation450 BeadChip array to measure DNA methylation levels in blood leukocytes on 485,577 CpG sites. Heritability was assessed using the variance components method implemented in the QTDT software, which partitions the variance into polygenic (G), common environmental (C), and non-shared environmental (E) effects. We computed maximal heritability, genetic heritability, and common environmental effect for all probes (12.7%, 8.2%, and 4.5%, respectively) and for statistically significant probes (81.8%, 26.9%, and 54.9%, respectively). Higher maximal heritability was observed in the Major Histocompatibility Complex region on chromosome 6. In conclusion, familial resemblances in DNA methylation levels are mainly attributable to genetic factors when considering the average across the genome, but common environmental effect plays an important role when considering statistically significant probes. Further epigenome-wide studies on larger samples combined with genome-wide genotyping studies are needed to better understand the underlying mechanisms of DNA methylation heritability.
Abbreviations
| CpG | = | Cytosine-phosphate-guanine |
| FDR | = | False discovery rate |
| MHC | = | Major Histocompatibility Complex |
| SNP | = | Single nucleotide polymorphism |
Introduction
Epigenetics is the study of mitotically and meiotically heritable DNA alterations affecting gene expression that are not mediated by changes in DNA sequence.Citation1 There are several epigenetic modifications, namely DNA methylation, histone modification, occupancy of chromatin factors, and changes in chromatin structure.Citation2 DNA methylation is the best-characterized epigenetic modification and involves the methylation of cytosine residues, mainly at cytosine-phosphate-guanine (CpG) dinucleotides.Citation3 DNA methylation, as other epigenetic modifications, can be altered by environmental conditions, such as diet and smoking habits, and thus affect the expression of various genes. The modification of DNA methylation by the environment may be implicated in a wide-range of cardiovascular risk factors, including hypertension, atherosclerosis, and inflammation.Citation4-6 Epigenetic modifications including DNA methylation have also the potential to convey the effects of environmental exposures transgenerationally.Citation7
There are 2 main forms of inheritance of epigenetic modifications: genetic inheritance and epigenetic inheritance via the gametes.Citation3 Genetic inheritance refers to the effect of DNA sequence on the epigenetic marks with the example of a single nucleotide polymorphism (SNP) located at, or nearby, a CpG site that can disrupt DNA methylation at this site.Citation8 Epigenetic inheritance via the gametes is the transmission of epigenetic marks, such as DNA methylation, across generations, which can occur during epigenetic reprogramming events.Citation9,10 However, demonstrations of epigenetic inheritance in humans remain rare.
Some epidemiological studies have demonstrated that prenatal exposure to the Dutch Famine from December 1944 to April 1945 increased the risk of developing cardiovascular disease later in life.Citation11 Some other transgenerational studies have reported that epigenetic modifications may persist for longer than a single generation. Indeed, a study by Kaati et al. reported the effect of grandparental food supply on the risk of grandchildren mortality due to type 2 diabetes.Citation12
Studies with twins have also demonstrated a greater similarity in DNA methylation levels in monozygotic twins compared to dizygotic twins.Citation13 Twin studies also reported an average estimated genetic heritability of DNA methylation (proportion of variance explained by additive genetic factors) between 12 and 18% in whole blood,Citation14,15 5% in placenta,Citation15 and 7% in human umbilical vascular endothelial cells.Citation15 These studies using promoter-specific genome-wide DNA methylation array (Illumina 27K) examined only a fraction of methylation variations. More recently, studies using genome-wide DNA methylation array (Illumina 450K) have reported an average genetic heritability of 19% in peripheral blood leukocytes and 19% in adipose tissue.Citation9,16
Transgenerational similarity in DNA methylation levels has also been reported.Citation13 Several genome-wide association studies with methylation quantitative trait loci (mQTL) in both cis and trans locations have confirmed the genetic heritability of DNA methylation.Citation14,17,18 However, it is difficult to understand to what extent the observed familial resemblance in DNA methylation is due to genetic heritability and/or common environmental effect.
Our objective is thus to quantify the contribution of genetic and common environmental effects in the familial resemblances in DNA methylation levels using a family based sample of 48 French Canadians from 16 families.
Results
Correlations of methylation levels between relative pairs
Average absolute correlations across all 472,494 probes were calculated between pairs from normalized methylation levels (). Average siblings correlation was 0.299 ± 0.211 (n = 13), mother-offspring correlation was 0.267 ± 0.183 (n = 26), parent-offspring correlation was of 0.243 ± 0.170 (n = 37), whereas the correlation between unrelated individuals was 0.029 ± 0.028 (n=1178). Comparisons of genome-wide methylation levels within pairs indicated that mother-offspring and parent-offspring pairs had more similar DNA methylation levels compared to unrelated individuals.
Table 1. Average absolute correlations across all probes of normalized methylation levels between relative pairs.
DNA methylation heritability analyses
We conducted heritability analyses to better characterize the similarity in genome-wide methylation levels between related individuals. shows estimates of maximal heritability, genetic heritability, and common environmental effect from the full general model. When considering all probes (n = 472,494), we obtained an average maximal heritability of 12.7%, a genetic heritability of 8.2%, and a common environmental effect of 4.5% (). shows the distribution of maximal heritability estimates for DNA methylation levels of all probes (n = 472,494) with heritability from 0 to 100% on the x-axis and the count (number of probes) on the y-axis. A total of 215,136 probes (45.5% of all probes) and 1,507 probes (0.32% of all probes) had an estimated maximal heritability of 0% and 100%, respectively.
Figure 1. Distribution of maximal heritability estimates for DNA methylation of a) all probes (n = 472,494), b) significant probes (n = 13,621), c) FDR-corrected significant probes (n = 105).
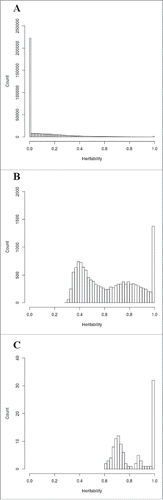
Table 2. Heritability estimates.
When considering the 13,621 probes showing a significant (P ≤ 0.05) familial effect, we obtained an average maximal heritability of 63.9%, a genetic heritability of 39.3%, and a common environmental effect of 24.6% (). When maximal heritability estimates of significant probes are plotted, the distribution shifts to the right, in accordance with the higher maximal heritability estimates ranging from 29.6 to 100% (). A total of 1,186 probes (8.71% of all significant probes) gave an estimated maximal heritability of 100%.
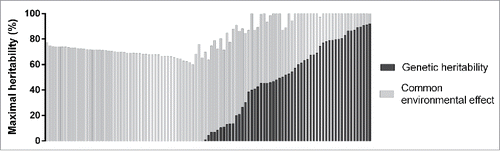
Lastly, the same analysis was repeated for probes showing a significant familial effect after False Discovery Rate (FDR) correction (FDR-corrected P ≤ 0.05). We obtained 105 FDR-corrected significant probes giving an average maximal heritability of 81.8%, a genetic heritability of 26.9%, and a common environmental effect of 54.9% (). shows the distribution of maximal heritability estimates for these probes with maximal heritability ranging from 60 to 100%. A total of 30 probes (28.6% of all FDR-corrected significant probes) had an estimated maximal heritability of 100%. The distribution of genetic heritability and common environmental effect for the 105 most significant probes is depicted in the . Genetic heritability ranged from 0 to 91.9% while common environmental effect ranged from 8 to 77.3%. A total of 48 probes had an estimated genetic heritability of 0%. On the other hand, majority of probes with a genetic component (51 out of 57 probes) were also found significant in the alternative genetic model (data not shown).
Figure 2. Distribution of genetic heritability and common environmental effect of FDR-corrected significant probes (n = 105).
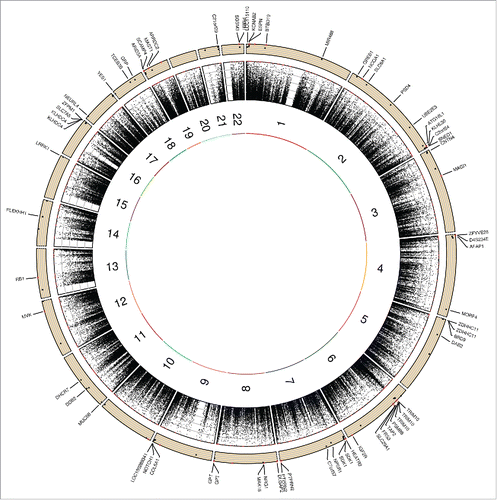
Moreover, shows Circos plot depicting the distribution of maximal heritability estimates across the genome. There is a visible higher maximal heritability on chromosome 6, which corresponds to the Major Histocompatibility Complex (MHC) region.Citation19 A total of 6 probes out of 105 were located in the MHC, and 5 were associated with genes (TAP2, TRIM10, and PSMB8). These 105 FDR-corrected significant probes were assigned to 69 genes as illustrated in .
Figure 3. Circos plot depicting the distribution of maximal heritability estimates across the genome. Moving from inner to outer circles, first circle represents chromosomes. Maximal heritability of all 472,494 probes has been represented in second circle as scatter plot (values ranging from 0 to 100%). Third circle represents maximal heritability of the 105 FDR-corrected significant probes as scatter plot with genes name in which probes are located. Note: only 4 of the 5 genes (ARID3A, ARRDC2, MAST1, SCAMP4, and ZNF235) on chromosome 19 are shown due to place limitation.
Pathways analyses
Ingenuity Pathway Analysis (IPA) revealed that 218 pathways were significantly overrepresented among genes (n = 5,625) of the 13,621 significant probes (Supplementary Table 1). Among these pathways, 15 were also overrepresented among genes (n = 69) of the 105 FDR-corrected significant probes (). These latter were related to signaling pathways (Antigen Presentation Pathway, Epithelial Adherens Junction Signaling, Cell Cycle: G1/S Checkpoint Regulation, Glioma Signaling, Pancreatic Adenocarcinoma Signaling, and Protein Ubiquitination Pathway), cholesterol metabolism [Superpathway of Cholesterol Biosynthesis, Cholesterol Biosynthesis I, Mevalonate Pathway I, Cholesterol Biosynthesis II (via 24,25-dihydrolanosterol), and Cholesterol Biosynthesis III (via Desmosterol)], alanine metabolism (Alanine Degradation III and Alanine Biosynthesis II), sugar nucleotides metabolism (GDP-mannose Biosynthesis), and cell structure biosynthesis (Colanic Acid Building Blocks Biosynthesis) (). Among those 15 pathways, only 2 were also overrepresented among genes (n = 680) of the 1,186 significant probes with a maximal heritability of 100% (Antigen Presentation Pathway and Cell Cycle: G1/S Checkpoint Regulation) (Supplementary Table 2).
Table 3. Overrepresented pathways identified among genes of FDR-corrected significant probes (n = 105).
Discussion
The aim of this study was to quantify the contribution of genetic and common environmental effects in the familial resemblance of DNA methylation levels in a family-based sample of 48 subjects from 16 families. Average absolute correlations between relative pairs revealed that DNA methylation levels of related individuals are more similar than DNA methylation levels of unrelated individuals. Similar absolute correlations were reported by McRae et al., suggesting that correlations of DNA methylation levels within family members are caused by an underlying genetic similarity.Citation9
In accordance with other studies, genetic factors across the genome seem to be the major determinant of familial resemblances in DNA methylation levels. Indeed, we obtained a ratio of 2-thirds for genetic heritability and one-third for common environmental effect for all probes (n = 472,494) and significant probes (n = 13,621). However, when considering only the most significant probes (n = 105), the ratio is reversed. In most significant probes, genetic component had an important contribution to familial resemblances in DNA methylation levels only in some probes. In the same manner, the majority of most significant probes with a genetic component in the full general model are found in the alternative genetic model. According to this alternative model, genetic heritability estimate for all probes was of 14.2%, which is slightly lower than estimates reported in other published studies. Indeed, McRae et al., using 614 individuals from 117 families of European descent, reported a genetic heritability of 19.9% in peripheral blood leukocytes.Citation9 Gruenberg et al. using 648 twins of European descent also reported a genetic heritability of 19% in adipose tissue.Citation16 Our genetic heritability estimate is more similar to the one reported by Garvin et al., which ranges from 2 to 16% in the MHC region in CD4+ lymphocytes of healthy monozygotic and dizygotic twins.Citation20 In this study, genetic heritability also varies by gene region types (CpG islands, 5′ ends of genes, conserved noncoding regions, and randomly selected regions).Citation20 Similarly, we also observed a variation in maximal heritability depending on the chromosomic and gene position, as depicted in . However, our results should be compared with caution considering that Gervin et al. only analyzed 1,760 CpG sites in the MHC region while we considered 472,494 CpG sites across the genome.Citation20 The difference in the types of cells (all leucocytes vs. CD4+ lymphocytes) should also be considered.
Regarding common environmental effect in all probes, we reported a higher percentage (4.5%) than McRae et al. (2.3%).Citation9 We can postulate that the homogenous environment to which subjects of our cohort are exposed may allow us to detect more significant common environmental effect. Indeed, all French Canadian subjects are Caucasians, lived in the same city (Quebec City), and have similar socio-economic characteristics and dietary patterns (data not shown). Moreover, dietary habits may affect DNA methylation levels of highly heritable probes. This association may be plausible since pathways related to cholesterol and alanine metabolism were significantly overrepresented among genes of the most significant probes (n = 105). Most importantly, the effect of common environmental component in those probes is non-negligible, since it contributes to different extent (8 to 77.3%) to maximal heritability in all 105 probes. The inclusion of the common environmental effect in the full general model thus allows us to detect more significant probes that contribute to better explain familial resemblances in DNA methylation levels.
Overall, we reported a maximal heritability of 12.7%, which is relatively low, but maximal heritability varies a lot from one CpG site to another (ranging from 0 to 100%) and several CpG sites (13,621 sites) show evidence of familial effect (maximal heritability ≥29.6%). Some CpG sites (105 sites) also show strong evidence of familial effect (maximal heritability ≥60%) even after FDR correction for multiple testing. We also observed highly heritable probes in the MHC region on chromosome 6, as reported by McRae et al.Citation9 Interestingly, they also reported that these probes located in the MHC had highly significant cis mQTL.
Finally, using pathways analysis, we identified overrepresented pathways among genes of significant and FDR-corrected significant probes. A total of 15 pathways were still overrepresented among genes of FDR-corrected probes, thus suggesting a strong overrepresentation of these pathways. Interestingly, among the 15 pathways, 5 were related to cholesterol metabolism. Studies in twins and family studies have demonstrated the strong heritability of cholesterol levels. Indeed, high-density lipoprotein cholesterol has an estimated heritability ranging from 40 to 60%.Citation21-23 In addition, the heritability of low-density lipoprotein peak particle diameter is 52% when considering age, body mass index (BMI), and plasma triglyceride concentrations.Citation24 Thus, this may suggest a potential link between strong DNA methylation heritability of some CpG sites within genes involved in cholesterol metabolism and the reported heritability of cholesterol levels. Obviously, we do not suggest a direct association but it sheds light on specific epigenetic factors that may contribute to the heritability of cholesterol levels. Moreover, the majority of overrepresented pathways among genes represented by the 1,186 probes with a maximal heritability of 100% were related to cellular immune response (Supplementary Table 2). This finding is in line with the fact that we observed a higher maximal heritability in the MHC region, which is known to contain genes involved in immune function.Citation25
The present study has its own limitations. The main one is the small sample size, which limits the statistical power required to detect significant genetic heritability and common environmental effect. Given the family design used in this study, the effects of common environment and epigenetic inheritance are highly intertwined and consequently difficult to estimate separately. McRae et al., using 117 families, failed to distinguish these 2 effects and reported that estimated common environmental effects may be inflated by potential epigenetic inheritance.Citation9 Therefore, separating common environmental effect and epigenetic inheritance will require a very large sample size.Citation9 In counterpart, as mentioned earlier, the homogeneity of the present study sample may allow us to detect more significant common environmental effect. Our small sample size may also explain why we observed such a large proportion of probes (45.5% of all probes) giving an estimated maximal heritability of 0%. In fact, a study by Grunberg et al. stated that a sample of 648 twins was not sufficient to obtain reliable heritability estimates of less than 10%.Citation16 Thus, our small sample size may affect the reliability of low values of heritability estimates and partly explain the large number of probes with an estimated maximal heritability of 0%. In addition, a SNP found in a probe location or nearby may cause methylation differences and impact the estimation of DNA methylation heritability.Citation8 A study by Lemire et al. demonstrated that there is extensive long-range regulation of CpG methylation associated with genetic variation.Citation26 The fact that CpG sites associated with a SNP were not excluded in the present study may affect heritability analysis and explain why we obtain 1,186 significant probes that give an estimated maximal heritability of 100%. To verify that, we conducted further analysis to investigate SNPs position in these probe locations using the 1,000 Genomes Project Phase 1 Version 3.Citation27 We found that 1,014 probes (85.5% of significant probes with a maximal heritability of 100%) have one or more SNPs within their location (defined as ± 25 base pairs from the CpG site). This suggests that the presence of SNPs at, or nearby, a CpG site may contribute to increase genetic heritability and therefore influence maximal heritability. However, we cannot confirm that SNPs ascertained in the 1,000 Genomes Project are present in our population. In addition, McRae et al. reported that the effect of SNPs within a probe location have limited effect on average genetic heritability across the genome.Citation9 Finally, DNA methylation is specific to the type of cell, tissue, locus, and developmental stages, which affects DNA methylation heritability estimates.Citation13 However, McRae et al. reported little effect of the cellular composition of blood samples on the heritability estimates for the majority of probes (genetic heritability reduced from 19.9% to 17.6%).Citation9 Other studies have also reported that DNA methylation patterns are globally correlated between 11 different somatic tissues.Citation28,29 However, it is important to consider cellular composition when interpreting and comparing DNA methylation heritability results.
In conclusion, in a family-based sample of 48 French Canadians, familial resemblances in blood leukocyte DNA methylation levels are mainly due to genetic factors when considering the average across the genome, but common environmental effect plays an important role when considering most significant probes. It would be interesting to verify possible associations between common environment effect such as dietary habits and methylation levels in highly heritable probes. Considering the small sample size upon which the present study is based, results should be viewed with caution. Further epigenome-wide studies on larger samples combined with genome-wide genotyping are needed to better understand the underlying mechanisms of DNA methylation heritability.
Patients and methods
Patients and design
A total of 48 subjects from 16 families were recruited in the Greater Quebec City metropolitan area, in Canada, as part of the GENERATION Study. Families were composed of 16 mothers, 6 fathers, and 26 children. To be eligible, parents had to be the biological parents of their child (or children). Families living under the same roof comprised at least the mother and one child aged between 8 and 18. Parents had to be in good general health, non-smokers, with BMI ranging between 18 and 35 kg/m2, and free of any metabolic conditions requiring treatment, although the use of Synthroid® (levothyroxine) or oral contraceptive was tolerated. Children also had to be non-smokers and in good general health. They were not eligible if using psycho-stimulators [Ritalin® (methylphenidate), Concerta® (methylphenidate), and Strattera® (atomoxetine)]. Parents and children were asked to complete several dietary, physical activity, medical history, and pregnancy questionnaires under the supervision of a registered dietitian during their visit at the Institute of Nutrition and Functional Foods (INAF). The experimental protocol was approved by the Ethics Committees of Laval University Hospital Research Center and Laval University.
Anthropometric measurements and blood samples
Body weight, waist girth, and height were measured according to the procedures recommended by the Airlie Conference.Citation30 Blood samples were collected from an antecubital vein into vacutainer tubes containing EDTA after 12-hour overnight fast and 48-hour alcohol abstinence. Plasma was separated by centrifugation (2,500 g for 10 min at 4°C) and blood leukocytes were collected.
DNA extraction and methylation analysis
Genomic DNA was extracted from blood leukocytes using the GenElute Blood Genomic DNA kit (Sigma-Aldrich, St. Louis, MO, USA) for all 48 samples. DNA was quantified using both NanoDrop Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and PicoGreen DNA methods. Methylation levels were measured using Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA). Bisulfite conversion and quantitative DNA methylation analysis were processed at the McGill University and Genome Quebec Innovation Center (Montreal, Canada). Illumina GenomeStudio software v2011.1 and the Methylation Module were used to analyze methylation data on 485,577 CpG sites. All samples were retained after quality control steps (bisulfite conversion, extension, staining, hybridization, target removal, negative and nonpolymorphic control probes). Methylation levels [β values varying from 0 to 1] were estimated as the proportion of total signal intensity from methylated-specific probe. Global normalization using control probes was performed in GenomeStudio. Probes with a detection P-value > 0.01 in more than 5 subjects (>10 % of all subjects) were removed, as well as probes on the X and Y chromosomes (to eliminate gender bias) and probes mapped to multiple chromosomes.Citation31 Thus, 472,494 probes were considered in the analysis.
Statistical analysis
R software v2.14.1 (R Foundation for Statistical Computing; http://www.r-project.org) Citation32 was used to compute the average absolute correlation of DNA methylation between relative pairs across all 472,494 probes. For heritability analysis, corrections were made for the effects of microarray, position on microarray, sex, age, age2, sex*age, and sex*age2, using a standard least squares model in JMP software v12. We used residuals from this model to compute heritability estimates using the variance component method implemented in QTDT v2.6.1.Citation33 We used a full general model in which the variance in methylation levels of each probe was partitioned into polygenic effects (Vg), common environmental effects shared by family members (Vc), and non-shared environmental effects unique to each individual (Ve). We tested this full general model against a null model of no familial resemblance in which Vg = Vc = 0. We then computed average maximal heritability, as the proportion of variance accounted by genetic and common environmental effects (Vg+Vc/Vg+Vc+Ve), average genetic heritability, as the proportion of variance accounted by genetics effects (Vg/Vg+Vc+Ve), and the average proportion of variance accounted by common environmental effects (Vc/Vg+Vc+Ve). Additionally, we computed heritability estimates in probes showing a significant familial effect (Vg and Vc significantly different from zero (P ≤ 0.05)). We computed FDR-corrected P-values to account for multiple testing. We also used an alternative genetic model in which the variance in methylation levels of each probe was partitioned into Vg and Ve. We then computed average genetic heritability, as the proportion of variance accounted by genetic effects (Vg/Vg+Ve). We used RCircos package in R software to make the Circos plot.Citation34 Finally, we used the IPA system (Ingenuity® Systems, www.ingenuity.com) to analyze pathways overrepresented among genes of significant and FDR-corrected significant probes. Using a right-tailed Fisher's exact test, IPA measured the likelihood that genes belong to specific overrepresented pathways. Pathway analysis was conducted with genes of significant probes (n = 13,621), significant probes with a maximal heritability of 100% (n = 1,186), and FDR-corrected significant probes (n = 105).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1232234_s02.zip
Download Zip (63.9 KB)Acknowledgments
We would like to thank Véronique Garneau and Christian Couture, who contributed to the success of this study. We also thank Catherine Raymond for the laboratory work.
Funding
This work was supported by the Canada Research Chair in Genomics Applied to Nutrition and Health. MCV is Tier 1 Canada Research Chair in Genomics Applied to Nutrition and Health. BLT is a recipient of a scholarship from Fonds de recherche du Québec – Santé (FRQS).
Related Research Data
References
- Waterland RA. Epigenetic mechanisms and gastrointestinal development. J Pediatrics 2006; 149:S137-42; PMID:17212956; http://dx.doi.org/10.1016/j.jpeds.2006.06.064
- Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA 2008; 299:1345-50; PMID:18349095; http://dx.doi.org/10.1001/jama.299.11.1345
- Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genetics 2012; 13:153-62; PMID:22290458; http://dx.doi.org/10.1038/nrm3288
- Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V, Carletto A, Pattini P, Corrocher R, Olivieri O. Epigenetic control of 11 β-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 2008; 199:323-7; PMID:18178212; http://dx.doi.org/10.1016/j.atherosclerosis.2007.11.029
- Turunen MP, Aavik E, Yla-Herttuala S. Epigenetics and atherosclerosis. Biochimica Et Biophys Acta 2009; 1790:886-91; PMID:19233248; http://dx.doi.org/10.1016/j.bbagen.2009.02.008
- Makar KW, Wilson CB. DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol 2004; 173:4402-6; PMID:15383570; http://dx.doi.org/10.4049/jimmunol.173.7.4402
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J, Team AS. Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genetics 2006; 14:159-66; PMID:16391557; http://dx.doi.org/10.1038/sj.ejhg.5201538
- Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res 2010; 20:883-9; PMID:20418490; http://dx.doi.org/10.1101/gr.104695.109
- McRae AF, Powell JE, Henders AK, Bowdler L, Hemani G, Shah S, Painter JN, Martin NG, Visscher PM, Montgomery GW. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol 2014; 15:R73; PMID:24887635; http://dx.doi.org/10.1186/gb-2014-15-5-r73
- Guibert S, Forne T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res 2012; 22:633-41; PMID:22357612; http://dx.doi.org/10.1101/gr.130997.111
- Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, van Montfrans GA, Michels RP, Bleker OP. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–45. Heart 2000; 84:595-8; PMID:11083734; http://dx.doi.org/10.1136/heart.84.6.595
- Kaati G, Bygren LO, Pembrey M, Sjostrom M. Transgenerational response to nutrition, early life circumstances and longevity. European J Hum Genetics 2007; 15:784-90; PMID:17457370; http://dx.doi.org/10.1038/sj.ejhg.5201832
- Bell JT, Spector TD. DNA methylation studies using twins: what are they telling us? Genome Biol 2012; 13:172; PMID:23078798; http://dx.doi.org/10.1186/gb-2012-13-10-172
- Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genetics 2012; 8:e1002629; PMID:22532803; http://dx.doi.org/10.1371/journal.pgen.1002629
- Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK, et al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res 2012; 22:1395-406; PMID:22800725; http://dx.doi.org/10.1101/gr.136598.111
- Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genetics 2013; 93:876-90; PMID:24183450; http://dx.doi.org/10.1016/j.ajhg.2013.10.004
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genetics 2010; 6:e1000952; PMID:20485568; http://dx.doi.org/10.1371/journal.pgen.1000952
- Drong AW, Nicholson G, Hedman AK, Meduri E, Grundberg E, Small KS, Shin SY, Bell JT, Karpe F, Soranzo N, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PloS One 2013; 8:e55923; PMID:23431366; http://dx.doi.org/10.1371/journal.pone.0055923
- Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Ann Rev Genomics Hum Genetics 2013; 14:301-23; PMID:23875801; http://dx.doi.org/10.1146/annurev-genom-091212-153455
- Gervin K, Hammero M, Akselsen HE, Moe R, Nygard H, Brandt I, Gjessing HK, Harris JR, Undlien DE, Lyle R. Extensive variation and low heritability of DNA methylation identified in a twin study. Genome Res 2011; 21:1813-21; PMID:21948560; http://dx.doi.org/10.1101/gr.119685.110
- Weissglas-Volkov D, Pajukanta P. Genetic causes of high and low serum HDL-cholesterol. J Lipid Res 2010; 51:2032-57; PMID:20421590; http://dx.doi.org/10.1194/jlr.R004739
- Qasim A, Rader DJ. Human genetics of variation in high-density lipoprotein cholesterol. Curr Atherosclerosis Reports 2006; 8:198-205; PMID:16767843; http://dx.doi.org/10.1007/s11883-006-0074-0
- Lusis AJ, Mar R, Pajukanta P. Genetics of atherosclerosis. Ann Rev Genomics Hum Genetics 2004; 5:189-218; PMID:15485348; http://dx.doi.org/10.1146/annurev.genom.5.061903.175930
- Bosse Y, Vohl MC, Despres JP, Lamarche B, Rice T, Rao DC, Bouchard C, Perusse L. Heritability of LDL peak particle diameter in the Quebec Family Study. Genetic Epidemiol 2003; 25:375-81; PMID:14639707; http://dx.doi.org/10.1002/gepi.10272
- Traherne JA. Human MHC architecture and evolution: implications for disease association studies. Int J Immunogenetics 2008; 35:179-92; PMID:18397301; http://dx.doi.org/10.1111/j.1744-313X.2008.00765.x
- Lemire M, Zaidi SH, Ban M, Ge B, Aissi D, Germain M, Kassam I, Wang M, Zanke BW, Gagnon F, et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat Commun 2015; 6:6326; PMID:25716334; http://dx.doi.org/10.1038/ncomms7326
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491:56-65; PMID:23128226; http://dx.doi.org/10.1038/nature11632
- Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, Yang AS. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genetics 2009; 18:4808-17; PMID:19776032; http://dx.doi.org/10.1093/hmg/ddp445
- Fan S, Zhang X. CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem Biophys Res Commun 2009; 383:421-5; PMID:19364493; http://dx.doi.org/10.1016/j.bbrc.2009.04.023
- Callaway C, Chumlea W, Bouchard C, Himes J, Lohman T, Martin A, Mitchell C, Mueller W, Roche A, Seefeldt V. Standardization of anthropomeric measurements : The Airlie (VA) Consensus Conference. In: TG. L, AF. R, R. M, eds. Champaign, IR, USA, 1988:39-80
- Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 2013; 6:4; PMID:23452981; http://dx.doi.org/10.1186/1756-8935-6-4
- Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria, 2013
- Abecasis GR, Cardon LR, Cookson WO. A general test of association for quantitative traits in nuclear families. Am J Hum Genetics 2000; 66:279-92; PMID:10631157; http://dx.doi.org/10.1086/302698
- Zhang H, Meltzer P, Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics 2013; 14:244; PMID:23937229; http://dx.doi.org/10.1186/1471-2105-14-244