
ABSTRACT
Genes adjacent to telomeres are subject to transcriptional repression mediated by an integrated set of chromatin modifying and remodeling factors. The telomeres of Saccharomyces cerevisiae have served as a model for dissecting the function of diverse chromatin proteins in gene silencing, and their study has revealed overlapping roles for many chromatin proteins in either promoting or antagonizing gene repression. The H3K4 methyltransferase Set1, which is commonly linked to transcriptional activation, has been implicated in telomere silencing. Set5 is an H4 K5, K8, and K12 methyltransferase that functions with Set1 to promote repression at telomeres. Here, we analyzed the combined role for Set1 and Set5 in gene expression control at native yeast telomeres. Our data reveal that Set1 and Set5 promote a Sir protein-independent mechanism of repression that may primarily rely on regulation of H4K5ac and H4K8ac at telomeric regions. Furthermore, cells lacking both Set1 and Set5 have highly correlated transcriptomes to mutants in telomere maintenance pathways and display defects in telomere stability, linking their roles in silencing to protection of telomeres. Our data therefore provide insight into and clarify potential mechanisms by which Set1 contributes to telomere silencing and shed light on the function of Set5 at telomeres.
Introduction
Altered chromatin dynamics at telomeres, the protective nucleiprotein structures at the ends of eukaryotic chromosomes, are linked to chromosomal instability, a hallmark of both cellular transformation and aging.Citation1,2 The telomeres of Saccharomyces cerevisiae have long served as a paradigm for understanding the role of diverse chromatin modifiers in organizing and regulating chromatin structure and gene expression.Citation3,4 Expression analysis of genes near telomeres (within the subtelomere) in budding yeast using both reporter gene assays and measures of endogenous gene expression have shown that these genes are subject to silencing,Citation5-8 a stable form of transcriptional repression that is gene and sequence independent and is associated with the formation of condensed chromatin analogous to higher eukaryotic heterochromatin. Despite being a model for chromatin regulation for many years, our understanding of gene expression control mechanisms at telomeres is still incomplete. Interestingly, systematic analyses of gene silencing at native telomeres have revealed that it does not occur to the same extent at all yeast telomeres.Citation8-10 Moreover, factors that promote transcriptional repression have been observed to be discontinuous across subtelomeric regions and, while telomere-adjacent genes are lowly expressed, the extent to which their expression is silenced is less than predicted.Citation8-13 Finally, many studies of yeast telomeric silencing have relied on genetic reporter systems, which have proven to display telomere nonspecific phenotypes in some cases,Citation11,12 highlighting the need for further analysis of endogenous gene expression at native yeast telomeres.
Silencing at yeast telomeres is primarily thought to be mediated by the Sir protein complex (Sir2, Sir3, and Sir4) and the subsequent deacetylation of H4K16 by the deacetylase Sir2. This region of hypoacetylation, and the presence of Sir proteins, creates an environment refractory to transcription.Citation4 The activity and localization of the Sir protein complex to transcriptionally-active chromatin is opposed by euchromatic factors, such as H4K16 acetylation catalyzed by Sas2,Citation14-16 and the histone variant Htz1.Citation17 Additionally, regulation of H4K5, K8, and K12 acetylation is also required for proper telomeric silencing.Citation18,19 The proposed role of these acetyl marks at telomeres is to contribute to the heterochromatin-euchromatin boundary that protects neighboring euchromatic genes from the spreading of silencing proteins.Citation14,20-23 In addition to this role, it has also been observed that H4K5ac and H4K12ac directly localize to silent chromatin at telomeres and they are important for heterochromatin formation.Citation21, 24
In addition to histone acetylation, methylation of histone H3 has been implicated in silencing of genes near telomeres. In particular, cells lacking the H3K4 methyltransferase Set1 have a well-established defect in telomeric silencing;Citation13,25-29 however, the precise mechanism by which Set1 promotes silencing is still unclear. Heterochromatic telomeres are largely devoid of H3K4 methylation,Citation26,30-32 although H3K4me3 is detected at heterochromatin-euchromatin boundary regions.Citation30 In set1Δ cells, the Sir complex has been reported beyond the heterochromatin-euchromatin boundary and at ectopic sites throughout the genome.Citation26,29 Also, Sir3, which directly binds chromatin, preferentially interacts with unmethylated nucleosomes.Citation26 These observations led to the proposal that Set1 and H3K4 methylation indirectly maintain Sir protein localization at telomeres by preventing their titration away from heterochromatin through H3K4me-mediated inhibition of Sir3 association with euchromatin.Citation26, 29, 33 This model predicts a decreased level of Sir proteins at telomeres in set1Δ yeast; however, different experiments have yielded conflicting results on Sir protein-telomeric chromatin interactions in set1Δ cells.Citation25,26,33 The loss of Set1 also manifests other defects at telomeres, including short telomeres,Citation27,34,35 and a decrease in telomere clustering at the nuclear periphery,Citation35,36 which have both been linked to defective silencing. Therefore, while proper silencing clearly depends on functional Set1, further investigation of the molecular consequences of the loss of Set1 at telomeres will likely reveal more details regarding the mechanism by which it promotes silencing.
We previously identified a novel histone methyltransferase in budding yeast, Set5, which targets lysines 5, 8, and 12 of the histone H4 tail.Citation37 These sites of modification were previously unknown to be methylated in any organism and, importantly, subsequent investigation uncovered that Set5 has a human ortholog, SMYD3, which also catalyzes methylation at H4K5.Citation38 Investigation of the role of these methyl marks at chromatin showed that combined deletion of SET5 and SET1 results in decreased fitness in response to cellular and genotoxic stresses.Citation37 Transcriptome analysis of set1Δ set5Δ cells revealed a synergistic de-repression of transcription in these cells, particularly at genes that are adjacent to telomeres. Set1 and Set5 also appeared to promote repression of transposable elements and neighboring genes. Together, these data indicate that both Set1 and Set5 contribute to repression of lowly transcribed genes; however, potential mechanisms by which Set5 contributes to gene repression are unknown, and the precise role of Set1 in telomeric gene repression is still unclear.
In this study, we investigated the contributions of both Set1 and Set5 to multiple aspects of telomeric chromatin that may contribute to gene repression. Our results indicate that Set1 and Set5-mediated silencing is largely independent of the Sir proteins and H4K16ac, and is more closely linked to defects in telomere maintenance. We further implicate a possible role for H4K5ac and, to a lesser extent, H4K8ac in Set1- and Set5-dependent telomeric silencing.
Materials and methods
Yeast strains and growth conditions
The genotypes for all Saccharomyces cerevisiae strains used in this study are listed in . Strains carrying gene deletions were constructed using standard PCR-based gene disruption methodsCitation39 and isogenic double and triple mutant strains were isolated following haploid mating, sporulation and tetrad dissection. All strains were confirmed by growth on the appropriate selective media and colony PCR using primers specific to individual gene deletions. Standard media conditions for rich media (YPD; 1% yeast extract, 1% peptone, 2% dextrose) and synthetic complete (SC) or dropout media (US Biological) were used as necessary.
Table 1. Yeast strains used in this study. All strains are derived from the BY4741 genetic background (shown for yEG230) unless otherwise indicated.
Quantitative reverse transcriptase PCR
Total RNA was extracted from 1.5 mL of an OD600 ∼0.6–0.8 culture of yeast cells using the Masterpure Yeast RNA Purification kit (Epicentre) following the manufacturer's instructions. Genomic DNA was eliminated using the Turbo DNA-free kit (Ambion) and cDNA was generated from 1 µg of total RNA using the iScript cDNA Synthesis kit (Biorad) containing both oligo dT and random hexamers to prime reverse transcription of mRNA. For quantitative PCR (qPCR) of transcript levels, 0.5 µL of the cDNA mixture was added to 1X iTAQ Universal SYBR Green Supermix (Biorad) with the appropriate gene specific primers (Table S1) in a 10 µL reaction. Real-time amplification was performed on a Biorad CFX384 Real-time Detection System. Three technical replicates were performed for each reaction, and a minimum of three biological replicates was performed for each experiment. Relative gene expression values were normalized to the control gene TFC1, whose expression is very stable.Citation40
Immunoblotting
Lysates for immunoblotting were prepared using 0.2 M NaOH treatment of harvested yeast cells,Citation41 subjected to SDS-PAGE and transferred to PVDF. Blots were probed with the following antibodies: rabbit anti-H3K4me3 (Active Motif, catalog no. 39159), rabbit anti-H3K4me2 (Active Motif, catalog no. 39141), rabbit-anti-H3 (Active Motif, catalog no. 39163). Imaging was performed using a Licor C-DiGit Chemiluminescent Western Blot Scanner.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described.Citation42,43 Briefly, 100 mL of mid-log phase yeast cultures were fixed with 1% formaldehyde for 15–45 min. Yeast lysates were generated and chromatin was sheared using either sonication or micrococcal nuclease digestion. Chromatin extracts were normalized to total protein content and antibody pre-bound to protein A/G magnetic beads (Pierce) or protein G magnetic beads (Life Technologies) was incubated in the extracts from 2 h to overnight with rotating at 4°C. Protein-DNA complexes were eluted using 1% SDS and 0.1 M NaHCO3, cross-links were reversed, samples were treated with proteinase K and RNaseA and subject to extraction with phenol-chloroform-isoamyl alcohol. DNA was precipitated using ethanol and qPCR was performed as described above using 0.5 μL of ChIP DNA per reaction and ChIP-specific primers (Table S1). Technical triplicates were performed for each qPCR reaction and two to five biological replicates were performed for each ChIP experiment. Percent input was calculated using the ΔΔCt method. The following antibodies were used for chIP experiments: rabbit-anti-H4K5ac (Abcam, catalog no. ab51997), rabbit anti-H4K8ac (EMD Millipore, catalog no. ABE534), rabbit anti-H4K12ac (EMD Millipore, catalog no. ABE532), rabbit-anti-H4K16ac (EMD Millipore, catalog no. 07–329), rabbit-anti-H4 (EMD Millipore, catalog no. 04–858), rabbit-anti-HA (Abcam, catalog no. ab9110).
RNA-sequencing analysis and Spearman's rank correlation
Previously performed RNA-sequencing (seq) experimentsCitation44 were reanalyzed for performing correlation analysis. Data sets analyzed contain raw sequence reads for two replicates of wt, set5Δ, set1Δ, and set1Δ set5Δ cells (GEO Series accession number GSE52086). A Bioconducter R pipeline was used to process and analyze the data.Citation45 Briefly, read quality control and trimming were performed using FastQC. Reads were mapped to the S. cereviaise reference genome using Rsubread, the data were normalized within the limma package using voom and differential gene expression was determined using EdgeR.Citation46, 47 Significantly differentially expressed (SDE) genes with a log2 fold-change (FC)>1.2, P < 0.05 are listed in Table S2. The SDE genes of each mutant were used to calculate Spearman's rank correlation coefficients relative to the 700 responsive regulatory mutants identified by Kemmeren et al.Citation48 in the R statistical environment. Gene ontology (GO) analysis was performed using FunSpecCitation49 and P-values were calculated using the hypergeometric test.
Canavanine resistance spontaneous mutation rate measurement
The spontaneous mutation rate of CAN1 was measured as described.Citation50, 51 Individual colonies were grown overnight at 30°C in 3 mL YPD. The OD600 of each culture was determined and 1×107 cells were plated on SC – arginine plates in the presence of 60 μg/mL canavanine. Serial dilutions of cells were also plated on SC – arginine to determine the total number of viable cells per culture. Plates were incubated for 3–4 d at 30°C prior to colony counting. The spontaneous Canr mutation rate and 95% confidence intervals were determined for two biological replicates of 10 colonies each using the MMS Maximum Likelihood Method calculated by the Fluctuation Analysis Calculator.Citation52
Transposition assays
PCR-based surveillance of transposition upstream of SUF16 was performed as previously described.Citation53 Briefly, single colonies were inoculated in 3 mL of YPD and grown at 20°C for 72 h with constant agitation. Genomic DNA was extracted using a standard phenol-chloroform based protocol. PCR was performed on 1.5 µg of genomic DNA using primers specific to Ty1 elements (Primer A in ) and the region downstream of SUF16 (in the SNR33 locus, Primer B), as described,Citation53,54 and indicated in Table S1. Amplification of PHO89 was used as a loading control.
Results
Gene silencing by Set1 and Set5 in subtelomeric regions
Set1 has been implicated in gene repression within subtelomeric regions using both targeted and genome-wide gene expression assays.Citation13,25-29,44 Previous transcriptomic analysis of cells lacking the methyltransferases Set1 and Set5 showed that the additional loss of Set5 exacerbated the silencing defect observed in set1Δ cells.Citation44 Consistent with previous results, we observed decreased repression of telomere-proximal genes at TEL07L, TEL02L, and TEL09R in set1Δ cells that was enhanced in the set1Δ set5Δ double mutants using quantitative RT-PCR (qRT-PCR) (). Our data show that individual genes within the subtelomeric region are variably dependent on either Set1 or Set5. For example, while expression of the telomere proximal genes COS12, YGL262W, and YPS6 is upregulated in set1Δ, and further upregulated in set1Δ set5Δ mutants, the expression of YBL113C (proximal to TEL02L) is not altered in set1Δ, but does appear to depend on both Set1 and Set5. These data agree with reports of variable and discontinuous degrees of silencing of subtelomeric genes at endogenous yeast telomeres,Citation8-10 and suggest that subtelomeric gene repression by Set1 and Set5 acts gene-specifically.
Figure 1. Set5 and Set1 promote gene silencing at telomeres. (A) Quantitative RT-PCR (qRT-PCR) of mRNA levels from genes located within subtelomeric regions of telomeres TEL07L, TEL02L, and TEL09R in wild type, set5Δ, set1Δ, and set1Δ set5Δ. The distance of the genes from the chromosome end is indicated below each gene name. For all qRT-PCR experiments, expression values are relative to the control housekeeping gene, TFC1. Error bars represent standard error of the mean (SEM) for a minimum of three biological replicates. (B) Immunoblot of whole cell extracts from COMPASS mutant strains used for qRT-PCR in . Blots were probed with anti-H3K4me3, anti-H3K4me2, and anti-H3 as a loading control. (C) qRT-PCR of TEL07L genes in wild type or set5Δ yeast combined with COMPASS mutants sdc1Δ and spp1Δ. (D) qRT-PCR of TEL07L genes in wild type or set5Δ yeast strains carrying either wild type H3K4 or the H3K4A mutant.
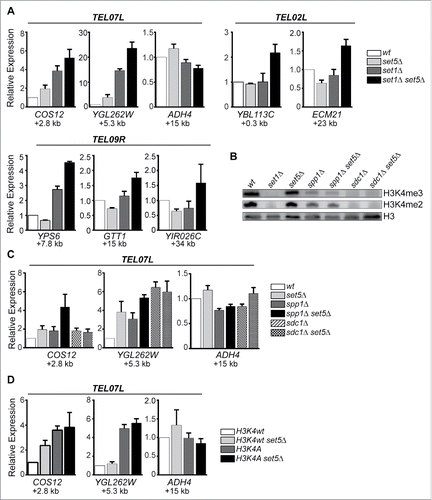
Set1 catalyzes mono-, di-, and tri-methylation at H3K4 as a member of the conserved COMPASS complex.Citation28,55,56 Other components of the COMPASS complex have previously been implicated in silencing at telomeric regions. For example, loss of the subunit Sdc1, which abrogates both H3K4me3 and H3K4me2, shows decreased silencing of reporter genes integrated near telomeres, and deletion of the Spp1 component, which is primarily required for H3K4me3, also shows silencing defects, although often to a lesser extent than sdc1Δ cells.Citation31, 57 (). Here, we used RT-qPCR to assess silencing of endogenous subtelomeric genes in the absence of COMPASS subunits and Set5. Our data show silencing defects in sdc1Δ and spp1Δ cells (); however, there was only minimal enhancement of this defect with the additional loss of Set5. These data are consistent with previous reports,Citation28,57,58 indicating that mutation of these COMPASS components results in relatively moderate silencing defects compare with set1Δ cells (compare scales in ), despite the loss of H3K4 methyl marks. Furthermore, mutation of H3K4 to alanine (H3K4A), which blocks methylation by Set1, also resulted in decreased silencing (), although we did not observe any enhancement of the silencing defect when SET5 is simultaneously deleted. Together, these results suggest that there may be additional factors that contribute to the silencing defect observed in set1Δ cells beyond the loss of the H3K4 methyl marks, and that the enhancement of the silencing defect when SET5 is missing may depend on this H3K4 methyl-independent pathway.
Telomeric gene derepression in set1Δ set5Δ is largely independent of Sir proteins
The Sir2/Sir3/Sir4 protein complex, and its associated role in promoting H4K16 deacetylation, has been identified as the primary factor mediating gene silencing at yeast telomeres.Citation4 However, a recent studyCitation8 used RNA-seq and ChIP-seq to define the genes that depend on an intact Sir complex for silencing and found that only 6% of subtelomeric genes rely on Sir proteins for silencing. Furthermore, Sir-mediated silencing was found to be discontinuous over telomeric regions, rather than present in large domains of chromatin.Citation8 To investigate the possible role of the Sir proteins in the derepression observed in cells lacking Set1 and Set5, we performed additional gene expression and chromatin immunoprecipitation (ChIP) analysis. First, we re-analyzed RNA-seq data sets from wild type, set5Δ, set1Δ, and set1Δ set5Δ cells using the limma package, which provides greater flexibility for downstream applications.Citation46,59 The log2 fold-change (logFC) relative to wild type was determined for two biological replicates of each mutant and lists of significantly differentially expressed (SDE; logFC>1.2, P < 0.05) genes were generated (Table S2). The SDE genes identified here show a similar profile to those previously reported,Citation44 with a majority of genes upregulated in the absence of either Set1 or Set1 and Set5, and more genes jointly regulated by Set1 and Set5 than Set1 alone (Fig. S1A,B). Additionally, a significant fraction of upregulated genes are enriched within 20 kb of a chromosome end in set5Δ, set1Δ, and set1Δ set5Δ cells (Fig. S1C). We analyzed the logFC of genes within 20 kb of the chromosome end at two telomeric regions, TEL07L and TEL09R, which have Set1- and Set5-dependent changes in gene expression (). TEL07L has a region of Sir-mediated silencing, as identified by Ellahi and Rine,Citation8 whereas TEL09R does not exhibit Sir-dependent silencing, despite Sir protein occupancy. Consistent with our qRT-PCR results (), both set1Δ and set1Δ set5Δ cells have increased derepression of these genes, with a larger defect in the double mutant, and set5Δ cells show an increase in depression of a smaller subset of telomeric genes. We also observed that derepression of genes in set1Δ and set1Δ set5Δ cells was strongest at telomere proximal genes, and fewer expression changes occurred at the more distal loci. Additionally, while the Sir-regulated genes of TEL07L are derepressed in the absence of Set1 and Set5, the region of derepression is much larger than the Sir-dependent region. Furthermore, upregulation of expression is clearly observed across TEL09R, which does not possess any Sir-regulated genesCitation8 ().
Figure 2. Telomeric gene derepression in the absence of Set1 and Set5 is not linked to Sir protein dependent silencing. (A) Log2 fold-change from RNA-seq of set5Δ, set1Δ, and set1Δ set5Δ cells in genes within 20 kb of the chromosome end for TEL07L, a SIR-dependent telomere, and TEL09R, a SIR-independent telomere. Beginning from the y-axis, all genes with available data are shown and arranged from most telomere proximal to telomere distal. Systematic names are indicated for all genes. Genes identified as regulated by Sir2, Sir3, and Sir48 are indicated in bold. (B) Chromatin immunoprecipitation (ChIP) using anti-HA antibodies of Sir3-HA in wt, set5Δ, set1Δ, and set1Δ set5Δ cells, as well as an untagged SIR3 control. qPCR was performed on immunoprecipitated DNA using primers targeting the positions indicated on the schematic of TEL07L. Genes within 20 kb of the chromosome end are shown as black boxes and primer positions are indicated with red lines. Primer sequences are listed in Table S2. The graph shows percent input for two biological replicates. Error bars indicate SEM. Inset graph shows percent input of Sir3-HA at the 1.5 kb position to more readily compare peak levels of Sir3-HA in the mutants.
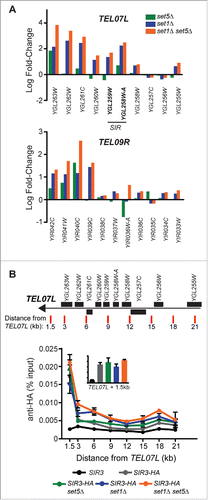
To further investigate the potential role of Sir-mediated repression in our single and double mutants, we performed ChIP of epitope-tagged Sir3 (Sir3-HA) and probed its localization across TEL07L (). A strain carrying an untagged version of Sir3 was used as a negative control. In all strains, Sir3-HA was most enriched closest to the telomere (1.5 kb from the chromosome end). We did not detect any statistically significant differences between Sir3-HA levels closest to the chromosome end (see bar graph inset in ), although changes in gene expression are observed in these regions (). Additionally, at 3 kb and 6 kb from TEL07L, there is a slight increase in Sir3-HA occupancy in both set1Δ and set1Δ set5Δ cells. These data are consistent with other published reports showing no or minimal changes in Sir protein occupancy at telomeres in set1Δ,25,33,35 and further suggest that the additional loss of Set5 does not alter Sir3 distribution at telomeric chromatin.
Sir-mediated silencing depends on the catalytic activity of Sir2, which deacetylates H4K16.Citation4 We therefore also performed ChIP of H4K16ac at the subtelomere of TEL07L using the same primer pairs as diagrammed in . For both the set1Δ and set1Δ set5Δ mutants, H4K16ac levels are largely similar to wild type across the region (). These data support the conclusion that Set1- and Set5-dependent gene silencing occurs independently of Sir protein binding and Sir2 catalytic activity.
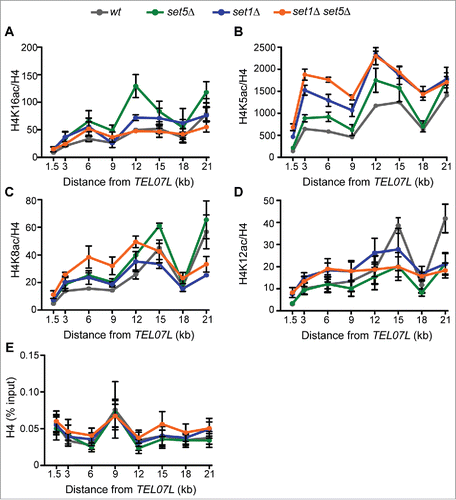
Figure 3. Loss of Set1 and Set5 results in subtelomeric hyperacetylation primarily at H4K5. ChIP of (A) H4K16ac, (B) H4K5ac, (C) H4K8ac and (D) H4K12ac in wt, set5Δ, set1Δ, and set1Δ set5Δ cells. qPCR of immunoprecipitated DNA was performed using primers as shown in . Percent input of H4 acetyl ChIPs was normalized to percent input of total H4 levels and the ratio is shown. (E) Percent input of H4 across the TEL07L region. The mean of two to five biological replicates is shown for each ChIP and error bars represent SEM.
The loss of Set1 and Set5 is accompanied by disrupted H4 acetylation patterns at subtelomeres
While H4K16ac is the primary mark implicated in silencing, regulation of acetylation at H4 K5, K8, and K12 is also required for proper silencing, and both H4K5ac and H4K12ac have been specifically implicated in heterochromatin formation at telomeres.Citation20,21,24 Due to the known role of Set1 in modulating histone acetylation at other loci,6061 and Set5-mediated methylation of these H4 sites, we tested the distribution of H4K5ac, H4K8ac, and H4K12ac across the TEL07L subtelomere in set5Δ, set1Δ, and set1Δ set5Δ cells by ChIP (). Our data revealed distinct patterns for the three marks in the mutant strains. Primarily, H4K5ac showed increases across the subtelomere in all three mutants, with set1Δ set5Δ cells being most distinct from set1Δ in the range 3 to 9 kb from the chromosome end (). Increased H4K5ac at telomeres has been previously demonstrated in set1Δ mutants,Citation60 although our results reveal a function for Set5 in maintaining H4K5 deacetylation at telomeres as well. In a distinct pattern from H4K5ac, H4K8ac was increased in telomere-proximal regions in the double mutants; however, set1Δ and set5Δ single mutants showed relatively mild increases and appeared largely similar to wild type (). And, finally, the distribution of H4K12ac showed few statistically significant distinctions from wild type in all mutants except at 21 kb from the chromosome end, in which H4K12ac levels were higher in wild type than either single or double mutants (). Furthermore, we did not detect alterations to H4 occupancy across the region in any of the mutant strains, (), indicating that histone levels and distribution are not likely to be altered in the mutants.
The H4 acetyl marks tested show unique patterns in the absence of Set1 and Set5, indicating that they have non-redundant functions in subtelomeric gene expression control, consistent with previous work.Citation20,21,24 Moreover, this suggests that the specific changes detected in these marks are unlikely to be byproducts of increased transcription in the region. Of the acetyl marks tested, the changes in H4K5ac most closely reflect the observed alterations in gene expression in the mutants, with set5Δ cells showing a mild phenotype, set1Δ cells with an intermediate phenotype, and set1Δ set5Δ having the greatest defect in gene derepression and H4K5ac levels. These data suggest that H4K5ac, and H4K8ac to a lesser extent, may be the primary contributors to altered subtelomeric gene expression patterns in cells lacking both Set1 and Set5.
Set1 and Set5 promote telomere maintenance pathways
To further identify pathways that contribute to gene derepression near telomeres in the absence of Set1 and Set5, we performed correlation analysis between the transcriptomes of our mutants and those of a panel of approximately 700 yeast strains carrying mutations in known or putative regulators of transcription and chromatin structure.Citation48 These transcriptomes were rigorously curated by Kemmeren et al.Citation48 using microarray data from a set of mutants representing approximately one-quarter of the yeast genome. Among these mutants, the authors defined approximately 700 “responsive” strains, which they characterized as reproducibly inducing a minimum of four genes showing significant changes in transcript levels (fold-change >1.7, P < 0.05). The RNA-seq data of set5Δ, set1Δ, and set1Δ set5Δ cells were obtained from cells grown under similar conditions and collected at mid-log phase,Citation44 as in the Kemmeren et al. data set. As described above, the limma algorithm was used to reanalyze the RNA-seq data sets since it was originally developed for analysis of microarray data and has since been updated for RNA-seq and has the capability of transforming RNA-seq-obtained read counts to log-scale data and incorporating them into a linear model.Citation59 This analysis pipeline is therefore more similar to that of microarrays, and cross-platform comparisons using limma have yielded high concordance between microarray and RNA-seq data sets.Citation62
We correlated the set5Δ, set1Δ, and set1Δ set5Δ RNA-seq data sets to the approximately 700 responsive yeast mutants in gene expression regulators using Spearman's rank correlation. Given the low number of significantly differentially expressed genes in set5Δ cells and a more limited role in regulating telomeric gene expression on its own (), we primarily analyzed the results for set1Δ and set1Δ set5Δ cells. Differentiating between these two data sets specifically allowed us to identify potential pathways contributing to the enhanced telomere silencing defect in set1Δ set5Δ relative to set1Δ alone. Correlation coefficients for all mutants are presented in Table S3. We classified the top 50 mutants most correlated with the set1Δ and set1Δ set5Δ mutants using gene ontology analysis. This revealed significant enrichment in chromatin silencing, telomere maintenance, and DNA repair pathways for both set1Δ and set1Δ set5Δ cells (). Although there were a few minor differences in P-values (e.g., mutants categorized as part of chromatin silencing pathways show more significant enrichment with set1Δ set5Δ cells), the overall set of enriched gene ontology categories was largely similar for both mutants.
Figure 4. set1Δ and set1Δ set5Δ mutants are most similar to mutants in chromatin silencing and telomere maintenance. (A) Gene ontology (GO) analysis of the top 50 most correlated mutants to set1Δ and set1Δ set5Δ cells. P-values, calculated by hypergeometric test for enrichment within the mutant data set used, are plotted for the most enriched GO categories. (B) Heatmap indicating Spearman's rank correlation coefficients for the top 20 mutants most correlated with set1Δ and set1Δ set5Δ. Mutants are manually curated into functional categories, indicated on the left by differently colored bars. Within each category, mutants are listed from most to least correlated with the set1Δ set5Δ cells.
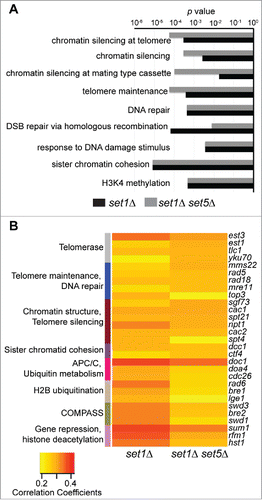
We then directly compared the correlation coefficients for the top 20 mutants most correlated with set1Δ and set1Δ set5Δ and identified the primary processes to which those mutants contribute (). As expected, we observed high correlation between set1Δ cells and mutants that have defects in H3K4 methylation, such as COMPASS complex components and rad6Δ and bre1Δ, required for H2B ubiquitination. These mutants, along with others associated with ubiquitin metabolism and the anaphase promoting complex/cyclosome (APC/C), showed lower correlation with set1Δ set5Δ cells, whereas mutants in telomere function and DNA repair pathways were more highly correlated with the double mutant cells than set1Δ single mutants. These data suggest that the transcriptome of set1Δ set5Δ cells is more similar to that of cells lacking telomerase components, such as EST1 and TLC1, or DNA repair and replication factors, such as MRE11 and MMS22, than is the transcriptome of set1Δ cells.
Many of the top correlated mutants are factors known to be important for telomere silencing, either through direct mechanisms, such as modification or remodeling of telomeric chromatin or transcriptional regulation, or through mechanisms that indirectly alter telomeric chromatin, such as telomere shortening or DNA repair. Interestingly, the only histone deacetylase (HDAC) identified in the top most correlated genes is HST1. Mutants lacking an Hst1 binding partner, Rfm1, and the DNA binding protein, Sum1, were also identified (). These proteins act as a complex to repress genes during vegetative growth normally required for the middle phase of sporulation,Citation63 and Sum1 and Hst1 have partially overlapping activity with the Sir complex to silence telomeric genes.Citation64-66 However, rfm1Δ cells have similar correlation values as hst1Δ and sum1Δ, and Rfm1 is not known to function at telomeres.Citation64 Also, the values for all three mutants are lower in set1Δ set5Δ cells than set1Δ cells, suggesting that the high correlation with these mutants is most likely due to Set1 role in the regulation of sporulation-specific genes,Citation67 and is independent of telomere silencing. Interestingly, other mutants in key genes implicated in chromatin-mediated telomeric silencing, such as SIR2, SIR3, and SIR4, have substantially lower correlation values than mutants in telomere structure and maintenance pathways (Table S3). These observations are consistent with our gene expression and ChIP data (), indicating that disruption of Sir proteins in the absence of Set1 and Set5 may not be the primary mechanism driving derepression of silent genes.
Our correlation data of the transcriptomes of cells lacking Set1 and Set5 suggest that set5Δ further sensitizes set1Δ cells to defects in telomere maintenance and stability. Telomere dysfunction is often linked to increased mutation rates and overall genomic instability in cells.Citation68 We tested the mutation rate of set5Δ, set1Δ, and set1Δ set5Δ by monitoring mutations in the CAN1 gene, which is just over 30 kb from TEL05L.Citation50, 51 CAN1 encodes an arginine permease and mutation within the gene allows growth in the presence of the otherwise toxic arginine analog, canavanine. Fluctuation analysis revealed that set1Δ cells have an increased mutation rate of CAN1 relative to wild type, and the mutation rate in set1Δ set5Δ is higher than the set1Δ single mutant (). These data suggest the loss of Set5 in set1Δ cells further exacerbates defects in genome stability.
Figure 5. Set1 and Set5 contribute to telomere stability. (A) Mutation rate of CAN1 was measured in the mutant strains as described.Citation50, 51 The mutation rate for two experiments of ten colonies each was determined using fluctuation analysis.Citation52 Mutation rate times 10−7 is shown with 95% confidence intervals. (B) Transposition of Ty1 elements upstream of the SUF16 locus was monitored by PCR as described.Citation53, 54 Schematic shows insertion sites upstream of SUF16 and the location of PCR primers. Predicted amplicon sizes range from 780 bp to 1,400 bp depending on the Ty1 insertion site. The mutant strains were grown at 20°C for 72 h to promote transposition events, genomic DNA was extracted and PCR was performed using Primers A and B and against the PHO89 gene as a loading control (Table S1).
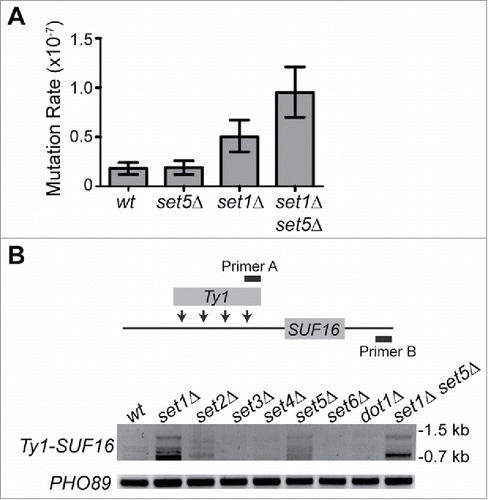
Previous data also indicated a combined role for Set1 and Set5 in the regulation of transposable elements.Citation44 A large fraction of transposable elements are encoded within telomeric regions, and increased transposition is often associated with telomere dysfunction.Citation69 We therefore monitored the transposition of a population of endogenous Ty1 transposable elements to an insertion hotspot upstream of SUF16 using a PCR-based assay,Citation53,54 in SET1- and SET5-mutant cells, as well as strains deleted for other known and putative histone methyltransferases (). Our data show increased transposition predominantly in the set1Δ cells, consistent with previous reportsCitation70 and set5Δ mutants also have increased transposition relative to wild type and the other methyltransferase mutants. Only set2Δ cells show a mild increase in transposition, indicating that this defect is specific to cells lacking Set1 or Set5 and further suggests they each contribute to multiple mechanisms promoting genomic stability.
Discussion
In this study, we investigated the combined role for Set1 and Set5 in gene silencing to identify pathways through which both enzymes act to repress gene expression near telomeres. The role for Set1 in telomere silencing has previously been investigated;Citation13,25-29 however, the precise defect in telomeric chromatin in the absence of Set1 is still unclear. Furthermore, Set5 is a newly-identified histone methyltransferase which catalyzes largely uncharacterized methylation marks, therefore insight into its potential role in silencing provides further understanding of the function of these marks in the genome. At native yeast telomeres, we uncovered a specific defect in H4K5ac levels and telomere maintenance pathways when both Set1 and Set5 are inactivated.
To investigate the genetic interaction between SET5 and the H3K4 methyl species catalyzed by Set1, we analyzed mutants in the COMPASS complex and cells expressing H3K4A. Our qRT-PCR analysis of TEL07L indicates that loss of other COMPASS components required for H3K4me3 and H3K4me2 both show less severe silencing defects than set1Δ, and there is little exacerbation of the silencing defect upon loss of Set5 (see differing scales in relative to ). These results are consistent with other reports showing mild to moderate silencing defects in the absence of COMPASS components Sdc1 and Spp1 relative to set1Δ mutantsCitation13,57,58,71 While the H3K4A mutant also has a less severe silencing defect than set1Δ (), this has been reported to be due to histone dosage effects in a similar strain;Citation13 however, we have not investigated this possibility in our strain background.
One possible interpretation of these data are that Set1 contributes to silencing in both H3K4 methylation-dependent and independent pathways, which has previously been proposed for S. pombe.Citation72 Interestingly, previous work also showed that telomeric gene repression mediated by the SET domain of Set1 depends on the DNA repair protein Mec3, whereas repression mediated by full-length Set1 does not depend on Mec3,Citation34 indicating that there may be SET domain-dependent and independent pathways by which Set1 promotes silencing. This is consistent with our observations regarding the partial dependence of the silent state on H3K4 methylation. Additionally, our investigation of set5Δ mutants and H3K4 methylation defective strains revealed that loss of Set5 does not further sensitize either sdc1Δ or H3K4A mutants to silencing defects, although we did observe a mild increase in expression in the spp1Δ set5Δ double mutant relative to either single mutant. However, we predict that this is most likely due to independent functions of Spp1,Citation73 as others have demonstrated that specific loss of trimethylated H3K4 has little effect on gene derepression.Citation13, 35, 58 Overall, these data imply that Set5 activity is more important for the H3K4 methylation-independent pathway for silencing telomeric genes; however, further genetic analysis is required to elucidate the factors critical to this pathway.
Set1- and Set1/Set5-mediated silencing does not rely on the Sir proteins
The localization of Set1 and H3K4 methyl species within euchromatin, the ability to suppress telomere silencing defects in set1Δ mutants by SIR3 overexpression and the detection of Sir proteins localized to euchromatin in the absence of Set1 has led to the hypothesis that Set1 promotes telomeric silencing by antagonizing Sir protein binding to euchromatin, consequently maintaining the pool of Sir proteins at telomeres.Citation26,29,33 While Sir protein spreading in set1Δ mutants is apparent,Citation26, 29, 35 it has been less clear whether there is a defect in Sir protein occupancy at telomeres in the absence of Set1.Citation25,35 Our data suggest that alterations to Sir3 binding or H4K16ac do not account for decreased silencing at telomeres in the absence of either Set1 and/or Set5 (). Furthermore, only minimal overlap was detected between Set1/Set5-repressed genes and a recently curated list of Sir-regulated genes at telomeres,Citation8 and genes at telomeres that do not show significant Sir-induced silencing do have increased expression in set5Δ, set1Δ and set1Δ set5Δ cells (see TEL09R in ). We also did not observe a high correlation between the transcriptomes of SIR mutants and those of either set1Δ or set1Δ set5Δ mutants (, Table S3). Therefore, while our results are not inconsistent with the model that H3K4 methylation within euchromatin acts as an anti-silencing mechanism, they do not support the conclusion that defective silencing at telomeres is due to local loss of Sir proteins. These data are similar to previous ChIP investigations in set1Δ, as well as cells lacking H2BK123Ub, a mark required for methylation by Set1.Citation25,35 Our data overall are in agreement with the broad notion that additional mechanisms beyond the activity of the Sir proteins are critical for gene repression near telomeres,Citation8,13 such as pathways linked to nucleosome density, telomere function, antisense transcription, other types of chromatin modification or as yet undiscovered interactions between chromatin modifiers.
In accordance with our finding that Set1- and Set5-mediated gene silencing likely relies on mechanisms independent of Sir protein activity and H4K16 deacetylation, we investigated other potential avenues of silencing. Given that Set5 is an H4 K5, K8, and K12 methyltransferase,Citation37 we predicted that it may have a role in modulating acetylation at these sites.Citation74 Our data show increasing levels of H4K5ac in set5Δ relative to wild type, in set1Δ relative to set5Δ, and in set1Δ relative to set1Δ set5Δ cells across TEL07L (), which largely mirrors the changes in gene expression observed in these mutants (). We observed a moderate increase in H4K8ac, but no significant changes in H4K12ac levels (). We also did not observe any changes in histone H4 levels across the subtelomere () Interestingly, unique roles for H4K5ac and H4K12ac at telomeres have previously been proposed. H4K12ac, and less so H4K5ac, are required for the establishment of heterochromatin at telomeres,Citation21 distinct from H4K8ac. Also, the action of the HDAC Rpd3 at the subtelomeric heterochromatin-euchromatin boundary has been proposed to counteract silencing by specifically deacetylating H4K5, in contrast to other acetyl marks.Citation20 Our data suggest that Set1 and Set5 do not have a significant impact on H4K12ac levels; however, modulation of H4K5ac levels in particular appears linked to the defect in telomere silencing in the absence of Set1 and Set5.
Functional roles of Set5 and Set1 in telomere maintenance
The wealth of genomic data available for yeast allowed us to further define potential pathways through which Set1 and Set5 may act to promote telomere silencing. Our correlation analysis of set1Δ and set1Δ set5Δ cells relative to a collection of mutants representing the primary regulators of gene expression in yeast revealed that mutants in telomere maintenance pathways and DNA repair were the main categories that distinguished set1Δ set5Δ cells from set1Δ cells. A genome-wide genetic interaction screen, or E-MAP (epistatic miniarray profile), previously conducted with both set1Δ and set5Δ mutants also showed that each mutant was enriched for negative genetic interactions with mutants in telomere maintenance and DNA repair and replication factors.Citation44 Increased mutation rate and genomic instability in set1Δ set5Δ cells () corroborate these genetic findings and suggest that the loss of Set5 further sensitizes set1Δ mutants to telomere damage.
While Set1 has previously been implicated in maintaining telomere structure,Citation27,34,35,55 our data indicate a role for Set5 in protecting telomeres in the absence of Set1. Given our observation of increased H4K5ac over the TEL07L subtelomere, one possible mechanism is that methylation of H4K5 by Set5 may block acetylation in the region, inhibiting the development of a euchromatic-like chromatin state. The lack of ChIP-grade antibodies against the H4 methyl species has precluded us from reliably determining whether Set5 acts directly at telomeres, or whether its activity elsewhere in the genome influences telomeres. However, based on the low abundance of the methyl marks in the genome under normal growth conditionsCitation37 we predict that Set5 is acting in a site-specific manner to promote proper chromatin structure and function at telomeres. Additional experiments will be required to dissect the mechanism by which Set5 role in telomere silencing and maintenance overlaps with that of Set1.
The transcriptomes of set1Δ set5Δ cells is most highly correlated with those of mutants whose primary defect is in telomere structure (e.g., EST1 and TLC1, both of which encode components of telomerase), suggesting that the aberrant silencing in these cells shares similar features to strains with telomere maintenance defects. We speculate that Set5 and Set1 have unique but overlapping roles in maintaining H4K5 and, to a lesser extent, H4K8 deacetylation in the region, thereby preventing the formation of a euchromatic-like state at telomeres. As described above, we predict that Set5 acts directly at telomeres to methylate H4K5 and K8 to inhibit acetylation, and Set1 may act indirectly through its roles in euchromatin, or directly at the heterochromatin-euchromatin boundary, where H3K4 methylation has been detectedCitation30 to maintain a deacetylated state, independently of Sir proteins. The loss of heterochromatin at telomeres is associated with increased genomic instability stemming from large deletions, de novo telomere additions and end-to-end fusions.Citation75,76 In addition, forced transcription through telomeres or disruption of silencing proteins leads to decreased telomere length and aberrant rates of telomere-dependent cellular senescence.Citation77-79 Although additional mechanistic details remain to be determined, the increased gene expression and euchromatic state of telomeric chromatin in the absence of Set1 and Set5 are most similar to cells with defects in telomere maintenance pathways, suggesting that the repressive environment promoted by Set1 and Set5 is important for generating protective chromatin states at telomeres to prevent instability. Further analysis of the specific functions for both Set1 and Set5 at telomeres will provide insight into their partially overlapping roles in gene silencing and telomere maintenance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1265712_supplementary_data.zip
Download Zip (392.9 KB)Acknowledgments
The authors acknowledge members of the Green lab for additional technical support, helpful discussions, and comments on the manuscript. Author contributions: A.G. and M.J. contributed equally to the work. M.J., A.G., A.G.-Y. and E.M.G performed genetic and molecular biology experiments. G.C., R.K., J.Q. and D.P. performed RNA-seq analysis and correlation analysis. E.M.G. conceived of the study and wrote the manuscript. The authors declare that they have no conflicts of interest with the contents of this article.
Funding
This work was supported in part by NIH grant R03AG052018 to E.M.G. and by an Undergraduate Biology and Mathematics NSF training grant number 1031420 to the Departments of Biological Sciences and Mathematics and Statistics at UMBC.
Related Research Data
References
- Brunet A, Berger SL. Epigenetics of aging and aging-related disease. J Gerontol A Biol Sci Med Sci 2014; 69 Suppl 1:S17-20; PMID:24833581; http://dx.doi.org/10.1093/gerona/glu042
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153:1194-217; PMID:23746838; http://dx.doi.org/10.1016/j.cell.2013.05.039
- Teixeira MT. Saccharomyces cerevisiae as a model to study replicative senescence triggered by telomere shortening. Front Oncol 2013; 3:101; PMID:23638436; http://dx.doi.org/10.3389/fonc.2013.00101
- Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 2003; 72:481-516; PMID:12676793; http://dx.doi.org/10.1146/annurev.biochem.72.121801.161547
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 1990; 63:751-62; PMID:2225075; http://dx.doi.org/10.1016/0092-8674(90)90141-Z
- Renauld H, Aparicio OM, Zierath PD, Billington BL, Chhablani SK, Gottschling DE. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev 1993; 7:1133-45; PMID:8319906; http://dx.doi.org/10.1101/gad.7.7a.1133
- Wyrick JJ, Holstege FC, Jennings EG, Causton HC, Shore D, Grunstein M, Lander ES, Young RA. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 1999; 402:418-21; PMID:10586882; http://dx.doi.org/10.1038/46567
- Ellahi A, Thurtle DM, Rine J. The Chromatin and Transcriptional Landscape of Native Saccharomyces cerevisiae Telomeres and Subtelomeric Domains. Genetics 2015; 200:505-21; PMID:25823445; http://dx.doi.org/10.1534/genetics.115.175711
- Loney ER, Inglis PW, Sharp S, Pryde FE, Kent NA, Mellor J, Louis EJ. Repressive and non-repressive chromatin at native telomeres in Saccharomyces cerevisiae. Epigenetics Chromatin 2009; 2:18; PMID:19954519; http://dx.doi.org/10.1186/1756-8935-2-18
- Pryde FE, Louis EJ. Limitations of silencing at native yeast telomeres. EMBO J 1999; 18:2538-50; PMID:10228167; http://dx.doi.org/10.1093/emboj/18.9.2538
- Takahashi YH, Schulze JM, Jackson J, Hentrich T, Seidel C, Jaspersen SL, Kobor MS, Shilatifard A. Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol Cell 2011; 42:118-26; PMID:21474073; http://dx.doi.org/10.1016/j.molcel.2011.03.006
- Rossmann MP, Luo W, Tsaponina O, Chabes A, Stillman B. A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol Cell 2011; 42:127-36; PMID:21474074; http://dx.doi.org/10.1016/j.molcel.2011.03.007
- Margaritis T, Oreal V, Brabers N, Maestroni L, Vitaliano-Prunier A, Benschop JJ, van Hooff S, van Leenen D, Dargemont C, Géli V, et al. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3′-end antisense transcription. PLoS Genet 2012; 8:e1002952; PMID:23028359; http://dx.doi.org/10.1371/journal.pgen.1002952
- Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet 2002; 32:370-7; PMID:12410229; http://dx.doi.org/10.1038/ng993
- Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet 2002; 32:378-83; PMID:12379856; http://dx.doi.org/10.1038/ng1017
- Hecht A, Strahl-Bolsinger S, Grunstein M. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature 1996; 383:92-6; PMID:8779721; http://dx.doi.org/10.1038/383092a0
- Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003; 112:725-36; PMID:12628191; http://dx.doi.org/10.1016/S0092-8674(03)00123-5
- Clarke AS, Samal E, Pillus L. Distinct roles for the essential MYST family HAT Esa1p in transcriptional silencing. Mol Biol Cell 2006; 17:1744-57; PMID:16436512; http://dx.doi.org/10.1091/mbc.E05-07-0613
- Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Côté J. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev 2003; 17:1415-28; PMID:12782659; http://dx.doi.org/10.1101/gad.1056603
- Zhou J, Zhou BO, Lenzmeier BA, Zhou JQ. Histone deacetylase Rpd3 antagonizes Sir2-dependent silent chromatin propagation. Nucleic Acids Res 2009; 37:3699-713; PMID:19372273; http://dx.doi.org/10.1093/nar/gkp233
- Zhou BO, Wang SS, Zhang Y, Fu XH, Dang W, Lenzmeier BA, Zhou JQ. Histone H4 lysine 12 acetylation regulates telomeric heterochromatin plasticity in Saccharomyces cerevisiae. PLoS Genet 2011; 7:e1001272; PMID:21249184; http://dx.doi.org/10.1371/journal.pgen.1001272
- Carmen AA, Milne L, Grunstein M. Acetylation of the yeast histone H4 N terminus regulates its binding to heterochromatin protein SIR3. J Biol Chem 2002; 277:4778-81; PMID:11714726; http://dx.doi.org/10.1074/jbc.M110532200
- Ladurner AG, Inouye C, Jain R, Tjian R. Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol Cell 2003; 11:365-76; PMID:12620225; http://dx.doi.org/10.1016/S1097-2765(03)00035-2
- Braunstein M, Sobel RE, Allis CD, Turner BM, Broach JR. Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol Cell Biol 1996; 16:4349-56; PMID:8754835; http://dx.doi.org/10.1128/MCB.16.8.4349
- Ng HH, Dole S, Struhl K. The Rtf1 component of the Paf1 transcriptional elongation complex is required for ubiquitination of histone H2B. J Biol Chem 2003; 278:33625-8; PMID:12876293; http://dx.doi.org/10.1074/jbc.C300270200
- Santos-Rosa H, Bannister AJ, Dehe PM, Géli V, Kouzarides T. Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatin. J Biol Chem 2004; 279:47506-12; PMID:15280381; http://dx.doi.org/10.1074/jbc.M407949200
- Nislow C, Ray E, Pillus L. SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol Biol Cell 1997; 8:2421-36; PMID:9398665; http://dx.doi.org/10.1091/mbc.8.12.2421
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U S A 2001; 98:12902-7; PMID:11687631; http://dx.doi.org/10.1073/pnas.231473398
- Venkatasubrahmanyam S, Hwang WW, Meneghini MD, Tong AH, Madhani HD. Genome-wide, as opposed to local, antisilencing is mediated redundantly by the euchromatic factors Set1 and H2A.Z. Proc Natl Acad Sci U S A 2007; 104:16609-14; PMID:17925448; http://dx.doi.org/10.1073/pnas.0700914104
- Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Vermeulen M, Mann M, Bähler J, Green RD, Kouzarides T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 2007; 449:928-32; PMID:17898715; http://dx.doi.org/10.1038/nature06160
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A 2002; 99:8695-700; PMID:12060701; http://dx.doi.org/10.1073/pnas.082249499
- Bryk M, Briggs SD, Strahl BD, Curcio MJ, Allis CD, Winston F. Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanism. Curr Biol 2002; 12:165-70; PMID:11818070; http://dx.doi.org/10.1016/S0960-9822(01)00652-2
- Katan-Khaykovich Y, Struhl K. Heterochromatin formation involves changes in histone modifications over multiple cell generations. EMBO J 2005; 24:2138-49; PMID:15920479; http://dx.doi.org/10.1038/sj.emboj.7600692
- Corda Y, Schramke V, Longhese MP, Smokvina T, Paciotti V, Brevet V, Gilson E, Géli V. Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nat Genet 1999; 21:204-8; PMID:9988274; http://dx.doi.org/10.1038/5991
- Leung A, Cajigas I, Jia P, Ezhkova E, Brickner JH, Zhao Z, Geng F, Tansey WP. Histone H2B ubiquitylation and H3 lysine 4 methylation prevent ectopic silencing of euchromatic loci important for the cellular response to heat. Mol Biol Cell 2011; 22:2741-53; PMID:21680712; http://dx.doi.org/10.1091/mbc.E11-05-0426
- Trelles-Sticken E, Bonfils S, Sollier J, Géli V, Scherthan H, de La Roche Saint-André C. Set1- and Clb5-deficiencies disclose the differential regulation of centromere and telomere dynamics in Saccharomyces cerevisiae meiosis. J Cell Sci 2005; 118:4985-94; PMID:16254243; http://dx.doi.org/10.1242/jcs.02612
- Green EM, Mas G, Young NL, Garcia BA, Gozani O. Methylation of H4 lysines 5, 8 and 12 by yeast Set5 calibrates chromatin stress responses. Nat Struct Mol Biol 2012; 19:361-3; PMID:22343720; http://dx.doi.org/10.1038/nsmb.2252
- Van Aller GS, Reynoird N, Barbash O, Huddleston M, Liu S, Zmoos AF, McDevitt P, Sinnamon R, Le B, Mas G, et al. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 2012; 7:340-3; PMID:22419068; http://dx.doi.org/10.4161/epi.19506
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14:953-61; PMID:9717241; http://dx.doi.org/10.1002/(SICI)1097-0061(199807)14:10%3c953::AID-YEA293%3e3.0.CO;2-U
- Teste MA, Duquenne M, François JM, Parrou JL. Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol Biol 2009; 10:99; PMID:19874630; http://dx.doi.org/10.1186/1471-2199-10-99
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast 2000; 16:857-60; PMID:10861908; http://dx.doi.org/10.1002/1097-0061(20000630)16:9%3c857::AID-YEA561%3e3.0.CO;2-B
- Meluh PB, Broach JR. Immunological analysis of yeast chromatin. Methods Enzymol 1999; 304:414-30; PMID:10372374; http://dx.doi.org/10.1016/S0076-6879(99)04025-2
- Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol 2005; 3:e328; PMID:16122352; http://dx.doi.org/10.1371/journal.pbio.0030328
- Martín GM, King DA, Green EM, Garcia-Nieto PE, Alexander R, Collins SR, Krogan NJ, Gozani OP, Morrison AJ. Set5 and Set1 cooperate to repress gene expression at telomeres and retrotransposons. Epigenetics 2014; 9:513-22; PMID:24442241; http://dx.doi.org/10.4161/epi.27645
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 2015; 12:115-21; PMID:25633503; http://dx.doi.org/10.1038/nmeth.3252
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15:R29; PMID:24485249; http://dx.doi.org/10.1186/gb-2014-15-2-r29
- Law CW, Alhamdoosh M, Su S, Smyth GK, Ritchie ME. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 2016; 5:1408; PMID:27441086; http://dx.doi.org/10.12688/f1000research.9005.1
- Kemmeren P, Sameith K, van de Pasch LA, Benschop JJ, Lenstra TL, Margaritis T, O'Duibhir E, Apweiler E, van Wageningen S, Ko CW, et al. Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell 2014; 157:740-52; PMID:24766815; http://dx.doi.org/10.1016/j.cell.2014.02.054
- Robinson MD, Grigull J, Mohammad N, Hughes TR. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics 2002; 3:35; PMID:12431279; http://dx.doi.org/10.1186/1471-2105-3-35
- Huang ME, Rio AG, Galibert MD, Galibert F. Pol32, a subunit of Saccharomyces cerevisiae DNA polymerase delta, suppresses genomic deletions and is involved in the mutagenic bypass pathway. Genetics 2002; 160:1409-22; PMID:11973297
- Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A 2003; 100:11529-34; PMID:12972632; http://dx.doi.org/10.1073/pnas.2035018100
- Hall BM, Ma CX, Liang P, Singh KK. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 2009; 25:1564-5; PMID:19369502; http://dx.doi.org/10.1093/bioinformatics/btp253
- Nyswaner KM, Checkley MA, Yi M, Stephens RM, Garfinkel DJ. Chromatin-associated genes protect the yeast genome from Ty1 insertional mutagenesis. Genetics 2008; 178:197-214; PMID:18202368; http://dx.doi.org/10.1534/genetics.107.082602
- Checkley MA, Nagashima K, Lockett SJ, Nyswaner KM, Garfinkel DJ. P-body components are required for Ty1 retrotransposition during assembly of retrotransposition-competent virus-like particles. Mol Cell Biol 2010; 30:382-98; PMID:19901074; http://dx.doi.org/10.1128/MCB.00251-09
- Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 2001; 20:7137-48; PMID:11742990; http://dx.doi.org/10.1093/emboj/20.24.7137
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci U S A 2002; 99:90-4; PMID:11752412; http://dx.doi.org/10.1073/pnas.221596698
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 2002; 277:10753-5; PMID:11805083; http://dx.doi.org/10.1074/jbc.C200023200
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 2005; 19:849-56; PMID:16168379; http://dx.doi.org/10.1016/j.molcel.2005.07.024
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47; PMID:25605792; http://dx.doi.org/10.1093/nar/gkv007
- Kim T, Buratowski S. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell 2009; 137:259-72; PMID:19379692; http://dx.doi.org/10.1016/j.cell.2009.02.045
- Morillon A, Karabetsou N, Nair A, Mellor J. Dynamic lysine methylation on histone H3 defines the regulatory phase of gene transcription. Mol Cell 2005; 18:723-34; PMID:15949446; http://dx.doi.org/10.1016/j.molcel.2005.05.009
- Wang C, Gong B, Bushel PR, Thierry-Mieg J, Thierry-Mieg D, Xu J, Fang H, Hong H, Shen J, Su Z, et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat Biotechnol 2014; 32:926-32; PMID:25150839; http://dx.doi.org/10.1038/nbt.3001
- McCord R, Pierce M, Xie J, Wonkatal S, Mickel C, Vershon AK. Rfm1, a novel tethering factor required to recruit the Hst1 histone deacetylase for repression of middle sporulation genes. Mol Cell Biol 2003; 23:2009-16; PMID:12612074; http://dx.doi.org/10.1128/MCB.23.6.2009-2016.2003
- Li M, Valsakumar V, Poorey K, Bekiranov S, Smith JS. Genome-wide analysis of functional sirtuin chromatin targets in yeast. Genome Biol 2013; 14:R48; PMID:23710766; http://dx.doi.org/10.1186/gb-2013-14-5-r48
- Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 1995; 9:2888-902; PMID:7498786; http://dx.doi.org/10.1101/gad.9.23.2888
- Hickman MA, Rusche LN. Substitution as a mechanism for genetic robustness: the duplicated deacetylases Hst1p and Sir2p in Saccharomyces cerevisiae. PLoS Genet 2007; 3:e126; PMID:17676954; http://dx.doi.org/10.1371/journal.pgen.0030126
- Sollier J, Lin W, Soustelle C, Suhre K, Nicolas A, Géli V, de La Roche Saint-André C. Set1 is required for meiotic S-phase onset, double-strand break formation and middle gene expression. EMBO J 2004; 23:1957-67; PMID:15071505; http://dx.doi.org/10.1038/sj.emboj.7600204
- Hackett JA, Feldser DM, Greider CW. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001; 106:275-86; PMID:11509177; http://dx.doi.org/10.1016/S0092-8674(01)00457-3
- Chan JE, Kolodner RD. A genetic and structural study of genome rearrangements mediated by high copy repeat Ty1 elements. PLoS Genet 2011; 7:e1002089; PMID:21637792; http://dx.doi.org/10.1371/journal.pgen.1002089
- Berretta J, Pinskaya M, Morillon A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev 2008; 22:615-26; PMID:18316478; http://dx.doi.org/10.1101/gad.458008
- Mueller JE, Canze M, Bryk M. The requirements for COMPASS and Paf1 in transcriptional silencing and methylation of histone H3 in Saccharomyces cerevisiae. Genetics 2006; 173:557-67; PMID:16582434; http://dx.doi.org/10.1534/genetics.106.055400
- Mikheyeva IV, Grady PJ, Tamburini FB, Lorenz DR, Cam HP. Multifaceted genome control by Set1 Dependent and Independent of H3K4 methylation and the Set1C/COMPASS complex. PLoS Genet 2014; 10:e1004740; PMID:25356590; http://dx.doi.org/10.1371/journal.pgen.1004740
- Acquaviva L, Drogat J, Dehé PM, de La Roche Saint-André C, Géli V. Spp1 at the crossroads of H3K4me3 regulation and meiotic recombination. Epigenetics 2013; 8:355-60; PMID:23511748; http://dx.doi.org/10.4161/epi.24295
- Green EM, Morrison AJ, Gozani O. New marks on the block: Set5 methylates H4 lysines 5, 8 and 12. Nucleus 2012; 3:335-9; PMID:22688645; http://dx.doi.org/10.4161/nucl.20695
- Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, Haughton M, Baird DM. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev 2007; 21:2495-508; PMID:17908935; http://dx.doi.org/10.1101/gad.439107
- Frias C, Pampalona J, Genesca A, Tusell L. Telomere dysfunction and genome instability. Front Biosci (Landmark Ed) 2012; 17:2181-96; PMID:22652771; http://dx.doi.org/10.2741/4044
- Maicher A, Kastner L, Dees M, Luke B. Deregulated telomere transcription causes replication-dependent telomere shortening and promotes cellular senescence. Nucleic Acids Res 2012; 40:6649-59; PMID:22553368; http://dx.doi.org/10.1093/nar/gks358
- Kozak ML, Chavez A, Dang W, Berger SL, Ashok A, Guo X, Johnson FB. Inactivation of the Sas2 histone acetyltransferase delays senescence driven by telomere dysfunction. EMBO J 2010; 29:158-70; PMID:19875981; http://dx.doi.org/10.1038/emboj.2009.314
- Sandell LL, Gottschling DE, Zakian VA. Transcription of a yeast telomere alleviates telomere position effect without affecting chromosome stability. Proc Natl Acad Sci U S A 1994; 91:12061-5; PMID:7991584; http://dx.doi.org/10.1073/pnas.91.25.12061
- Krishnamoorthy T, Chen X, Govin J, Cheung WL, Dorsey J, Schindler K, Winter E, Allis CD, Guacci V, Khochbin S, et al. Phosphorylation of histone H4 Ser1 regulates sporulation in yeast and is conserved in fly and mouse spermatogenesis. Genes Dev 2006; 20:2580-92; PMID:16980586; http://dx.doi.org/10.1101/gad.1457006