
ABSTRACT
Aberrations in the epigenetic landscape are a hallmark of cancer. Alterations in enzymes that are “writers,” “erasers,” or “readers” of histone modification marks are common. Bromodomains are “readers” that bind acetylated lysines in histone tails. Their most important function is the regulation of gene transcription by the recruitment of different molecular partners. Moreover, proteins containing bromodomains are also epigenetic regulators, although little is known about the specific function of these domains. In recent years, there has been increasing interest in developing small molecules that can target specific bromodomains. First, this has helped clarify biological functions of bromodomain-containing proteins. Secondly, it opens a new front for combatting cancer. In this review we will describe the structures and mechanisms associated with Bromodomain and Extra-Terminal motif (BET) inhibitors and non-BET inhibitors, their current status of development, and their promising role as anti-cancer agents.
Introduction
The different cells of an organism contain the same DNA sequence but they are able to differentiate and maintain different phenotypes that express different biological functions. These processes are possible due to epigenetics. The term “epigenetics” was coined by C.H. Waddington in 1939Citation1 and later defined as heritable changes in gene expression that are not due to any alteration in the DNA sequence.Citation2 Aberrancies in epigenetic regulation are frequent in cancer, the best known epigenetic mark being DNA methylation. In general terms, cancer cells undergo global hypomethylation of their DNA, and localized hypermethylation of some gene promoters, specifically in tumor-suppressor genes.Citation3
Apart from changes in DNA methylation, covalent modification of histones is an important mechanism in the epigenetic landscape. Histones can undergo several types of modification, the most common being phosphorylation, acetylation, methylation, ubiquitination, and sumoylation.Citation4 Disruption of normal histone modification patterns is common in cancer, as are mutation and deregulation of the enzymes responsible for adding, removing, or recognizing these histone marks.Citation5
Lysine acetylation is one of the main modifications occurring in histone tails and it has been widely studied in the context of the histone code. Acetylation of chromatin has generally been associated with its open state and transcriptional activation, although more recent studies have found some acetylation marks to be responsible for the compaction of chromatin,Citation6 protein stability,Citation4 and the regulation of protein-protein interactions.
ϵ-N-acetylation of lysine residues on the amino-terminal tails of histones is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). The former act as “writers,” adding acetyl groups, the latter are “erasers,” which remove these acetyl marks. These enzymes are often aberrantly expressed in cancer, suffering mutations and being subjected to other deregulation mechanisms. For example, in the HAT group, we can find inactivating mutations in CBP (KAT3A) and p300 (KAT3B) of B cell lymphomas,Citation7 monoallelic loss of KAT5 in human lymphomas, breast and head and neck cancers,Citation8 and homozygous deletion of KAT6B in small cell lung cancer.Citation9 Additionally, HDAC deregulation results in the silencing of tumor-suppressor genes or overexpression of oncogenes. For example, HDAC1, HDAC3 and HDAC6 are overexpressed in tumors.Citation10 These studies have provided the basis for the development of HATCitation11 and HDAC inhibitors, some of which had already proved successful in clinical oncology.Citation10 However, they have two common weaknesses: a lack of efficacy and a lack of specificity.
Bromodomains (BRDs) are “readers” of acetyl marks in histone tails, targeting chromatin-modifying enzymes and other protein machinery to specific sites in the chromatin, thus regulating gene transcription. They appear to be a potential druggable epigenetic target, which has encouraged the discovery and development of several small-molecule inhibitors in recent years.
In this review we will summarize the bromodomain inhibitors discovered so far, focusing on their molecular mechanisms in cancer and their developmental status. For convenience, we will classify them as BET and non-BET inhibitors.
Bromodomains
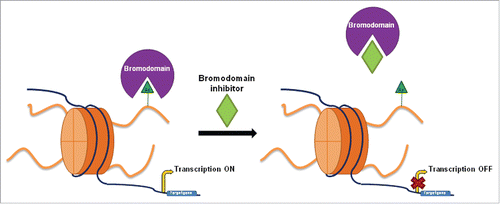
Bromodomains are a family of evolutionarily conserved motifs identified for the first time in the early 1990s in the brahma gene of Drosophila melanogaster.Citation12 BRDs bind the acetylated lysines in histone tails, the recognition of the acetyl group being decisive for the recruitment of other chromatin factors and transcriptional machinery, and thereby the regulation of gene transcription ().
Figure 1. Overview of bromodomain inhibition. Bromodomains recognize acetylation marks in histone tails and recruit transcriptional machinery promoting target gene transcription, such as in the case of c-MYC. Bromodomain inhibitors prevent interaction between the bromodomain and the acetyl group, causing the downregulation of certain genes. Bromodomains play a key role in gene transcription regulation.
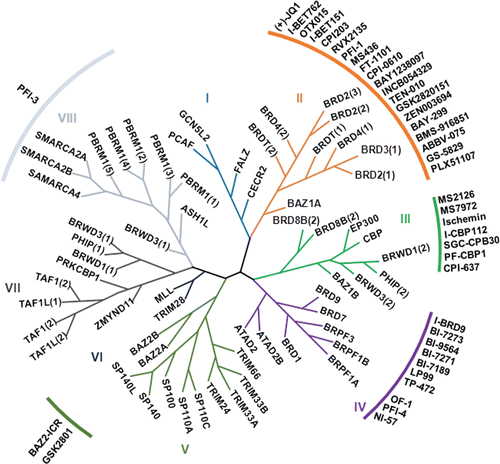
A total of 61 bromodomains were found in 46 different proteins of the human proteome.Citation13 They are classified into eight subfamilies on the basis of their structure (). They share a highly conserved structure consisting of a left-handed bundle of four α-helices (αZ, αA, αB, αC), linked by flexible loop regions known as ZA and BC loops, which are variable in sequence and charge and which form the acetyl binding site. Despite the variations in the loop regions of the various bromodomains, the amino acid residues responsible for the recognition of the acetyl lysine are highly conserved, asparagine (Asn) and tyrosine (Tyr) residues being present where a specific hydrogen bond and water-mediated interactions, respectively, occur with the acetyl group.Citation14 The cooperative binding of two acetyl groups by a single bromodomain has also been reported in BRD3, BRD4Citation13 and BRDT.Citation15 Some structural studiesCitation16 suggest the possibility of binding di-acetylated peptides by two bromodomains in tandem, such as in the transcription-initiation factors TAF1 and TAF1L, in which two bromodomains are separated by sequences of fewer than 20 residues.Citation13
Figure 2. Structure-based phylogeny of the human bromodomains and their inhibitors. There are 61 bromodomains in 46 bromodomain-containing proteins. Roman numerals indicate the eight major structural classes. The phylogenetic tree is derived from data obtained Filipakoupoulos at al. (ref 17). The specific inhibitors described in this review are indicated next to the corresponding bromodomain.
The hydrophobic nature of the acetylated lysine-binding pocket of the bromodomain, which is optimal for the interaction of the charge-neutralized acetylated lysine, and the fact that the strength of this protein-protein interaction is comparatively low, make these domains particularly targetable by small molecules that interfere with this interaction.Citation17 In recent years, numerous discoveries about the potential of new bromodomain inhibitors have been published.
BET bromodomains
The bromodomain and extra terminal (BET) family has been thoroughly investigated. It is made up of BRD2, BRD3, BRD4, and BRDT, all of which are ubiquitously expressed, except for BRDT, which is only expressed in testis. Their main features are two bromodomains in tandem (BD1 and BD2) in the N-terminal and a C-terminal extra-terminal (ET) domain.
BRD4 and BRD2 play an important role in transcription elongation by recruiting the positive transcription elongation factor complex (P-TEFb) through its BRDs to acetylated chromatin.Citation18 P-TEFb is composed of cyclin-dependent kinase-9 (CDK9) and its activator, cyclin T. Efficient transcription requires the phosphorylation of the C-terminal repeat domain (CTD) of RNA polymerase II (RNAP II). It has been reported that BRD4 recruits P-TEFb to acetylated chromatin and plays a role in activating the CDK9 kinase subunit, whereby it acts as an important transcription regulator. In addition, BRD4 controls the release of active P-TEFb from its inactive complex with HEXIM1 protein and 7S snRNA. Recruitment of P-TEFb by BRD4 is crucial for transcriptional initiation and elongation, and for the expression of genes controlling cell proliferation.Citation18
BET proteins are highly involved in cancer, directly regulating the expression of certain cancer-related genes, such as c-MYC. Avoiding the binding of BET proteins at the MYC locus with BET inhibitors leads to a reduction in cell proliferation.Citation19
BRD4 regulates NF-κβ-dependent genes, preventing degradation of Rel A, which maintains NF-κβ activity, and so plays an important role in NF-κβ-driven cancers.Citation20 In breast cancer, BRD3/4 interacts with WHSC1, promoting ESR1 transcription and thereby contributing to tamoxifen resistance in ER-positive breast cancer.Citation21
The BET family also functions as cell cycle regulators. BRD4 is important in regulating the expression of genes required for M to early G1 phase transition,Citation18,22 while BRD2 provides a scaffold on the chromatin for recruiting the key transcriptional cell cycle-regulatory genes E2F1 and E2F2. BRD2 can also interact with the SWI/SNIF complex, regulating the expression of genes such as cyclin D1 (CCND1).
BRD4 is a global regulator of gene transcription, so its inhibition would be expected to cause the global downregulation of gene activity. However, BRD4 inhibition only downregulates few hundred genes, most of which are very important in tumorigenesis. The molecular basis of this selectivity can be explained by the fact that BRD4, in addition to occupying gene promoters, has a strong preference for enhancers and super-enhancers, the latter frequently being present in key genes of hematological and solid tumors, such as MYC.Citation23
Other bromodomains
As well as BET bromodomains, BRDs are present in other proteins that play important roles in the epigenetic landscape and in cancer development.
Histone methyltransferase ASH1L and the mixed-lineage leukemia (MLL) gene product contain one BRD. Both are associated with actively transcribed genes.Citation24 The BRPF (bromodomain and PHD finger-containing) family consist of BRPF1, BRPF2 and BRPF3, which function as scaffolds for assembling HAT complexes of the MOZ/MORF family. Bromodomains are also present in transcriptional coactivators like tripartite motif-containing proteins (TRIMS) and TBP-associated factors (TAFs).Citation17
ATPase family, AAA domain containing 2 (ATAD2) contains an ATPase region and a bromodomain located in the C-terminal. ATAD2 is overexpressed in a wide variety of cancers, such as colorectal,Citation25 gastric,Citation26 endometrial,Citation27 cervical,Citation28 and ovarianCitation29 cancers, in which it increases cell proliferation. Its overexpression is already used as a poor prognosis marker. ATAD2 is an E2F target regulated by the pRb-E2F pathway, and is important for access to the S phase of the cell cycle. ATAD2 is localized in the same chromosome arm as MYC, and is co-amplified with it in several tumors. In fact, it contributes to tumor development by binding the MYC oncogene and stimulating its transcriptional activity.Citation30 ATAD2 is also associated with other oncogenic transcription factors, like the androgen receptor (AR)Citation31 and estrogen receptors (ER).Citation32 ATAD2 expression is itself stimulated by androgens and estrogens and simultaneously works as a coactivator of AR and ER, contributing to carcinogenesis in prostate and breast cancers.Citation31,32 Little is known about the specific function of the ATAD2 bromodomain, but its importance in cancer development means there is increasing interest in finding bromodomain inhibitors for it. This can help elucidate its function and establish it as a novel target for anti-cancer drug development.
BAZ2A and BAZ2B are members of the BAZ protein family. They contain a PHD finger located near a homologous bromodomain in the C-terminal part of the protein.Citation33 BAZ2A (also known as TIP5) is a component of the NoRC nuclear remodeling complex, which is essential for rRNA silencing.Citation34 Additionally, BAZ2A is overexpressed in prostate cancer and is important for maintaining cell growth. BAZ2A directly interacts with EZH2 to epigenetically silence genes repressed in metastasis, playing a different role that is independent of rRNA.Citation35 Very little is known about the function of BAZ2B, apart from the recently published histone binding preferences of its bromodomain.Citation36
Several HATs also contain bromodomains that are simultaneously “writers” and “readers” of acetyl groups. The p300/CBP-associated factor PCAF (also known as KAT2B) acetylates histones H3 and H4,Citation37 regulating the expression of several genes, like insulin, and several transcription factors, including p53,Citation38 FOX1,Citation39 and p27,Citation40 that alter various important molecular pathways in cancers such as glioblastoma and medulloblastoma.Citation41 However, little is known about the specific function of the PCAF bromodomain. There is heightened interest in finding specific inhibitors against it, because it will probably cause the loss of PCAF acetylation function. In fact, PCAF bromodomain inhibitors have already been developed for the treatment of HIV. They work by preventing the interaction between the PCAF bromodomain and acetylated HIV Tat proteinCitation42 which is a potent inhibitor of HIV replication.Citation43 In the context of cancer, several molecules targeting PCAF bromodomain are being developed,Citation43,44 but they have not been clinically tested yet.
CREBBP (also known as CBP and KAT3A), the cAMP response binding protein, and its highly homologous EP300 (also known as p300 or KAT3B), adenoviral E1 binding protein, are two HATs that, in addition to the HAT domain, contain a CREB binding domain, several zinc finger domains, a plant homology domain (PHD) and a bromodomain. These bromodomains bind the acetylated lysine 382 of p53. This interaction is required for p53 recruitment of CREBBP after DNA damage, a step which is crucial for p53 transcriptional activation of p21 in cell cycle arrest.Citation45 CPB/p300 is a transcriptional coactivator that binds to several transcription factors and acetylates specific sites in chromatin (reviewed by Dancy et al.Citation46), causing its relaxation and subsequent transcriptional activation.Citation47 These proteins also acetylate some transcription factors, modulating their activity. Besides that, CBP and EP300 play a key role in the development of many human cancers (reviewed by Iyer et al.Citation48). Acetyltransferase activity is critical for the function of CBP/EP300, so some inhibitors are known to disrupt their catalytic domain.Citation49 There is now great interest in the development of specific bromodomain inhibitors for CBP/EP300.
Bromodomain-containing protein 9 (BRD9) is a known component of the chromatin-remodeling BAF SNF/SWI complex. Little is known about its function but a role in cancer has been described. A recent study reported that AML cells require BRD9 to sustain MYC transcription and thereby increase proliferation.Citation50 Bromodomain-containing protein 7 (BRD7) is very similar to BRD9 and is a subunit of PBAF SWI/SNF. Several studies have described BRD7 as a tumor suppressor gene,Citation51 whose expression is partially or completely suppressed in several types of cancer, such as colorectal cancer,Citation52 ovarian cancer,Citation53 hepatocellular carcinoma,Citation54 small-cell lung cancer,Citation55 endometrial carcinoma,Citation56 and breast cancer, in which it acts as a partner of BRCA1.Citation57 Due to the close homology between the BRD9 and BRD7 bromodomains, co-inhibitors exist.
Bromodomain inhibitors
BET inhibitors
The first known inhibitors of the BET bromodomain family were (+)-JQ1, reported by the Structural Genomics Consortium (SGC) and the Dana-Faber Cancer Institute,Citation58 and I-BET762, reported by GlaxoSmithKline (GSK).Citation59,60
When thieno-triazolo-1,4-diazepine (JQ1) () was first reported, it was tested in NUT midline carcinoma (NMC).Citation58 BRD4 forms a fusion protein with nuclear protein in testis (NUT) protein, BRD4-NUT oncoprotein, which plays a key role in the differentiation and proliferation of this aggressive squamous cell carcinoma.Citation61 JQ1 binds competitively to acetyl-lysine binding motifs and displaces the BRD4 fusion oncoprotein from chromatin, producing squamous differentiation and specific anti-proliferative effects in xenograft models and cell lines of NMC.Citation58
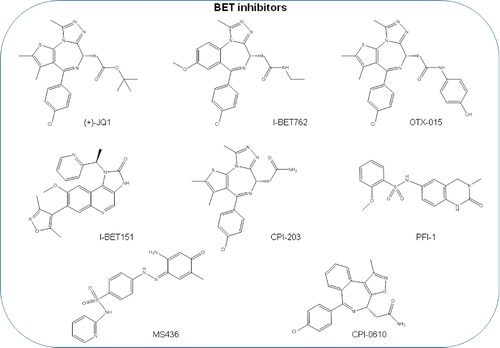
Figure 3. BET bromodomain inhibitor molecules. (+)-JQ1, I-BET762, OTX015, I-BET151, CPI203, PFI-1, MS436, CPI-0610 chemical structures are shown. RVX2135, FT-1101, BAY1238097, INCB054329, TEN-010, GSK2820151, ZEN003694, BAY-299, BMS-986158, ABBV-075, GS-5829, and PLX51107 are BET bromodomain inhibitors that are under clinical trial and whose structure has not been disclosed.
After the first report, a large number of studies were published showing the efficacy of JQ1 in hematological malignanciesCitation62-66 and in a variety of solid tumors such as glioblastoma,Citation67,68 medulloblastoma,Citation69-71 hepatocellular carcinoma,Citation72,73 colon cancer,Citation74,75 pancreatic cancer,Citation76-78 prostate cancer,Citation79-82 lung cancer,Citation83-87 and breast cancer.Citation21,88-94
The MYC oncoprotein regulates transcription and alters cell proliferation in acute myeloid leukemia,Citation62 Burkitt's lymphoma,Citation63 multiple myeloma,Citation64 and B cell acute lymphoblastic leukemia,Citation65 diffuse large B cell lymphoma.Citation66 In these hematological cancers, BET inhibition by JQ1 downregulates MYC transcription and genome-wide MYC-dependent target genes, promoting cell cycle arrest and cellular senescence. Even though MYC downregulation is the most common and important effect of BET bromodomain inhibition in hematological malignancies, other genes and mechanisms are also affected. For example, in acute lymphoblastic leukemia, JQ1 causes a significant deletion of interleukin 7 receptor gene (IL7R),Citation65 and in diffuse large B cell lymphoma it leads to the significant downregulation of MYC and E2F1 target genesCitation66 and an alteration of others such as POU2AF1 (which encodes the OCA-B transcriptional coactivator protein), BCL6, IRF8, and PAX5.Citation66
In the case of solid tumors, the treatment of a panel of a group of genetically heterogeneous glioblastoma (GBM) samples with JQ1-induced G1 cell cycle arrest and apoptosis and produces expression changes in key genes such as c-MYC, p21, hTERT, BCL−2, and BCL− XL.Citation67 Moreover, the common mutation, EGFRvIII, of the epidermal growth factor receptor (EGFR) sensitizes GBM cells to this drug. EGFRvIII regulates c-MYC levels through SOX9- and FOXG1-mediated regulation of BRD4.Citation68 In MYC-amplified medulloblastoma, JQ1 reduced cell viability and downregulated its expression, causing inhibition of MYC-associated transcriptional targets.Citation69 The treatment also altered the expression of cell cycle genes and p53 signaling.Citation70 Additionally, JQ1 promoted senescence in medulloblastoma cells by activating cell cycle kinase inhibitors and reducing E2F activity. JQ1 also attenuated stem cell signaling in MYC-driven medulloblastoma.Citation71
BRD4 is overexpressed in hepatocellular carcinoma (HCC). Treatment with JQ1 induced G1 cell cycle arrest by repressing MYC expression and caused the upregulation of p27 and the pro-apoptotic gene BIM.Citation72 Furthermore, inhibition of BRD4 by JQ1 in liver cancer repressed E2F2 cell cycle regulation, E2F2 overexpression being a marker of poor prognosis in HCC patients.Citation73
A recent study in colon cancer stated that BRD4 plays a key role in proliferation, JQ1 having an anti-growth effect, especially in tumors characterized as having the CpG island methylator phenotype (CIMP).Citation74 This is due to the presence of a CIMP-associated super-enhancer that regulates MYC transcription and from which CCAT1, a long noncoding RNA colon cancer-associated transcript 1, is transcribed, which significantly increases sensitivity to JQ1.Citation74 Another study reported that combination of JQ1 with arsenic sulfide (As4S4) in gastric and colon cancer cells inhibited BRD4 and c-MYC, synergistically activating p53.Citation75
In pancreatic ductal adenocarcinoma (PDAC), JQ1 inhibited tumor progression in patient-derived xenograft models, reducing CDC25B expression, a regulator of cell cycle progression, independently of MYC.Citation76 By contrast, a study in PDAC mouse modelsCitation77 found that tumor reduction by JQ1 was due to MYC activity inhibition together with inflammatory signals. Additionally, combination of the HDAC inhibitor SAHA and JQ1 produced upregulation of p57, a pro-apoptotic gene whose transcription is repressed by MYC and usually silenced in PDAC.Citation77 Resistance to JQ1 has also been studied in pancreatic cancer cells.Citation78 The main mechanism was the increased expression of JQ1 target genes that remain dependent on MYC, which, in turn, was co-regulated by GLI2.Citation78
Androgen receptor (AR) is a key element in castration-resistant prostate cancer (CRPC) progression, partly due to its overexpression.Citation79 Therapies targeting AR signaling have shown limitations creating a need for identifying new therapies. In 2014, Asangani et al. reported JQ1 potential activity in CRPCCitation80 showing that JQ1 reduces the levels of AR target gene transcription in AR-positive cells, inhibiting BRD4 localization in AR target loci. Prostate tumor xenograft mice treated with JQ1 also showed significant tumor reduction.Citation80 JQ1 has been broadly studied in an attempt to find a means of overcoming resistance to endocrine-based therapies in prostate cancer (PCa).Citation81 The expression of androgen receptor variants (AR-Vs) is associated with resistance to AR-targeting endocrine therapies. JQ1 downregulates AR-V transcription and protein expression, inhibiting cell growth driven by AR-Vs in vitro and in vivo.Citation81 Another study revealed that JQ1 inhibits the interaction between BRD4 and ERG, a transcription factor aberrantly upregulated in prostate cancer. This interaction seems to be essential for ERG-mediated cell invasion in this type of cancer.Citation82 All these studies support the potential activity of BET inhibitors in prostate cancer. In fact, clinical trials with I-BET762, GSK525762, ZEN003694, GS-5829, and OTX015 to treat castration-resistant prostate cancer are currently underway ().
Table 1. BET inhibitors
In non-small cell lung cancer (NSCLC), JQ1 was found to be effective in several lung adenocarcinoma cell lines due to its suppression of the oncogenic transcription factor FOSL1 and its targets.Citation83 JQ1 treatment also produced tumor regression in NSCLC KRAS mutant mice by inhibiting MYC function. However, KRAS and, simultaneously, liver kinase B1- (LKB1-) mutated mice were not sensitive to JQ1.Citation84 Nevertheless, human lung adenocarcinoma tumors with loss of wild type LKB1 have poor outcome and high invasiveness mediated by upregulation of MYC via an increase in MZF1 expression and can be overcome by treatment with JQ1.Citation85 Small cell lung cancer (SCLC) cell lines are especially sensitive to growth inhibition by JQ1, not due to changes in MYC expression, but because of the downregulation of the lineage-specific transcription factor achaete-scute homolog-1 (ASCL1), which is overexpressed in more than 50% of SCLC patients.Citation86 JQ1 disrupts the interaction between BRD4 and ASCL1 enhancer.Citation87
Several studies were made concerning JQ1 treatment in standard therapy-resistant breast cancer models. For example, in tamoxifen-resistant breast cancer, JQ1 demonstrated anti-tumor activity in mouse models and a synergistic activity with fulvestrant, an ER degrader.Citation21 Tamoxifen has been the first-line treatment for estrogen receptor alpha (ERα)-positive breast tumors, although resistance to this drug is very common.Citation88 JQ1 effect stems from its capacity to suppress the classic ERα signaling pathway. WHSC1, a histone H3K36 methyltransferase, is a positive regulator of ERα signaling in breast cancer that is recruited to the ERα gene by BRD3/4. JQ1 inhibits this interaction, slowing the rate of cell growth in tamoxifen-resistant breast cancer cells.Citation21
Another study reported that JQ1 blocks the transition of RNA polymerase II from initiation to elongation induced by estrogen (E2), establishing that BET proteins are mediators of E2-induced transcriptional activation.Citation89 Upregulation of MYC driven by BRD4 was observed in everolimus-resistant ER+ breast cancers. Depletion of MYC resensitized cells to everolimus, and JQ1 in combination with this drug decreased tumor growth in xenograft models.Citation90 Moreover, the combination of JQ1 with HDAC inhibitors such as mocetinostat resulted in a greater reduction in cell viability for triple-negative and ER+ breast cancer cells.Citation91 This synergistic effect was associated with the decreased expression of genes that play a key role in cell cycle progression and a significant increase in the expression of genes from the ubiquitin-specific protease (USP17) family, which were able to diminish the activity of the RAS/MAPK pathway, resulting in reduced cell viability.Citation91 In vitro and in vivo triple-negative breast cancer (TNBC) models were found to be more highly sensitive to BET inhibitors than to more resistant luminal lines. JQ1 treatment results in growth inhibition by cell cycle arrest in G1 and apoptosis. Similar results were obtained treating TNBC xenografts.Citation92 Data show that JQ1 selective disruption of super-enhancer-associated genes alters transcriptional pathways involved in cell proliferation, tumor invasion and survival.Citation92 Another study in TNBC reported that treatment with JQ1 resulted in a reduction of transcription factors like the DEP domain containing 1 (DEPDC), Forkhead box M1 (FOXM1), and Lim domain only 4 (LM04). It also suggests that BET inhibitors could be a new targeted therapy for TNBC, the most aggressive form of breast cancer, for which treatment is currently limited to chemotherapy.Citation93 Moreover, TNBC is the breast cancer subtype most frequently associated with hypoxia.Citation95 JQ1 modulated 44% of hypoxia-responsive genes,Citation94 most of which were downregulated, including carbonic anhydrase 9 (CA9) and vascular endothelial growth factor A (VEGF-A). This study concluded that BET inhibitors jointly target angiogenesis and the hypoxic response, making it an effective anti-tumor combination.Citation94
At the same time as the apparition of JQ1, another diazepine-based compound, I-BET762 (GSK525762) (), was independently reported by GSK.Citation60 I-BET762, like JQ1, arrests the growth of NMC-malignant cells. GSK started clinical trials for the treatment of this malignancy that were subsequently extended to other types of cancer, such as small cell lung cancer, non-small cell lung cancer, colorectal cancer, neuroblastoma, castration-resistant prostate cancer, triple-negative breast cancer, estrogen receptor-positive breast cancer, and MYCN driven solid tumor subjects (). I-BET762 has also been reported to exert antiproliferative effects and to induce apoptosis in multiple myeloma in vitro and in vivo.Citation96 It is also effective in prostate cancer, whereby tumor growth is reduced by inhibiting MYC.Citation97
OTX015 (MK-8628) () is a BRD2/3/4 inhibitor under evaluation in dose-finding studies for solid tumors as glioblastoma multiform. OTX015 showed a stronger anti-proliferative effect than JQ1, and a significant anti-tumoral effect in GBM mouse models has recently been reported.Citation98 Treatment with OTX015 leads to cell growth inhibition, cell cycle arrest and apoptosis in acute leukemia cell lines, indicating that OTX015 and JQ1 have similar effects in leukemic cells.Citation99 There are currently six clinical trials underway with OTX015 in GBM, hematological malignancies and several other solid tumors like NUT midline carcinoma, TNBC, castration-resistant prostate cancer and pancreatic ductal carcinoma ().
I-BET151 (GSK1210151A) () was reported in 2011 as being a novel BET bromodomain inhibitor that had proved its efficacy in mixed-lineage leukemia (MLL) cell lines at inducing apoptosis and cell cycle arrest, due in part to a decrease in the BCL-2, MYC, and CDK6 genes through the inhibition of BRD3/BRD4.Citation100 It also exhibits potent anti-myeloma activity through transcriptional repression of MYC by avoiding P-TEFb chromatin occupation, and upregulation of HEXIM1, which is an inhibitor of P-TEFb.Citation96 I-BET151 was reported to be an alternative treatment for myeloproliferative neoplasms driven by constitutively active JAK2 kinaseCitation101 and glioblastoma, in which it inhibits proliferation by arresting cell cycle progression.Citation102 It was also found to be effective for treating NPM1-mutated acute myeloid leukemia.Citation103 In melanoma cells, on the one hand, I-BET151 induces BIM-dependent apoptosis and cell cycle arrest,Citation104 while, on the other controlling NF-κB activity.Citation105
CPI203 () is a BET bromodomain inhibitor that has shown synergetic activity with drugs that have already been clinically approved for cancer treatment. Multiple myeloma cells resistant to bortezomib and melphalan treated with CPI203 plus bortezomib increased the apoptosis level and decreased the proliferation rate.Citation106 Furthermore, CPI203 demonstrated its efficacy in pancreatic neuroendocrine tumors (PanNET) by downregulating MYC expression, producing G1 cell cycle arrest and almost completely inhibiting cell proliferation. It also enhanced the antitumor effects of rapamycin in PanNET, attenuating rapamycin-induced AKT activation, which is a major limitation of rapamycin therapy.Citation107 CPI203 synergistic antitumor activity with lenalidomide, reported in bortezomib-resistant mantle cell lymphoma, caused simultaneous MYC and IRF4 downregulation and apoptosis induction.Citation108
RVX2135 inhibits proliferation and induces apoptosis in Myc-induced murine lymphoma, affecting many transcription factor networks.Citation109
PFI-1 () was reported as being a BET inhibitor for BRD2 and BRD4 that showed anti-proliferative efficiency in leukemic cell lines. Treatment with PFI-1 produced cell cycle arrest in G1, downregulation of MYC expression and induction of apoptosis. Unlike other BET inhibitors, PFI-1 caused significant downregulation of Aurora B kinase, suggesting a potential synergism between MYC and Aurora B, two potent oncogenes that could be targeted simultaneously with this inhibitor.Citation110
MS436 () is another BRD4 inhibitor that was reported to inhibit NF-Kβ-directed production of nitric oxide and pro-inflammatory cytokine interleukin-6 in murine macrophages.Citation111 However, since first being reported in 2013, no further studies have been published concerning this molecule.
FT-1101,Citation112 CPI-0610113 () are novel BET inhibitors that were reported recently and are undergoing clinical trials in hematological cancers. BAY 1238097114 and INCB054329115 are being clinically trialed for several solid tumors and hematological malignancies. Also currently the subject of clinical trials are TEN-010,Citation116 for NUT-midline carcinoma and advanced solid tumors, GSK2820151, for subjects with advanced or recurrent solid tumors, ZEN003694, which is for metastatic castration-resistant prostate cancer, and BMS-986158, ABBV-075, GS-5829, and PLX51107, for several types of cancer ().
Finally, BAY-299 is an inhibitor of BRD1 and the second bromodomain of TAF1 reported on the SGC webpage, as a collaboration with Bayer.Citation117
Non-BET inhibitors
BET-family inhibitors have been extensively studied in recent years, but far less attention has been paid to the other bromodomains. Given that the latter are frequently in proteins with other epigenetic functions, such as HAT activity, finding non-BET inhibitors will allow a better understanding of them. This will also help identify new druggable targets for treating diseases like cancer ().
Table 2. Non-BET inhibitors
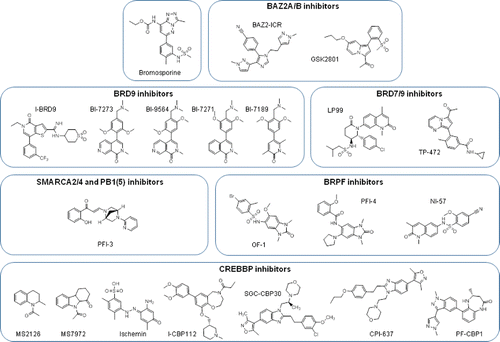
In this context, BromosporineCitation118 () is a multi-bromodomain inhibitor, which has been made available by the SGC. It acts as a broad-spectrum bromodomain inhibitor and can be useful for studying biological functions.
Figure 4. Non-BET bromodomain inhibitor molecules. Chemical structures of non-BET inhibitors, clustered according to the specific bromodomains in which they act. Bromosporine is a multibromodomain inhibitor.
The ATAD2 bromodomain is known to be difficult to drug.Citation119 GSK reported the first micromolar inhibitor of ATAD2, but it was not selective for the BET family.Citation120 This compound was then optimized to improve its activity to sub-nanomolar and 100-fold BET selectivity.Citation121 Studies to improve this compound are being conducted but have not yet been published.
Although the homologous bromodomains BAZ2A and BAZ2B also have low predicted druggability,Citation119 to date, two structurally distinct selective inhibitors have been reported, BAZ2-ICR and GSK2801 ().Citation122,123
BAZ2-ICRCitation122 was published in 2015 by the Institute of Cancer Research. It is suitable for use in vitro and in vivo and is available to the scientific community for the purpose of further investigating the biological role of BAZ2A/B. GSK2801123 was investigated as a collaboration between GSK and SGC as found to be a potent, selective bromodomain inhibitor of BAZ2A and BAZ2B that acts in a competitive binding model with acetyl-lysines. It is also suitable for treatments in vitro and in animal models.Citation123
BRD9 is a component of the chromatin remodeling complex SNF/SWI BAF but its biological function has not yet been fully elucidated.Citation124 Its potential role in disease has encouraged research on inhibitors, and several BRD9 chemical probes have been reported so far. Filippakopoulos and colleagues described the 9-H purine scaffold as a template to be used for further studies involving inhibition of BRD9.Citation125 Subsequently, GSK in collaboration with the University of Strathclyde reported I-BRD9126 (), a selective BRD9 inhibitor with more than 700-fold selectivity over the BET family and 200-fold selectivity over the highly homologous BRD7.Citation126 The SGC and the University of Oxford published LP99 (), a potent and selective inhibitor for both BRD9 and BRD7 bromodomains, which plays a role in regulating pro-inflammatory cytokine secretion.Citation127 Two other BRD9 inhibitors, BI-7273 and BI-9564, were described by Boehringer Ingelheim and the SGC ().Citation128 These compounds exert antitumor activity in an AML xenograft model.Citation128 Moreover, BI-7271, BI-7273, and BI-7189 BRD9 inhibitors () suppressed the proliferation of AML cells.Citation50 The SGC website has recently added TP-472 (), a new inhibitor of the BRD9/7 bromodomain that is also suitable for in vivo treatments.Citation129
Inhibitors of BRPF family bromodomains are also being investigated in order to elucidate their biological role and druggability. In 2014, GSK reported a series of selective drugs for the BRPF1 bromodomain. These are benzimidazolone compounds that showed 100- and >1000-fold selectivity over BRPF2 and BRPF3, respectively.Citation130 The SGC disclosed OF-1 on its website (), describing that it inhibits BRPF1B/2/3 bromodomains, and has good selectivity against other bromodomains, although the closest off target is BRD4 (39-fold selectivity).Citation131 The SGC in collaboration with Pfizer has reported PFI-4 (), a selective BRPF1B bromodomain inhibitor, and an isoform of BRPF1 that binds to acetylated histones in the opposite manner to isoform BRPF1B.Citation132 NI-57 () is another compound listed on the SGC webpage that was discovered by them in collaboration with University College London, and which selectively inhibits all members of the BRPF family. The closest off target is BRD9 (32-fold selectivity), but is very selective against other bromodomains, including those of the BET family.Citation133 Recently, a dual inhibitor of BRPF1 and TRIM24 was reported, which exerts good selectivity over other bromodomains.Citation134
SMARCA4 (BRG1), SMARCA2, and PB1 are bromodomain-containing proteins that are also members of the SWI/SNF chromatin-remodeling complexes. Loss of function of SMARCA4135 and other alterations in components of the SWI/SNF complex are associated with cancer development. PB1 (BAF180) contains six bromodomains that are frequently mutated in cancer.Citation136 The SMARCA4 bromodomain is involved in DNA damage repair.Citation137 The SGC reported the development of PFI-3 (), a bromodomain inhibitor of SMARCA2/4 and PB1(5),Citation138,139 which showed high selectivity over other bromodomains. However, subsequent studies with PFI-3 determined that the ATPase catalytic domain of SMARCA2/4 has an indispensable role in cancer growth, whereas bromodomain inhibition is not crucial for the process, thereby highlighting the importance of the ATPase domain in these proteins as a significant therapeutic target.Citation140 A recent study reported the optimization of a compound inhibiting the second and fifth bromodomains of PB1 more selectively than SMARCA2/4, and another one inhibiting PB1(5).Citation141
CREBBP bromodomain inhibitors are the second most thoroughly studied group, after the BET family inhibitors. The first attempts to do so involved inhibiting the CREBBP-p53 interaction using the compounds MS2126 and MS7972 ().Citation142 Treatment of U2OS cells with previously stimulated DNA damage resulted in a dramatic decrease in p53 levels.Citation142 Ischemin () was reported by the same groupCitation143 to be a bromodomain inhibitor of CREBBP that is also able to inhibit this interaction and consequently alter the expression of p53 target genes involved in apoptosis and DNA damage repair.Citation143 I-CBP112 () was reported by SGC and GSK as being a novel compound for CREBBP and P300 bromodomains with nanomolar activity and good selectivity.Citation144 This compound impaired the disease-initiating self-renewal leukemic cells in vitro and in vivo.Citation144 A recent study of I-CBP112145 reported that this compound stimulates acetylation activity by CREBBP/P300 acting as an activator of these HATs, contributing to its anti-proliferative effect in cancer. It is also suggested that activation activity of I-CBP112 could help restore the balance of acetylation levels in tumors that experience CREBBP/P300 loss of function due to mutations, although the mechanism and networks involved in tumors affected by this activation are not yet fully understood.Citation145 SGC-CBP30 () was reported by the SGC and the University of Oxford.Citation146 This compound is selective for CREBBP/P300 bromodomains but is not suitable for use in vivo because it is metabolized so quickly. SGC-CBP30 was reported to suppress the Th17 response that is critical to a variety of human autoimmune diseases.Citation147 Moreover, I-CBP112 and SGC-CBP30 suppress the lymphocyte-specific transcription factor IRF4, which is crucial for the viability of myeloma cells, and consequently targets c-MYC.Citation148 PF-CBP1 () is another selective inhibitor of the CREBBP/P300 bromodomain, which downregulates a number of inflammatory genes in macrophages and the RSG4 gene in neuronsCitation149 Finally, another CREBBP/P300 bromodomain inhibitor, CPI-637 (),Citation150 was recently reported to have high potency in vitro and selectivity over other bromodomains.
Targeting protein acetylation beyond bromodomain inhibition and cancer: HAT activation and other human diseases
Apart from cancer, bromodomains are key transcriptional regulators in diabetes, inflammation and cardiovascular diseases (reviewed by DenisCitation151 and Nicholas DA et al.Citation152), and are considered potentially druggable to treat these disorders.
RVX-208 (RVX000222; apabelaton) was developed by Resverlogix Corporation to treat atherosclerotic cardiovascular diseases.Citation153 Its use is currently being investigated in several clinical trials for atherosclerosis, coronary syndromes and Alzheimer disease.Citation154,155 RVX-208 is an orally available BET bromodomain inhibitor which selectively inhibits BRD2.Citation156 It increases apolipoprotein AI (ApoA-I) gene transcription and leads to production of high density lipoprotein (HDL) cholesterol levels in vitro and in vivo,Citation157 resulting in the stimulation of reverse cholesterol transport. It also represses inflammatory and atherosclerotic pathways that contribute directly to cardiovascular risk.Citation158 RVX-208 is also being tested in patients with pre-diabetes, demonstrating effects in HDL lipidome and glucose metabolism that may protect against the development of type 2 diabetes.Citation159
Furthermore, some of the bromodomain inhibitors studied for cancer therapies have also been tested in diabetes and inflammatory diseases. For example, I-BET151 suppressed the development of type 1 diabetes in mice.Citation160 Another study suggests that BET inhibitors may be useful to treat diabetic patients resistant to insulin because it was observed that JQ1 increases insulin secretion in vitro and decreased intracellular triglyceride stores in cells.Citation161 In inflammation, JQ1 was reported to suppress psoriasis-like skin inflammation in mice by modulating RORC/IL-17A pathway.Citation162 In addition, JQ1163 and I-BET151164 prevented synovial inflammation in rheumatoid arthritis synovial fibroblasts. I-BET151 also regulates IL-6 production, decreasing the early symptoms in a multiple sclerosis mouse model.Citation165 I-BET762 was also reported to have high anti-inflammatory potential by regulating the expression of key inflammatory genes.Citation59 Moreover, a recent study described that inhibition of BET bromodomain also suppresses vascular inflammation by inhibiting NF-kB and MAPK activation.Citation166
Aberrant protein acetylation is common in cancer as well as in other disorders such as neurological and cardiac disease. Besides bromodomain inhibition, HAT activation emerged as other potential strategy in treating diseases through modulation of protein acetylation levels and consequent gene expression regulation. Several positive modulators of HATs have been identified in the last few years. CTPB, an anacardic acid derivative, was reported as a selective activator of p300 (KAT3B) HAT activity but not PCAF (KAT2B), which enhanced HAT-dependent transcriptional activation.Citation167 Posterior studies reported long chain alkylidenemalonates (LoCAMs) as a novel class of HAT modulators with a powerful apoptotic effect.Citation168 Important to highlight in this family of compounds are the unique properties of pentadecylidenemalonate 1b (SPV106), that leads to PCAF (KAT2B) acetylation activity increase, together with its inhibitory properties against CBP (KAT3A) and p300 (KAT3B).Citation168,169 This inspired the synthesis of SVP106 analogs that feature different levels of activity modulation for KAT2B and KAT3B simultaneously.Citation170 SPV106 has been successfully used in neurologicalCitation171 and cardiologicalCitation172,173 studies where the acetylation activity of KAT2B was investigated. Moreover, in cardiac mesenchymal cells, SPV106 recovered differentiation and proliferation in patients with diabetes.Citation174 Additionally, TTK21 was reported as an activator of CBP (KAT3A) and p300 (KAT3B), which was able to promote neurogenesis and increased long-term memory in adult mice.Citation175 These HAT positive modulators may be useful for a better understanding the biology of HATs and also may open new treatment strategies for cancer as well as for other diseases frequent in developed societies like cardiac and brain disease.
Concluding remarks
Since the discovery of the high potency of (+)-JQ1 and I-BET762 in NMC and subsequently in other types of cancer, a large number of bromodomain inhibitors have been developed. Currently, several clinical trials of BET inhibitors, representing a new family of compounds for cancer-targeted therapy, are underway. Discovery of non-BET inhibitors will also help elucidate the possible role(s) of bromodomain-containing proteins in cancer, and reveal new druggable targets for combating this disease. The problems of specificity should be overcome in the near future, but bromodomain inhibitors already show great potential as new drugs in cancer therapy. Progress in this field of research should reveal new molecular targetable pathways, and even novel biomarkers that predict bromodomain-inhibitor sensitivity, which would represent a significant step toward personalized medicine.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Waddington CH. Preliminary notes on the development of the wings in normal and mutant strains of Drosophila. Proc Natl Acad Sci USA 1939; 25(7):299-307; PMID:16577903; https://doi.org/10.1073/pnas.25.7.299
- Holliday R. The inheritance of epigenetic defects. Science 1987; 238(4824):163-70; PMID:3310230; https://doi.org/10.1126/science.3310230
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358(11):1148-59; PMID:18337604; https://doi.org/10.1056/NEJMra072067
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128(4):693-705; PMID:17320507; https://doi.org/10.1016/j.cell.2007.02.005
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev 2009; 23(7):781-783; PMID:19339683; https://doi.org/10.1101/gad.1787609
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311(5762):844-7; PMID:16469925; https://doi.org/10.1126/science.1124000
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011; 471(7337):189-95; PMID:21390126; https://doi.org/10.1038/nature09730
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007; 448(7157):1063-7; PMID:17728759; https://doi.org/10.1038/nature06055
- Simo-Riudalbas L, Perez-Salvia M, Setien F, Villanueva A, Moutinho C, Martinez-Cardus A, Moran S, Berdasco M, Gomez A, Vidal E, et al. KAT6B Is a tumor suppressor histone H3 lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Res 2015; 75(18):3936-45; PMID:26208904; https://doi.org/10.1158/0008-5472.CAN-14-3702
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5(9):769-84; PMID:16955068; https://doi.org/10.1038/nrd2133
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol 2010; 17(5):471-82; PMID:20534345
- Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, Kennison JA. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992; 68(3):561-72; PMID:1346755; https://doi.org/10.1016/0092-8674(92)90191-E
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012; 149(1):214-31; PMID:22464331; https://doi.org/10.1016/j.cell.2012.02.013
- Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007; 26(37):5521-7; PMID:17694091; https://doi.org/10.1038/sj.onc.1210618
- Moriniere J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009; 461(7264):664-8; PMID:19794495; https://doi.org/10.1038/nature08397
- Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett 2012; 586(17):2692-704; PMID:22710155; https://doi.org/10.1016/j.febslet.2012.04.045
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 2014; 13(5):337-56; PMID:24751816; https://doi.org/10.1038/nrd4286
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005; 19(4):535-45; PMID:16109377; https://doi.org/10.1016/j.molcel.2005.06.029
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146(6):904; PMID:21889194; https://doi.org/10.1016/j.cell.2011.08.017
- Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, Nair S, Chen LF. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene 2014; 33(18):2395-404; PMID:23686307; https://doi.org/10.1038/onc.2013.179
- Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, Lanz R, He B, Wang L, Fu X, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res 2014; 24(7):809-19; PMID:24874954; https://doi.org/10.1038/cr.2014.71
- Yang Z, He N, Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol 2008; 28(3):967-76; PMID:18039861; https://doi.org/10.1128/MCB.01020-07
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013; 153(2):320-34; PMID:23582323; https://doi.org/10.1016/j.cell.2013.03.036
- Mohan M, Lin C, Guest E, Shilatifard A. Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer 2010; 10(10):721-8; PMID:20844554; https://doi.org/10.1038/nrc2915
- Hou M, Huang R, Song Y, Feng D, Jiang Y, Liu M. ATAD2 overexpression is associated with progression and prognosis in colorectal cancer. Jpn J Clin Oncol 2016; 46(3):222-7; PMID:26819280; https://doi.org/10.1093/jjco/hyv195
- Zhang M, Zhang C, Du W, Yang X, Chen Z. ATAD2 is overexpressed in gastric cancer and serves as an independent poor prognostic biomarker. Clin Transl Oncol 2016; 18(8):776-81; PMID:26527032; https://doi.org/10.1007/s12094-015-1430-8
- Shang P, Meng F, Liu Y, Chen X. Overexpression of ANCCA/ATAD2 in endometrial carcinoma and its correlation with tumor progression and poor prognosis. Tumour Biol 2015; 36(6):4479-85; PMID:25934333; https://doi.org/10.1007/s13277-015-3089-8
- Zheng L, Li T, Zhang Y, Guo Y, Yao J, Dou L, Guo K. Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol Rep 2015; 33(5):2337-44; PMID:25813398; https://doi.org/10.3892/or.2015.3867
- Wan WN, Zhang YX, Wang XM, Liu YJ, Zhang YQ, Que YH, Zhao WJ. ATAD2 is highly expressed in ovarian carcinomas and indicates poor prognosis. Asian Pacific J Cancer Prev 2014; 15(6):2777-83; PMID:24761900; https://doi.org/10.7314/APJCP.2014.15.6.2777
- Ciro M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J, et al. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res 2009; 69(21):8491-8; PMID:19843847; https://doi.org/10.1158/0008-5472.CAN-09-2131
- Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung HJ, Chen HW. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res 2009; 69(8):3339-46; PMID:19318566; https://doi.org/10.1158/0008-5472.CAN-08-3440
- Zou JX, Revenko AS, Li LB, Gemo AT, Chen HW. ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification. Proc Natl Acad Sci U S A 2007; 104(46):18067-72; PMID:17998543; https://doi.org/10.1073/pnas.0705814104
- Jones MH, Hamana N, Nezu J, Shimane M. A novel family of bromodomain genes. Genomics 2000; 63(1):40-5; PMID:10662543; https://doi.org/10.1006/geno.1999.6071
- Guetg C, Lienemann P, Sirri V, Grummt I, Hernandez-Verdun D, Hottiger MO, Fussenegger M, Santoro R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J 2010; 29(13):2135-46; PMID:20168299; https://doi.org/10.1038/emboj.2010.17
- Gu L, Frommel SC, Oakes CC, Simon R, Grupp K, Gerig CY, Bar D, Robinson MD, Baer C, Weiss M, et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat Genet 2015; 47(1):22-30; PMID:25485837; https://doi.org/10.1038/ng.3165
- Philpott M, Yang J, Tumber T, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Keates T, Felletar I, Ciulli A, et al. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol Biosyst 2011; 7(10):2899-908; PMID:21804994; https://doi.org/10.1039/c1mb05099k
- Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem 1999; 274(3):1189-92; PMID:9880483; https://doi.org/10.1074/jbc.274.3.1189
- Kim JY, Lee KS, Seol JE, Yu K, Chakravarti D, Seo SB. Inhibition of p53 acetylation by INHAT subunit SET/TAF-Ibeta represses p53 activity. Nucleic Acids Res 2012; 40(1):75-87; PMID:21911363; https://doi.org/10.1093/nar/gkr614
- Ge X, Jin Q, Zhang F, Yan T, Zhai Q. PCAF acetylates {beta}-catenin and improves its stability. Mol Biol Cell 2009; 20(1):419-27; PMID:18987336; https://doi.org/10.1091/mbc.E08-08-0792
- Perez-Luna M, Aguasca M, Perearnau A, Serratosa J, Martinez-Balbas M, Jesus Pujol M, Bachs O. PCAF regulates the stability of the transcriptional regulator and cyclin-dependent kinase inhibitor p27 Kip1. Nucleic Acids Res 2012; 40(14):6520-33; PMID:22547391; https://doi.org/10.1093/nar/gks343
- Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M, Helin K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res 2013; 73(20):6323-33; PMID:23943798; https://doi.org/10.1158/0008-5472.CAN-12-4660
- Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, Verdin E, Zhou MM. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol Cell 2002; 9(3):575-86; PMID:11931765; https://doi.org/10.1016/S1097-2765(02)00483-5
- Wang Q, Wang R, Zhang B, Zhang S, Zheng Y, Wang Z. Small organic molecules targeting PCAF bromodomain as potent inhibitors of HIV-1 replication. Med Chem Comm 2013; 4(4):737-40; https://doi.org/10.1039/C3MD20376J
- Chaikuad A, Lang S, Brennan PE, Temperini C, Fedorov O, Hollander J, Nachane R, Abell C, Muller S, Siegal G, et al. Structure-based identification of inhibitory fragments targeting the p300/CBP-associated factor bromodomain. J Med Chem 2016; 59(4):1648-53; PMID:26731131; https://doi.org/10.1021/acs.jmedchem.5b01719
- Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand Sanchez R, Zeleznik-Le NJ, Ronai Z, Zhou MM. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol Cell 2004; 13(2):251-63; PMID:14759370; https://doi.org/10.1016/S1097-2765(03)00528-8
- Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev 2015; 115(6):2419-52; PMID:25594381; https://doi.org/10.1021/cr500452k
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996; 87(5):953-9; PMID:8945521; https://doi.org/10.1016/S0092-8674(00)82001-2
- Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene 2004; 23(24):4225-31; PMID:15156177; https://doi.org/10.1038/sj.onc.1207118
- Arif M, Pradhan SK, Thanuja GR, Vedamurthy BM, Agrawal S, Dasgupta D, Kundu TK. Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J Med Chem 2009; 52(2):267-77; PMID:19086895; https://doi.org/10.1021/jm800657z
- Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, Bader G, McConnell D, Pearson M, Gerstberger T, et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol 2016; 12(9):672-9; PMID:27376689; https://doi.org/10.1038/nchembio.2115
- Yu X, Li Z, Shen J. BRD7: a novel tumor suppressor gene in different cancers. Am J Transl Res 2016; 8(2):742-8; PMID:27158366
- Wu WJ, Hu KS, Chen DL, Zeng ZL, Luo HY, Wang F, Wang DS, Wang ZQ, He F, Xu RH. Prognostic relevance of BRD7 expression in colorectal carcinoma. Eur J Clin Invest 2013; 43(2):131-40; PMID:23215825; https://doi.org/10.1111/eci.12024
- Park YA, Lee JW, Kim HS, Lee YY, Kim TJ, Choi CH, Choi JJ, Jeon HK, Cho YJ, Ryu JY, et al. Tumor suppressive effects of bromodomain-containing protein 7 (BRD7) in epithelial ovarian carcinoma. Clin Cancer Res 2014; 20(3):565-75; PMID:24198243; https://doi.org/10.1158/1078-0432.CCR-13-1271
- Chen CL, Wang Y, Pan QZ, Tang Y, Wang QJ, Pan K, Huang LX, He J, Zhao JJ, Jiang SS, et al. Bromodomain-containing protein 7 (BRD7) as a potential tumor suppressor in hepatocellular carcinoma. Oncotarget 2016; 7(13):16248-61; PMID:26919247; https://doi.org/10.18632/oncotarget.7637
- Li D, Yang Y, Zhu G, Liu X, Zhao M, Li X, Yang Q. MicroRNA-410 promotes cell proliferation by targeting BRD7 in non-small cell lung cancer. FEBS Lett 2015; 589(17):2218-23; PMID:26149213; https://doi.org/10.1016/j.febslet.2015.06.031
- Park YA, Lee JW, Choi JJ, Jeon HK, Cho Y, Choi C, Kim TJ, Lee NW, Kim BG, Bae DS. The interactions between MicroRNA-200c and BRD7 in endometrial carcinoma. Gynecol Oncol 2012; 124(1):125-33; PMID:22015043; https://doi.org/10.1016/j.ygyno.2011.09.026
- Harte MT, O'Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT, Mullan PB, Harkin DP. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res 2010; 70(6):2538-47; PMID:20215511; https://doi.org/10.1158/0008-5472.CAN-09-2089
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature 2010; 468(7327):1067-73; PMID:20871596; https://doi.org/10.1038/nature09504
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010; 468(7327):1119-23; PMID:21068722; https://doi.org/10.1038/nature09589
- Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem 2013; 56(19):7501-15; PMID:24015967; https://doi.org/10.1021/jm401088k
- French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2008; 27(15):2237-42; PMID:17934517; https://doi.org/10.1038/sj.onc.1210852
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011; 478(7370):524-8; PMID:21814200; https://doi.org/10.1038/nature10334
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ, 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 2011; 108(40):16669-74; PMID:21949397; https://doi.org/10.1073/pnas.1108190108
- Soodgupta D, Pan D, Cui G, Senpan A, Yang X, Lu L, Weilbaecher KN, Prochownik EV, Lanza GM, Tomasson MH. Small molecule MYC inhibitor conjugated to integrin-targeted nanoparticles extends survival in a mouse model of disseminated multiple myeloma. Mol Cancer Ther 2015; 14(6):1286-94; PMID:25824336; https://doi.org/10.1158/1535-7163.MCT-14-0774-T
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012; 120(14):2843-52; PMID:22904298; https://doi.org/10.1182/blood-2012-02-413021
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013; 24(6):777-90; PMID:24332044; https://doi.org/10.1016/j.ccr.2013.11.003
- Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res 2013; 19(7):1748-59; PMID:23403638; https://doi.org/10.1158/1078-0432.CCR-12-3066
- Liu F, Hon GC, Villa GR, Turner KM, Ikegami S, Yang H, Ye Z, Li B, Kuan S, Lee AY, et al. EGFR Mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell 2015; 60(2):307-18; PMID:26455392; https://doi.org/10.1016/j.molcel.2015.09.002
- Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res 2014; 20(4):912-25; PMID:24297863; https://doi.org/10.1158/1078-0432.CCR-13-2281
- Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, Speleman F, Althoff K, Schafers S, Schramm A, et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013; 4(11):2080-95; PMID:24231268; https://doi.org/10.18632/oncotarget.1534
- Venkataraman S, Alimova I, Balakrishnan I, Harris P, Birks DK, Griesinger A, Amani V, Cristiano B, Remke M, Taylor MD, et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 2014; 5(9):2355-71; PMID:24796395; https://doi.org/10.18632/oncotarget.1659
- Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP, Ma XX, Zhang G, Li J, Yan B, Tang HW, et al. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 2016; 7(3):2462-74; PMID:26575167; https://doi.org/10.18632/oncotarget.6275
- Hong SH, Eun JW, Choi SK, Shen Q, Choi WS, Han JW, Nam SW, You JS. Epigenetic reader BRD4 inhibition as a therapeutic strategy to suppress E2F2-cell cycle regulation circuit in liver cancer. Oncotarget 2016; 7(22):32628-40; PMID:27081696; https://doi.org/10.18632/oncotarget.8701
- McCleland ML, Mesh K, Lorenzana E, Chopra VS, Segal E, Watanabe C, Haley B, Mayba O, Yaylaoglu M, Gnad F, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest 2016; 126(2):639-52; PMID:26752646; https://doi.org/10.1172/JCI83265
- Zhang L, Tong Y, Zhang X, Pan M, Chen S. Arsenic sulfide combined with JQ1, chemotherapy agents, or celecoxib inhibit gastric and colon cancer cell growth. Drug Des Devel Ther 2015; 9:5851-62; https://doi.org/10.2147/DDDT.S92943
- Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, Heslin MJ, Arnoletti JP, Richardson JH, Chen D, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016; 35(7):833-45; PMID:25961927; https://doi.org/10.1038/onc.2015.126
- Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, Lofgren SM, Kuschma T, Hahn SA, Vangala D, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med 2015; 21(10):1163-71; PMID:26390243; https://doi.org/10.1038/nm.3952
- Kumar K, Raza SS, Knab LM, Chow CR, Kwok B, Bentrem DJ, Popovic R, Ebine K, Licht JD, Munshi HG. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep 2015; 5:9489; PMID:25807524; https://doi.org/10.1038/srep09489
- Klocker H, Culig Z, Kaspar F, Hobisch A, Eberle J, Reissigl A Bartsch G. Androgen signal transduction and prostatic carcinoma. World J Urol 1994; 12(2):99-103; PMID:8087144; https://doi.org/10.1007/BF00184245
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014; 510(7504):278-82; PMID:24759320; https://doi.org/10.1038/nature13229
- Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, Raj GV, Tilley WD, Dehm SM. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res 2015; 43(12):5880-97; PMID:25908785; https://doi.org/10.1093/nar/gkv262
- Blee AM, Liu S, Wang L, Huang H. BET bromodomain-mediated interaction between ERG and BRD4 promotes prostate cancer cell invasion. Oncotarget 2016; 7(25):38319-32; PMID:27223260; https://doi.org/10.18632/oncotarget.9513
- Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A 2012; 109(47):19408-13; PMID:23129625; https://doi.org/10.1073/pnas.1216363109
- Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, Gao Y, Cheng KA, Cohoon TJ, Qi J, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res 2013; 19(22):6183-92; PMID:24045185; https://doi.org/10.1158/1078-0432.CCR-12-3904
- Tsai LH, Wu JY, Cheng YW, Chen CY, Sheu GT, Wu TC, Lee H. The MZF1/c-MYC axis mediates lung adenocarcinoma progression caused by wild-type lkb1 loss. Oncogene 2015; 34(13):1641-9; PMID:24793789; https://doi.org/10.1038/onc.2014.118
- Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44(10):1104-10; PMID:22941188; https://doi.org/10.1038/ng.2396
- Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, Peterson R, Schweizer L, Purandare A, Ross-Macdonald P, et al. Sensitivity of small cell lung cancer to BET inhibition Is mediated by regulation of ASCL1 gene expression. Mol Cancer Ther 2015; 14(10):2167-74; PMID:26253517; https://doi.org/10.1158/1535-7163.MCT-15-0037
- Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003; 22(47):7316-39; PMID:14576841; https://doi.org/10.1038/sj.onc.1206937
- Sengupta S, Biarnes MC, Clarke R, Jordan VC. Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res Treat 2015; 150(2):265-78; PMID:25721606; https://doi.org/10.1007/s10549-015-3319-1
- Bihani T, Ezell SA, Ladd B, Grosskurth SE, Mazzola AM, Pietras M, Reimer C, Zinda M, Fawell S, D'Cruz CM. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget 2015; 6(4):2407-20; PMID:25537515; https://doi.org/10.18632/oncotarget.2964
- Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 2015; 6(32):33623-35; PMID:26378038; https://doi.org/10.18632/oncotarget.5601
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016; 529(7586):413-7; PMID:26735014; https://doi.org/10.1038/nature16508
- Perez-Pena J, Serrano-Heras G, Montero JC, Corrales-Sanchez V, Pandiella A, Ocana A. In silico analysis guides selection of BET inhibitors for triple-negative breast cancer treatment. Mol Cancer Ther 2016; 15(8):1823-33; PMID:27256375; https://doi.org/10.1158/1535-7163.MCT-16-0004
- da Motta LL, Ledaki I, Purshouse K, Haider S, De Bastiani MA, Baban D, Morotti M, Steers G, Wigfield S, Bridges E, et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2016; PMID:27292261; https://doi.org/10.1038/onc.2016.184 Oncogene 2016:1-11
- Bernardi R, Gianni L. Hallmarks of triple negative breast cancer emerging at last? Cell Res 2014; 24(8):904-5; PMID:24810303; https://doi.org/10.1038/cr.2014.61
- Chaidos A, Caputo V, Gouvedenou K, Liu B, Marigo I, Chaudhry MS, Rotolo A, Tough DF, Smithers NN, Bassil AK, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood 2014; 123(5):697-705; PMID:24335499; https://doi.org/10.1182/blood-2013-01-478420
- Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, McHugh CF, Vessella R, Creasy CL, Tummino PJ, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013; 4(12):2419-29; PMID:24293458; https://doi.org/10.18632/oncotarget.1572
- Berenguer-Daize C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, Bekradda M, MacKenzie S, Rezai K, Lokiec F, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer 2016; 139(9):2047-55; PMID:27388964; https://doi.org/10.1002/ijc.30256
- Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015; 6(19):17698-712; PMID:25989842; https://doi.org/10.18632/oncotarget.4131
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478(7370):529-33; PMID:21964340; https://doi.org/10.1038/nature10509
- Wyspianska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, Robson S, Rioja I, Li J, Wiese M, et al. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia 2014; 28(1):88-97; PMID:23929215; https://doi.org/10.1038/leu.2013.234
- Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, Graham RM, Allen B, Sarkaria JN, Komotar RJ, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014; 9(4):611-20; PMID:24496381; https://doi.org/10.4161/epi.27906
- Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, Cannizzaro E, Osaki H, Wiese M, Putwain S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 2014; 28(2):311-20; PMID:24220271; https://doi.org/10.1038/leu.2013.338
- Gallagher SJ, Mijatov B, Gunatilake D, Tiffen JC, Gowrishankar K, Jin L, Pupo GM, Cullinane C, Prinjha RK, Smithers N, et al. The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J Invest Dermatol 2014; 134(11):2795-805; PMID:24906137; https://doi.org/10.1038/jid.2014.243
- Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, Jin L, Pupo G, Cullinane C, McArthur GA, et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res 2014; 27(6):1126-37; PMID:24924589; https://doi.org/10.1111/pcmr.12282
- Siegel MB, Liu SQ, Davare MA, Spurgeon SE, Loriaux MM, Druker BJ, Scott EC, Tyner JW. Small molecule inhibitor screen identifies synergistic activity of the bromodomain inhibitor CPI203 and bortezomib in drug resistant myeloma. Oncotarget 2015; 6(22):18921-32; PMID:26254279; https://doi.org/10.18632/oncotarget.4214
- Wong C, Laddha SV, Tang L, Vosburgh E, Levine AJ, Normant E, Sandy P, Harris CR, Chan CS, Xu EY. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis 2014; 5:e1450; PMID:25299775; https://doi.org/10.1038/cddis.2014.396
- Moros A, Rodriguez V, Saborit-Villarroya I, Montraveta A, Balsas P, Sandy P, Martinez A, Wiestner A, Normant E, Campo E, et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014; 28(10):2049-59; PMID:24721791; https://doi.org/10.1038/leu.2014.106
- Bhadury J, Nilsson LM, Muralidharan SV, Green LC, Li Z, Gesner EM, Hansen HC, Keller UB, McLure KG, Nilsson JA. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci U S A 2014; 111(26):E2721-30; PMID:24979794; https://doi.org/10.1073/pnas.1406722111
- Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P, Bunnage ME, Owen DR, et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res 2013; 73(11):3336-46; PMID:23576556; https://doi.org/10.1158/0008-5472.CAN-12-3292
- Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, Zhou MM. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem 2013; 56(22):9251-64; PMID:24144283; https://doi.org/10.1021/jm401334s
- Millan DS, Alvarez Morales MA, Barr KJ, Cardillo D, Collis A, Dinsmore CJ, Escobedo JA, Foley KP, Herbertz T, Hubbs S, et al. FT-1101: A structurally distinct pan-BET bromodomain inhibitor with activity in preclinical models of hematologic malignancies. Poster presented at: 57th Annual Meeting & Exposition of American Society of Hematology (ASH); 2015 Dec 5–8; Orlando, USA
- Albrecht BK, Gehling VS, Hewitt MC, Vaswani RG, Cote A, Leblanc Y, Nasveschuk CG, Bellon S, Bergeron L, Campbell R, et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J Med Chem 2016; 59(4):1330-9; PMID:26815195; https://doi.org/10.1021/acs.jmedchem.5b01882
- Lejeune P, Sugawara T, Gelato KA, Ellinger-Ziegelbauer H, Fernandez-Montalvan AE, Schmees N, Siegel S, Weinmann H, Gekeler V, Walter AO, et al. BAY 1238097, a novel BET inhibitor with strong efficacy in hematological tumor models. Poster presented at: 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22; Philadelphia, USA. https://doi.org/10.1158/1538-7445.AM2015-3524
- Liu PCC, Liu XM, Stubbs MC, Maduskuie T, Sparks R, Zolotarjova N, Li J, Wen X, Favata M, Feldman P, et al. Discovery of a novel BET inhibitor INCB054329. Poster presented at: 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22; Philadelphia, USA. https://doi.org/10.1158/1538-7445.AM2015-3523
- Shapiro GI, Dowlati A, LoRusso P, Eder JP, Anderson A, Do KT, Kagey MH, Sirard C, Bradner JE, Landau SB. Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC). Poster presented at: AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2015 Nov 5–9; Boston, USA. https://doi.org/10.1158/1535-7163.TARG-15-A49
- Structural Genomics Consortium (SGC). BAY-299 A probe for BRD1 and TAF1 [Accessed 2016, Sept 7]. http://www.thesgc.org/chemical-probes/BAY-299
- Structural Genomics Consortium (SGC). Bromosporine [Accessed 2016, Sept 6]. http://www.thesgc.org/chemical-probes/Bromosporine
- Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem 2012; 55(17):7346-59; PMID:22788793; https://doi.org/10.1021/jm300346w
- Demont EH, Chung CW, Furze RC, Grandi P, Michon AM, Wellaway C, Barrett N, Bridges AM, Craggs PD, Diallo H, et al. Fragment-based discovery of low-micromolar ATAD2 bromodomain inhibitors. J Med Chem 2015; 58(14):5649-73; PMID:26155854; https://doi.org/10.1021/acs.jmedchem.5b00772
- Bamborough P, Chung CW, Furze RC, Grandi P, Michon AM, Sheppard RJ, Barnett H, Diallo H, Dixon DP, Douault C, et al. Structure-based optimization of naphthyridones into potent ATAD2 bromodomain inhibitors. J Med Chem 2015; 58(15):6151-78; PMID:26230603; https://doi.org/10.1021/acs.jmedchem.5b00773
- Drouin L, McGrath S, Vidler LR, Chaikuad A, Monteiro O, Tallant C, Philpott M, Rogers C, Fedorov O, Liu M, et al. Structure enabled design of BAZ2-ICR, a chemical probe targeting the bromodomains of BAZ2A and BAZ2B. J Med Chem 2015; 58(5):2553-9; PMID:25719566; https://doi.org/10.1021/jm501963e
- Chen P, Chaikuad A, Bamborough P, Bantscheff M, Bountra C, Chung CW, Fedorov O, Grandi P, Jung D, Lesniak R, et al. Discovery and characterization of GSK2801, a selective chemical probe for the bromodomains BAZ2A and BAZ2B. J Med Chem 2016; 59(4):1410-24; PMID:25799074; https://doi.org/10.1021/acs.jmedchem.5b00209
- Middeljans E, Wan X, Jansen PW, Sharma V, Stunnenberg HG, Logie C. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS One 2012; 7(3):e33834; PMID:22442726; https://doi.org/10.1371/journal.pone.0033834
- Picaud S, Strocchia M, Terracciano S, Lauro G, Mendez J, Daniels DL, Riccio R, Bifulco G, Bruno I, Filippakopoulos P. 9H-purine scaffold reveals induced-fit pocket plasticity of the BRD9 bromodomain. J Med Chem 2015; 58(6):2718-36; PMID:25703523; https://doi.org/10.1021/jm501893k
- Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, Chung CW, Dittmann A, Drewes G, Drewry DH, et al. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J Med Chem 2016; 59(4):1425-39; PMID:25856009; https://doi.org/10.1021/acs.jmedchem.5b00256
- Clark PG, Vieira LC, Tallant C, Fedorov O, Singleton DC, Rogers CM, Monteiro OP, Bennett JM, Baronio R, Muller S, et al. LP99: Discovery and synthesis of the first selective BRD7/9 bromodomain inhibitor. Angew Chem Weinheim Bergstr Ger 2015; 127(21):6315-9; PMID:27346896; https://doi.org/10.1002/ange.201501394
- Martin LJ, Koegl M, Bader G, Cockcroft XL, Fedorov O, Fiegen D, Gerstberger T, Hofmann MH, Hohmann AF, Kessler D, et al. Structure-based design of an in vivo active selective BRD9 inhibitor. J Med Chem 2016; 59(10):4462-75; PMID:26914985; https://doi.org/10.1021/acs.jmedchem.5b01865
- Structural Genomics Consortium (SGC). TP-472 A BRD9/7 Probe [Accessed 2016, Sept 6]. http://www.thesgc.org/chemical-probes/TP-472
- Demont EH, Bamborough P, Chung CW, Craggs PD, Fallon D, Gordon LJ, Grandi P, Hobbs CI, Hussain J, Jones EJ, et al. 1,3-dimethyl benzimidazolones are potent, selective inhibitors of the BRPF1 bromodomain. ACS Med Chem Lett 2014; 5(11):1190-5; PMID:25408830; https://doi.org/10.1021/ml5002932
- Structural Genomics Consortium (SGC). OF-1 A chemical probe for BRPF bromodomains [Accessed 2016, Sept 6]. http://www.thesgc.org/chemical-probes/OF-1
- Structural Genomics Consortium (SGC). PFI-4 A chemical probe for BRPF1B [Accessed 2016, Sept 7]. http://www.thesgc.org/chemical-probes/PFI-4
- Structural Genomics Consortium (SGC). NI-57 A chemical probe for BRPF bromodomains [Accessed 2016, Sept 7]. http://www.thesgc.org/chemical-probes/NI-57
- Bennett J, Fedorov O, Tallant C, Monteiro O, Meier J, Gamble V, Savitsky P, Nunez-Alonso GA, Haendler B, Rogers C, et al. Discovery of a chemical tool inhibitor targeting the bromodomains of TRIM24 and BRPF. J Med Chem 2016; 59(4):1642-7; PMID:25974391; https://doi.org/10.1021/acs.jmedchem.5b00458
- Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 2008; 29(5):617-22; PMID:18386774; https://doi.org/10.1002/humu.20730
- Brownlee PM, Chambers AL, Oliver AW, Downs JA. Cancer and the bromodomains of BAF180. Biochem Soc Trans 2012; 40(2):364-9; PMID:22435813; https://doi.org/10.1042/BST20110754
- Kwon SJ, Lee SK, Na J, Lee SA, Lee HS, Park JH, Chung JK, Youn H, Kwon J. Targeting BRG1 chromatin remodeler via its bromodomain for enhanced tumor cell radiosensitivity in vitro and in vivo. Mol Cancer Ther 2015; 14(2):597-607; PMID:25504753; https://doi.org/10.1158/1535-7163.MCT-14-0372
- Fedorov O, Castex J, Tallant C, Owen DR, Martin S, Aldeghi M, Monteiro O, Filippakopoulos P, Picaud S, Trzupek JD, et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci Adv 2015;1(10):e1500723; PMID:26702435; https://doi.org/10.1126/sciadv.1500723
- Structural Genomics Consortium (SGC). PFI-3 A selective chemical probe for SMARCA bromodomains [Accessed 2016, Sept 6]: http://www.thesgc.org/chemical-probes/PFI-3
- Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res 2015; 75(18):3865-78; PMID:26139243; https://doi.org/10.1158/0008-5472.CAN-14-3798
- Sutherell CL, Tallant C, Monteiro OP, Yapp C, Fuchs JE, Fedorov O, Siejka P, Muller S, Knapp S, Brenton JD, et al. Identification and development of 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one inhibitors targeting bromodomains within the switch/sucrose nonfermenting complex. J Med Chem 2016; 59(10):5095-101; PMID:27119626; https://doi.org/10.1021/acs.jmedchem.5b01997
- Sachchidanand , Resnick-Silverman L, Yan S, Mutjaba S, Liu WJ, Zeng L, Manfredi JJ, Zhou MM. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem Biol 2006; 13(1):81-90; PMID:16426974; https://doi.org/10.1016/j.chembiol.2005.10.014
- Borah JC, Mujtaba S, Karakikes I, Zeng L, Muller M, Patel J, Moshkina N, Morohashi K, Zhang W, Gerona-Navarro G, et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem Biol 2011; 18(4):531-41; PMID:21513889; https://doi.org/10.1016/j.chembiol.2010.12.021
- Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, Olzscha H, Monteiro O, Martin S, Philpott M, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res 2015; 75(23):5106-19; PMID:26552700; https://doi.org/10.1158/0008-5472.CAN-15-0236
- Zucconi BE, Luef B, Xu W, Henry RA, Nodelman IM, Bowman GD, Andrews AJ, Cole PA. Modulation of p300/CBP acetylation of nucleosomes by bromodomain ligand I-CBP112. Biochemistry 2016; 55(27):3727-34; PMID:27332697; https://doi.org/10.1021/acs.biochem.6b00480
- Hay DA, Fedorov O, Martin S, Singleton DC, Tallant C, Wells C, Picaud S, Philpott M, Monteiro OP, Rogers CM, et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc 2014; 136(26):9308-19; PMID:24946055; https://doi.org/10.1021/ja412434f
- Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA, Martinez FO, Al-Mossawi MH, de Wit J, Vecellio M, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci U S A 2015; 112(34):10768-73; PMID:26261308; https://doi.org/10.1073/pnas.1501956112
- Conery AR, Centore RC, Neiss A, Keller PJ, Joshi S, Spillane KL, Sandy P, Hatton C, Pardo E, Zawadzke L, et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. Elife 2016; 5:pii:e10483; PMID:26731516; https://doi.org/10.7554/eLife.10483
- Chekler EL, Pellegrino JA, Lanz TA, Denny RA, Flick AC, Coe J, Langille J, Basak A, Liu S, Stock IA, et al. Transcriptional profiling of a selective CREB binding protein bromodomain inhibitor highlights therapeutic opportunities. Chem Biol 2015; 22(12):1588-96; PMID:26670081; https://doi.org/10.1016/j.chembiol.2015.10.013
- Taylor AM, Cote A, Hewitt MC, Pastor R, Leblanc Y, Nasveschuk CG, Romero FA, Crawford TD, Cantone N, Jayaram H, et al. Fragment-based discovery of a selective and cell-active benzodiazepinone CBP/EP300 bromodomain inhibitor (CPI-637). ACS Med Chem Lett 2016; 7(5):531-6; PMID:27190605; https://doi.org/10.1021/acsmedchemlett.6b00075
- Denis GV. Bromodomain coactivators in cancer, obesity, type 2 diabetes, and inflammation. Discov Med 2010; 10(55):489-99; PMID:21189220
- Nicholas DA, Andrieu G, Strissel KJ, Nikolajczyk BS Denis GV. BET bromodomain proteins and epigenetic regulation of inflammation: implications for type 2 diabetes and breast cancer. Cell Mol Life Sci 2016; PMID:27491296; https://doi.org/10.1007/s00018-016-2320-0
- Rvx 208. Drugs R D 2011; 11(2):207-13; PMID:21679009; https://doi.org/10.2165/11595140-000000000-00000
- Nicholls SJ, Puri R, Wolski K, Ballantyne CM, Barter PJ, Brewer HB, Kastelein JJ, Hu B, Uno K, Kataoka Y, et al. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am J Cardiovasc Drugs 2016; 16(1):55-65; PMID:26385396; https://doi.org/10.1007/s40256-015-0146-z
- ClinicaTrials. gov. [Accessed 2016, Nov 9]. https://clinicaltrials.gov/">https://clinicaltrials.gov/">https://clinicaltrials.gov/
- Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci U S A 2013; 110(49):19754-9; PMID:24248379; https://doi.org/10.1073/pnas.1310658110
- Bailey D, Jahagirdar R, Gordon A, Hafiane A, Campbell S, Chatur S, Wagner GS, Hansen HC, Chiacchia FS, Johansson J, et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol 2010; 55(23):2580-9; PMID:20513599; https://doi.org/10.1016/j.jacc.2010.02.035
- Gilham D, Wasiak S, Tsujikawa LM, Halliday C, Norek K, Patel RG, Kulikowski E, Johansson J, Sweeney M, Wong NC. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 2016; 247:48-57; PMID:26868508; https://doi.org/10.1016/j.atherosclerosis.2016.01.036
- Siebel AL, Trinh SK, Formosa MF, Mundra PA, Natoli AK, Reddy-Luthmoodoo M, Huynh K, Khan AA, Carey AL, van Hall G, et al. Effects of the BET-inhibitor, RVX-208 on the HDL lipidome and glucose metabolism in individuals with prediabetes: A randomized controlled trial. Metabolism 2016; 65(6):904-14; PMID:27173469; https://doi.org/10.1016/j.metabol.2016.03.002
- Fu W, Farache J, Clardy SM, Hattori K, Mander P, Lee K, Rioja I, Weissleder R, Prinjha RK, Benoist C, et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and beta cells. Elife 2014; 3:e04631; PMID:25407682; https://doi.org/10.7554/eLife.04631
- Deeney JT, Belkina AC, Shirihai OS, Corkey BE Denis GV. BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic beta-Cell. PLoS One 2016; 11(3):e0151329; PMID:27008626; https://doi.org/10.1371/journal.pone.0151329
- Nadeem A, Al-Harbi NO, Al-Harbi MM, El-Sherbeeny AM, Ahmad SF, Siddiqui N, Ansari MA, Zoheir KM, Attia SM, Al-Hosaini KA, et al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation. Pharmacol Res 2015; 99:248-57; PMID:26149470; https://doi.org/10.1016/j.phrs.2015.06.001
- Xiao Y, Liang L, Huang M, Qiu Q, Zeng S, Shi M, Zou Y, Ye Y, Yang X, Xu H. Bromodomain and extra-terminal domain bromodomain inhibition prevents synovial inflammation via blocking IkappaB kinase-dependent NF-kappaB activation in rheumatoid fibroblast-like synoviocytes. Rheumatology (Oxford) 2016; 55(1):173-84; PMID:26324948; https://doi.org/10.1093/rheumatology/kev312
- Klein K, Kabala PA, Grabiec AM, Gay RE, Kolling C, Lin LL, Gay S, Tak PP, Prinjha RK, Ospelt C, et al. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis 2016; 75(2):422-9; PMID:25467295; https://doi.org/10.1136/annrheumdis-2014-205809
- Barrett E, Brothers S, Wahlestedt C Beurel E. I-BET151 selectively regulates IL-6 production. Biochim Biophys Acta 2014; 1842(9):1549-55; PMID:24859008; https://doi.org/10.1016/j.bbadis.2014.05.013
- Huang M, Zeng S, Zou Y, Shi M, Qiu Q, Xiao Y, Chen G, Yang X, Liang L, Xu H. BET bromodomain suppression inhibits vascular inflammation by blocking NF-kappaB and MAPK activation. Br J Pharmacol 2016; 174(1):101-115; PMID:27774624; https://doi.org/10.1111/bph.13657
- Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. The Journal of biological chemistry 2003; 278(21):19134-40; PMID:12624111; https://doi.org/10.1074/jbc.M301580200
- Milite C, Castellano S, Benedetti R, Tosco A, Ciliberti C, Vicidomini C, Boully L, Franci G, Altucci L, Mai A, et al. Modulation of the activity of histone acetyltransferases by long chain alkylidenemalonates (LoCAMs). Bioorg Med Chem 2011; 19(12):3690-701; PMID:21292492; https://doi.org/10.1016/j.bmc.2011.01.013
- Sbardella G, Castellano S, Vicidomini C, Rotili D, Nebbioso A, Miceli M, Altucci L, Mai A. Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorg Med Chem Lett 2008; 18(9):2788-92; PMID:18434144; https://doi.org/10.1016/j.bmcl.2008.04.017
- Castellano S, Milite C, Feoli A, Viviano M, Mai A, Novellino E, Tosco A, Sbardella G. Identification of structural features of 2-alkylidene-1,3-dicarbonyl derivatives that induce inhibition and/or activation of histone acetyltransferases KAT3B/p300 and KAT2B/PCAF. ChemMedChem 2015; 10(1):144-57; PMID:25333655; https://doi.org/10.1002/cmdc.201402371
- Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, Meyers D, Mukherjee C, Sbardella G, Castellano S, et al. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. J Neurosci 2012; 32(35):11930-41; PMID:22933779; https://doi.org/10.1523/JNEUROSCI.0178-12.2012
- Colussi C, Scopece A, Vitale S, Spallotta F, Mattiussi S, Rosati J, Illi B, Mai A, Castellano S, Sbardella G, et al. P300/CBP associated factor regulates nitroglycerin-dependent arterial relaxation by N(epsilon)-lysine acetylation of contractile proteins. Arterioscler Thromb Vasc Biol 2012; 32(10):2435-43; PMID:22859492; https://doi.org/10.1161/ATVBAHA.112.254011
- Colussi C, Rosati J, Straino S, Spallotta F, Berni R, Stilli D, Rossi S, Musso E, Macchi E, Mai A, et al. Nepsilon-lysine acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc Natl Acad Sci U S A 2011; 108(7):2795-800; PMID:21282606; https://doi.org/10.1073/pnas.1013124108
- Vecellio M, Spallotta F, Nanni S, Colussi C, Cencioni C, Derlet A, Bassetti B, Tilenni M, Carena MC, Farsetti A, et al. The histone acetylase activator pentadecylidenemalonate 1b rescues proliferation and differentiation in the human cardiac mesenchymal cells of type 2 diabetic patients. Diabetes 2014; 63(6):2132-47; PMID:24458358; https://doi.org/10.2337/db13-0731
- Chatterjee S, Mizar P, Cassel R, Neidl R, Selvi BR, Mohankrishna DV, Vedamurthy BM, Schneider A, Bousiges O, Mathis C, et al. A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J Neurosci 2013; 33(26):10698-712; PMID:23804093; https://doi.org/10.1523/JNEUROSCI.5772-12.2013