
ABSTRACT
microRNAs (miRs) are small noncoding RNAs that regulate/fine tune many cellular protein networks by targeting mRNAs for either degradation or translational inhibition. Dicer, a type III endoribonuclease, is a critical component in miR biogenesis and is required for mature microRNA production. Abnormal Dicer expression occurs in numerous cancer types and correlates with poor patient prognosis. Recent reports have demonstrated that epigenetic agents, including histone deacetylase inhibitors (HDACi), may regulate Dicer and miR expression. HDACi are a class of epigenetic agents used to treat cancer, viral infections, and inflammatory disorders. However, little is known regarding the epigenetic regulation of miR biogenesis and function. We therefore investigated whether clinically successful HDACi modulated Dicer expression and found that Panobinostat, a clinically approved HDACi, enhanced Dicer expression via posttranscriptional mechanisms. Studies using proteasome inhibitors suggested that Panobinostat regulated the proteasomal degradation of Dicer. Further studies demonstrated that Panobinostat, despite increasing Dicer protein expression, decreased Dicer activity. This suggests that Dicer protein levels do not necessarily correlate with Dicer activity and mature miR levels. Taken together, we present evidence here that Panobinostat posttranscriptionally regulates Dicer/miR biogenesis and suggest Dicer as a potential therapeutic target in cancer.
Introduction
Cancer development is a complex process involving multiple factors including, but not limited to, oncogene activation, silencing of tumor suppressor genes, resistance to apoptosis, and immune escape.Citation1 Recent work has demonstrated that epigenetic dysregulation is crucial to cancer development and growth.Citation2 Abnormal histone tail posttranslational modifications alter acetylation status and result in irregular gene expression.Citation3 Several clinical drugs aimed at targeting these epigenetic abnormalities in cancer are available. Histone deacetylase inhibitors (HDACi) are a relatively new class of epigenetic modifying drugs developed to treat cancer.Citation4 Tumor cells have a higher sensitivity to HDACi treatment compared with non-transformed cells due to enhanced cell cycle progression and altered tumor suppressor/oncogene expression.Citation5 HDACi treatment in cancer harnesses this sensitivity by selectively inducing tumor cell apoptosis, differentiation, and/or cell cycle inhibition.Citation6
Previous work from our lab demonstrated that HDACi treatment of tumor cells enhanced immune gene expression and tumor immunogenicity.Citation7-11 Identification of the mechanisms underlying immune modulation by HDACi treatment thus becomes an important question. A landmark study recently showed that a functional immune system is required for the beneficial effects of HDACi treatment.Citation12 However, HDACi may also have several negative effects on the immune system, including expansion of immunosuppressive cell populations such as T-regs and MDSC.Citation13,14 Furthermore, innate immune cells become suppressed upon exposure to HDACi.Citation15 Current HDACi treatment goals are to induce apoptosis and differentiation in tumor cells. In addition to negative effects on the immune system, unwanted side effects and toxicity have been reported.Citation16 Therefore, it is important to fully understand HDACi treatment effects.
microRNAs (miRs) are ∼22 nucleotide noncoding RNAs that contribute to posttranscriptional gene regulation.Citation17 Their biogenesis is a tightly regulated process that involves numerous evolutionarily conserved factors. Dicer, an RNase III endoribonuclease, is responsible for cleaving the pre-miR into a mature miR duplex in the cytoplasm and is indispensable to most miR biogenesis.Citation18 Upon completion of miR maturation, the mature miR duplex is unwound and the guide strand is loaded onto the RNA-induced silencing complex (RISC).Citation17 The RISC is responsible for bringing the miR to its target mRNAs, where it binds to the 3′ UTR and leads to translation inhibition or degradation of the target mRNA.Citation19 Dicer is essential for proper embryonic development, as well as organogenesis in vertebrates. An embryonic knockout of Dicer is lethal in mice.Citation20 Furthermore, Dicer has a substantial role in immune cell development and function.Citation21 A potential role for Dicer in oncogenesis was identified by the observation that diminished miR processing augmented tumorigenesis and cellular transformation in lung cancer.Citation22 Moreover, several cancer subtypes possess abnormal Dicer expression, correlating with increased disease staging and poor patient prognosis/overall survival.Citation23
One HDACi in particular, Panobinostat, has achieved some success in cancer and is FDA approved for treating multiple myeloma.Citation24 Panobinostat inhibits class I (HDAC 1, 2, 3, 8), class II (HDAC 4, 5, 6, 7, 9), and class IV (HDAC 11) HDACs and is one of the most robust pan-HDACi used clinically.Citation25 However, it remains unknown if Panobinostat treatment affects Dicer and miR expression. The studies reported here were designed to determine if clinically relevant HDACi, including Panobinostat, regulate Dicer and miR expression in tumor cells. We examined the consequence of Panobinostat treatments on Dicer expression and activity, while extending the studies to chemically similar HDACi. Lastly, we investigated the mechanism by which Panobinostat regulates Dicer protein expression and activity. We demonstrate that Panobinostat regulates Dicer expression and its activity in a posttranscriptional manner and that Dicer regulation by HDACi could potentially be used clinically to treat certain diseases.
Results
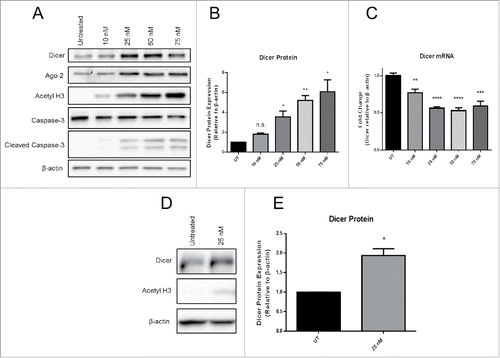
We sought to determine whether Panobinostat, a HDACi used in cancer treatment, regulates Dicer expression in tumor cells. JAR choriocarcinoma cells possess epigenetically silenced immune genes that serve as a model for immune escape. JAR cells were treated in vitro with increasing doses of Panobinostat for 24 h. We found that Panobinostat significantly enhanced Dicer protein expression in a dose dependent manner ( and ). Since HDACi promote the acetylation of histones and non-histone substrates, we used acetylated histone H3 to represent treatment efficacy. Panobinostat treatment enhanced acetylated histone H3 levels that correlated with increased Dicer protein expression. This suggests a direct connection between chromatin status and Dicer protein expression. An important issue is that HDACi can induce apoptosis in cells depending on the dose and treatment duration. The presence of cleaved Caspase-3 was measured to identify whether apoptosis was occurring following Panobinostat treatment. However, only low levels of cleaved Caspase-3 were detected after treatment (). Therefore, apoptosis likely does not play a role in Dicer enhancement by Panobinostat. Furthermore, Dicer was identified as a potential Caspase substrate, which suggests that apoptosis would have the opposite effect on Dicer mRNA expression.Citation26 Surprisingly, Dicer mRNA expression was significantly decreased by Panobinostat, implying that Dicer was likely being posttranscriptionally regulated (). Argonaute-2, a member of the RNA-induced silencing complex (RISC), was also enhanced at the protein level by Panobinostat (). This suggests that multiple steps in the miR pathway are epigenetically regulated. To investigate whether chemically similar HDACi to Panobinostat regulate Dicer, we tested a second hydroxamate, Trichostatin A (TSA). TSA is one of the most well characterized HDACi, but has not been used extensively in recent clinical trials. A dose of TSA that induced proportionate cell viability to 25 nM Panobinostat, as detected by trypan blue viability, was used to treat JAR cells in vitro for 24 h. Similar to Panobinostat, TSA significantly enhanced Dicer protein expression (and ). To confirm that Dicer regulation by Panobinostat and TSA was not specific to JAR cells, the above experiments were repeated using HeLa cells, a cervical carcinoma. A comparable significant enhancement in Dicer protein expression was obtained from HeLa cells treated with Panobinostat for 24 h (data not shown). Furthermore, TSA also significantly enhanced Dicer protein expression in HeLa cells (data not shown). These data demonstrate that Dicer protein expression is enhanced by the epigenetic agents Panobinostat and TSA.
Figure 1. Panobinostat posttranscriptionally enhances Dicer protein expression. A. JAR cells treated with increasing doses of Panobinostat for 24 h were harvested, lysed, and probed for expression of Dicer, Argonaute-2, acetylated histone 3, Caspase-3, cleaved Caspase-3, and β-actin via Western blot. Data represent three independent experiments. B. Data from A quantified from three independent experiments. C. RNA was harvested from the previous cell treatments and Dicer and β-actin mRNA levels were assessed via quantitative real-time RT-PCR. D. JAR cells treated with 25 nM Trichostatin A were harvested and probed for expression of Dicer, acetylated histone 3, and β-actin via Western blot. The data represent three independent experiments. E. Data from D quantified from three independent experiments. Error bars ± SEM, *P < 0.05, **P < 0.005, ***P < 0.0005.
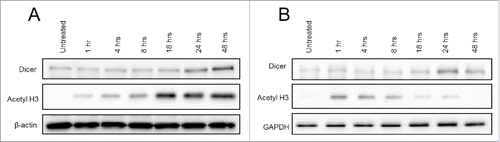
To further characterize Dicer protein regulation by Panobinostat, JAR cells were treated with 25 nM Panobinostat over a 48 h time course. An enhancement of Dicer protein expression was first detected at 24 h of treatment, suggesting the increase was occurring between 18–24 h post-treatment (). However, acetylated histone H3 was first observed 1 h post-treatment, and peaked 18 h post-treatment. This implies that Panobinostat rapidly inhibits deacetylase activity without measurable effects on Dicer protein expression. Dicer mRNA levels did not increase at any point during the time course (data not shown). Similar results were obtained using TSA; however, histone H3 acetylation following TSA treatment did not endure as long as that observed with Panobinostat (). Consistent with our data demonstrating that Dicer mRNA is decreased by Panobinostat, these results suggest that Panobinostat can posttranscriptionally regulate Dicer.
Figure 2. Dicer protein expression over 48 h of Panobinostat treatment. A. JAR cells treated with 25 nM Panobinostat over the course of 48 h were harvested and probed for expression of Dicer, acetylated histone 3, and β-actin via Western blot. B. JAR cells treated with 25 nM Trichostatin A over the course of 48 h were harvested and probed for expression of Dicer, acetylated histone 3, and GAPDH via Western blot. The data represent two independent experiments.
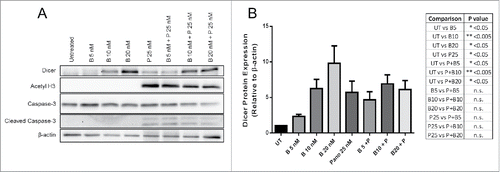
Since Dicer protein expression was enhanced by Panobinostat and TSA, Panobinostat was chosen for use in subsequent studies because it gave a greater enhancement of Dicer protein expression and due to its clinical relevance. HDACi treatment can alter proteasomal degradation of various proteins, either enhancing or inhibiting their degradation.Citation27-32 Therefore, we investigated whether the increase in Dicer protein expression by Panobinostat was a result of diminished proteasomal degradation. Bortezomib, a well-known and clinically relevant inhibitor of proteasomal degradation, was chosen for these studies. JAR cells were treated with Bortezomib, Panobinostat, or both for 24 h. As expected, Panobinostat significantly enhanced Dicer protein expression (). We also found that Bortezomib significantly increased Dicer protein expression, suggesting that Dicer is degraded by the proteasome (). Surprisingly, there was no significant difference between Panobinostat and Bortezomib combination treatments compared with the single agent treatments (). This suggests that Dicer protein expression was not synergistically enhanced by the Panobinostat and Bortezomib combination and that both drugs may regulate Dicer expression through similar mechanisms ().
Figure 3. Panobinostat reduces the proteasomal degradation of Dicer. A. JAR cells treated for 24 h with Bortezomib, Panobinostat, or Bortezomib + Panobinostat were harvested and probed for expression of Dicer, acetylated histone 3, Caspase-3, cleaved Caspase-3, and β-actin via Western blot. The data represent three independent experiments. B. Data from A quantified from three independent experiments combined and the statistics are displayed in the table. Error bars ± SEM, *P < 0.05, **P < 0.005.
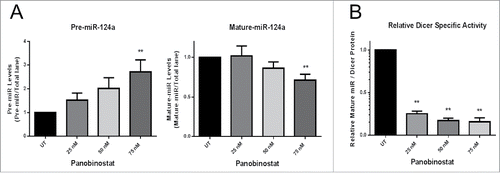
Although Dicer protein expression was enhanced by either Panobinostat or Bortezomib treatments, it is important to determine how these changes affect Dicer-dependent miR biogenesis. Therefore, we established an in vitro Dicer activity assay utilizing radiolabeled pre-miR-124a to quantify Dicer activity in protein lysates. Untreated JAR cell protein lysates were titrated to demonstrate the linearity in both pre-miR cleavage and mature-miR production (Supp. Fig. 1A and B). The radiolabeled pre-miR-124a Dicer cleavage sites are known, which allowed us to predict specific cleavage products. Recombinant Human Dicer was incubated with radiolabeled pre-miR-124a and demonstrated specific cleavage of the pre-miR by Dicer (Supp. Fig. 1C). Surprisingly, JAR cells treated with Panobinostat for 24 h exhibited significantly less Dicer activity, as determined by more pre-miR-124a template remaining and less mature miR-124a produced (). Dicer's relative specific activity was calculated using the ratio of mature-miR produced to normalized Dicer protein expression in the cell lysate. There was a significant reduction in Dicer's relative specific activity by Panobinostat treatments (). These results demonstrate that Panobinostat treated cells have reduced Dicer activity despite higher Dicer protein expression.
Figure 4. Panobinostat decreases Dicer activity. A. Protein lysates (20 µg) from JAR cells +/− 24 h of Panobinostat treatment were used in the Dicer activity assay and the amount of pre-miR-124a and mature-miR-124a were quantified. B. Dicer's relative specific activity was calculated using the ratio of mature-miR-124a produced to Dicer protein expression in a lysate. The data represent three independent experiments. Error bars ± SEM, **P < 0.005.
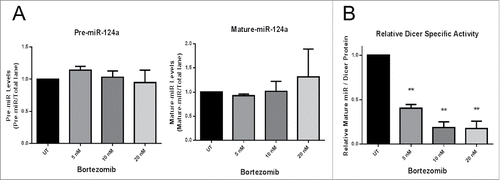
Since Panobinostat treated cells had enhanced Dicer protein expression, but reduced Dicer activity, we next sought to determine if Bortezomib had similar effects. As demonstrated earlier, Bortezomib significantly enhanced Dicer protein expression (). Unexpectedly, there was no significant difference in the amount of mature miR-124a produced or pre-miR-124a template remaining in assays with the JAR cell protein lysates +/− Bortezomib treatment (). However, Dicer's relative specific activity was significantly reduced by Bortezomib (). These data further suggest that Dicer protein expression does not reflect activity levels, as two separate treatments that increase Dicer protein expression (Panobinostat and Bortezomib) inhibit its activity.
Figure 5. Bortezomib decreases Dicer activity. A. 20 µg of protein lysates from JAR cells +/− 24 h of Bortezomib treatment were used in the Dicer activity assay and the amount of pre-miR-124a and mature-miR-124a were quantified. B. Dicer's relative specific activity was calculated using the ratio of mature-miR-124a produced to Dicer protein expression in a lysate. The data represent three independent experiments. Error bars ± SEM, **P < 0.005.
Discussion
To date, numerous studies have demonstrated that abnormal Dicer expression exists in various cancers and correlates with advanced staging, poor prognosis, and reduced overall survival.Citation23 The data presented here demonstrates that Dicer protein levels do not correlate with mature miR production. Therefore, it will be important to determine if patient tumor samples recapitulate this discrepancy between Dicer expression and activity. Assaying Dicer activity in patient samples could potentially clarify this conundrum. If Dicer protein levels do not correlate with activity in patient tumors, then a new layer of dysregulated miR biogenesis could be added to the already complex nature of cancer. A recent report has suggested that Dicer may be a potential therapeutic target to enhance multiple sclerosis patient response to treatment.Citation33 Determining Dicer activity levels in patients with various diseases will be important to understand their disease and stratify them into different treatment groups.
Several clinical trials involving HDACi have been completed or are ongoing.Citation34-36 Positive HDACi clinical trial results have been observed mainly in non-solid tumors, although encouraging results were recently obtained in solid tumors.Citation34-36 Despite this, the cellular effects of HDACi are largely unknown. Better understanding HDACi cellular impacts could explain their success in treating certain cancers compared with others. One contribution could be their ability to regulate Dicer and miRs. Future studies designed to determine whether other clinically relevant HDACi and non-HDACi drugs affect Dicer expression and activity could allow for better selectivity in targeting cancer subtypes. Additionally, it will be important to demonstrate Dicer regulation by HDACi using in vivo models, as our studies are limited to in vitro systems. Determining Dicer and miR regulation in HDACi treated patient tumors will also be essential to confirm these studies.
An important consideration is that Dicer transcript levels do not always correlate with Dicer protein expression, suggesting potential posttranscriptional regulation of Dicer expression.Citation37-39 The strong association between Dicer and chromatin status further complicates this matter. For example, the formation of heterochromatin requires Dicer expression.Citation40,41 Additionally, siRNA mediated knockdown of Dicer induced histone acetylation and promoted heterochromatin switching to euchromatin.Citation40 Hypoxia, through the inhibition of H3K27me3 demethylases KDM6A/B, was able to epigenetically silence the DICER promoter and reduce Dicer expression in breast cancer cells.Citation42 In colon cancer cells, epigenetically silenced genes are re-expressed upon loss of Dicer expression, suggesting a role for Dicer in the epigenetic regulation of tumor suppressor genes through regulation of CpG hypermethylation.Citation43 Collectively, these and other studies show that Dicer is differentially regulated through epigenetic mechanisms and plays an important role in epigenetic regulation.
Here, we demonstrated that Panobinostat regulates Dicer protein expression posttranscriptionally, potentially through reduced proteasomal degradation of Dicer protein. However, the mechanism involved in Panobinostat reduction of Dicer activity remains unknown and warrants further investigation. We believe that Panobinostat may be inducing a posttranslational modification on Dicer protein that could affect its activity and expression levels. We performed several experiments investigating whether Panobinostat treatment resulted in Dicer protein acetylation; however, these experiments were unsuccessful due to technical limitations and will require alternative approaches. Similarly, Bortezomib also increases Dicer protein expression, while reducing Dicer activity levels. This suggests that both Bortezomib and Panobinostat could be altering ubiquitination of Dicer protein, resulting in higher expression levels, but reducing its activity. Future studies utilizing mass spectrometry to analyze potential posttranslational modifications of Dicer protein will be needed to fully dissect Dicer protein modifications.
Currently, little is known regarding the mechanisms regulating Dicer expression under basal conditions. This presents a major gap of knowledge in Dicer biology. Distinct pathways, including autophagy, apoptosis, and hypoxia can regulate Dicer expression.Citation26,42,44-46 Additionally, transcription factors such as MITF, SOX4, and NF-κB interact with the DICER promoter to regulate its expression.Citation47-49 Since Dicer levels are frequently altered in disease states, determining the mechanisms of Dicer regulation and dysregulation will be of paramount importance. Additionally, Dicer can have various binding partners that influence its activity levels, including TRBP and PACT.Citation18 It will be important to determine how HDACi treatment affects their ability to interact with Dicer, their regulation of Dicer activity, and their expression levels. Furthermore, we showed that Argonaute-2 was enhanced by Panobinostat treatment. As Argonaute-2 is a vital member of the RNA-induced silencing complex (RISC), this suggests that miR function, in addition to miR biogenesis, could be regulated by HDACi treatment. Previously, it was shown that HDACi treatment could rapidly alter miR expression levels, either increasing or decreasing certain subsets of microRNA.Citation50-53 Future studies should include dissecting how and why HDACi differentially regulate the expression of miR subsets. Investigating whether different classes of HDACi affect miR expression similarly may lead to a better understanding of HDACi treatments and the epigenetic regulation of miRs. HDACi treatments may also modulate the half-life of certain miRs, resulting in altered expression levels.
Taken together, we have demonstrated that (1) Panobinostat enhances Dicer primarily through posttranscriptional mechanisms; (2) Panobinostat potentially regulates the proteasomal degradation of Dicer; (3) Panobinostat enhances Dicer protein expression, but decreases Dicer activity; (4) Dicer protein levels do not always correlate with mature miR production/expression levels. These new data contribute to a growing understanding of Dicer expression, regulation, and function critical to elaborating its biology and its clinical potential.
Materials and methods
Cell culture and reagents
The human choriocarcinoma cell line JAR and human cervical carcinoma cell line HeLa were purchased from American Type Culture Collection (ATCC) and cultured according to ATCC's instructions. Trichostatin A (Cat #203–17,561, Wako Biochemical) treatments were 25 nM. Panobinostat (Cat #S1030, Selleck Chemicals) treatments were between 10–75 nM. Bortezomib (Cat #S1013, Selleck Chemicals) treatments were between 5–20 nM.
Western blotting
For whole protein extracts, cells were washed with Phosphate Buffered Saline (PBS) (Cat #14,190–250, Life Technologies), harvested, pelleted at 450 × g for 7 min, and washed once with PBS. The pellets were lysed on ice for 60 min using RIPA lysis buffer (Cat #R0278, Sigma-Aldrich) supplemented with protease inhibitor cocktail (Cat #P8340, Sigma-Aldrich), phosphatase inhibitor cocktail (Cat #78,420, Thermo Scientific), and 1 mM dithiothreitol (Cat #D9779, Sigma-Aldrich). The extract was then centrifuged at 10,000 × g for 10 min and supernatants collected. Protein concentrations were determined with the Micro BCA Assay Kit (Cat #23,235, Pierce). Protein lysates (20 μg) were heated for 5 min at 95° C in SDS sample buffer plus 0.13 M dithiothreitol (Cat #D9779, Sigma-Aldrich), separated on 7% [for Dicer], 10% [for Argonaute-2], or 15% [for β-actin, acetyl-Histone H3, Caspase-3, GAPDH] SDS-PAGE gels and transferred to Immun-blot LF PVDF membranes (Cat # 162–0177, BioRad). Membranes were blocked using 5% non-fat dry milk in TTBS (Tween 20 Tris-Buffered Saline) followed by addition of primary antibodies. Antibodies used were anti-Dicer (Cat #3,363, Cell Signaling Technology), anti-Caspase-3 (Cat #9,662, Cell Signaling Technology), anti-Argonaute-2 (Cat #2,897, Cell Signaling Technology), anti-GAPDH (Cat #2,118, Cell Signaling Technology), anti-β-actin (Cat #8,227, Abcam), anti-acetyl-Histone H3 (Cat #06–599, Millipore), and goat anti-rabbit IgG-horseradish peroxidase (Cat #W4011, Promega). Blots were developed with a West Pico Chemiluminescent Kit (Cat #34,080, Pierce) and imaged with a Chemidoc MP imager (BioRad).
In vitro Dicer activity assay
We designed and optimized an in vitro assay to test Dicer activity in cellular protein lysates. First, we generated a pre-miR-124a template using PCR. The primers used were 5′-CAGCCCCATTCTTGGC-3′ [forward] and 5′-TAATACGACTCACTATAGGGAGGCCTCTCT-3′ [reverse]. Next, we used the MEGAscript T7 transcription kit (Cat #AM1334, Ambion) to synthesize a α-32P-UTP (Cat #NEG007C001MC, PerkinElmer) radiolabeled pre-miR-124a. We then performed reactions using the radiolabeled pre-miR-124a and recombinant Human Dicer (Cat #T520001, Genlantis) to demonstrate specific cleavage of the pre-miR by Dicer. We also confirmed our assay's specificity using Dicer loss of function cell lines. 20 µg of protein lysates from JAR cells +/− Panobinostat treatment or JAR cells +/− Bortezomib treatment were then incubated for 16 h with the radiolabeled pre-miR-124a. Once the reactions were complete, the samples were run on a denaturing Urea PAGE gel and specific mature-miR-124a cleavage products were quantified by phosphorimaging (Molecular Dynamics Storm).
Real-time quantitative RT-PCR
Total RNA was isolated using the mirVana kit (Cat #AM1560, Ambion) and 2 µg was used for reverse transcription with Superscript II (Cat #18,064–014, Invitrogen) plus oligo dT. Real-time PCR was performed as previously described on an ABI7900HT (Applied Biosystems).Citation11 Amplification of cDNA samples was performed with either Taqman PCR Master Mix (Cat #RT-QP2×02+10, Eurogentec) or SYBR Green Master Mix (Cat #4,913,850,001, Roche) according to the manufacturer's protocol. Primers used included human β-actin 5′-GGAGCAATGATCTTGATCTT-3′ [forward] and 5′- CCTTCCTGGGCATGGAGTCCT-3′ [reverse], human Dicer 5′-GTACGACTACCACAAGTACTTC-3′ [forward] and 5′-ATAGTACACCTGCCAGACTGT-3′ [reverse]. Relative expression levels were determined by the ΔΔCt method as previously described.Citation11
Statistical analysis
Statistical analysis was performed with GraphPad version 6.05 software. Results are shown as mean ± standard error of the mean (SEM). Comparison of results from Western blots, qPCR, and Dicer activity assays were performed using the two-tailed unpaired t-test. Analysis of the combination treatments () was validated with ANOVA followed by Fisher's LSD test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1267886_s02.pptx
Download MS Power Point (296.1 KB)Funding
This work was supported by the NIH NCI grant RO1-CA124971 and by M&T Bank. The study also used Roswell Park Cancer Institute's shared resources funded by NCI grant P30 CA016056. The authors thank our laboratory members for their support and helpful discussions.
Related Research Data
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 2013; 13:497-510; PMID:23760024; http://dx.doi.org/10.1038/nrc3486
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693-705; PMID:17320507; http://dx.doi.org/10.1016/j.cell.2007.02.005
- Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 2014; 13:673-91; PMID:25131830; http://dx.doi.org/10.1038/nrd4360
- Nalabothula N, Carrier F. Cancer cells' epigenetic composition and predisposition to histone deacetylase inhibitor sensitization. Epigenomics 2011; 3:145-55; PMID:21743813; http://dx.doi.org/10.2217/epi.11.12
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002; 1:287-99; PMID:12120280; http://dx.doi.org/10.1038/nrd772
- Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother 2008; 57:647-54; PMID:18046553; http://dx.doi.org/10.1007/s00262-007-0402-4
- Khan AN, Magner WJ, Tomasi TB. An epigenetic vaccine model active in the prevention and treatment of melanoma. J Transl Med 2007; 5:64-75; PMID:18070359; http://dx.doi.org/10.1186/1479-5876-5-64
- Magner WJ, Tomasi TB. Apoptotic and necrotic cells induced by different agents vary in their expression of MHC and costimulatory genes. Mol Immunol 2005; 42:1033-42; PMID:15829293; http://dx.doi.org/10.1016/j.molimm.2004.09.030
- Khan AN, Magner WJ, Tomasi TB. An epigenetically altered tumor cell vaccine. Cancer Immunol Immunother 2004; 53:748-54; PMID:14997346; http://dx.doi.org/10.1007/s00262-004-0513-0
- West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res 2013; 73:7265-76; PMID:24158093; http://dx.doi.org/10.1158/0008-5472.CAN-13-0890
- Rosborough BR, Castellaneta A, Natarajan S, Thomson AW, Turnquist HR. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol 2012; 91:701-9; PMID:22028329; http://dx.doi.org/10.1189/jlb.0311119
- Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 2007; 13:1299-307; PMID:17922010; http://dx.doi.org/10.1038/nm1652
- Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011; 117:1205-17; PMID:20956800; http://dx.doi.org/10.1182/blood-2010-05-284711
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014; 124:30-9; PMID:24382387; http://dx.doi.org/10.1172/JCI69738
- Zhang Z, Convertini P, Shen M, Xu X, Lemoine F, de la Grange P, Andres DA, Stamm S. Valproic acid causes proteasomal degradation of DICER and influences miRNA expression. PLoS One 2013; 8:e82895; PMID:24358235; http://dx.doi.org/10.1371/journal.pone.0082895
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15:509-24; PMID:25027649; http://dx.doi.org/10.1038/nrm3838
- Kurzynska-Kokorniak A, Koralewska N, Pokornowska M, Urbanowicz A, Tworak A, Mickiewicz A, Figlerowicz M. The many faces of Dicer: the complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res 2015; 43:4365-80; PMID:25883138; http://dx.doi.org/10.1093/nar/gkv328
- Pratt AJ, MacRae IJ. The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem 2009; 284:17897-901; PMID:19342379; http://dx.doi.org/10.1074/jbc.R900012200
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet 2003; 35:215-7; PMID:14528307; http://dx.doi.org/10.1038/ng1253
- Devasthanam AS, Tomasi TB. Dicer in immune cell development and function. Immunol Invest 2014; 43:182-95; PMID:24303839; http://dx.doi.org/10.3109/08820139.2013.863557
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 2007; 39:673-7; PMID:17401365; http://dx.doi.org/10.1038/ng2003
- Pellegrino L, Jacob J, Roca-Alonso L, Krell J, Castellano L, Frampton AE. Altered expression of the miRNA processing endoribonuclease Dicer has prognostic significance in human cancers. Expert Rev Anticancer Ther 2013; 13:21-7; PMID:23259424; http://dx.doi.org/10.1586/era.12.150
- Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the Treatment of Multiple Myeloma. Clin Cancer Res 2015; 21:4767-73; PMID:26362997; http://dx.doi.org/10.1158/1078-0432.CCR-15-0530
- Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 2009; 280:233-41; PMID:19344997; http://dx.doi.org/10.1016/j.canlet.2009.02.019
- Lu J, Yang C, Chen M, Ye DY, Lonser RR, Brady RO, Zhuang Z. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc Natl Acad Sci U S A 2011; 108:21200-5; PMID:22160715; http://dx.doi.org/10.1073/pnas.1119181109
- Kim SH, Kang HJ, Na H, Lee MO. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res 2010; 12:R22; PMID:20388208; http://dx.doi.org/10.1186/bcr2562
- Scognamiglio A, Nebbioso A, Manzo F, Valente S, Mai A, Altucci L. HDAC-class II specific inhibition involves HDAC proteasome-dependent degradation mediated by RANBP2. Biochim Biophys Acta 2008; 1783:2030-8; PMID:18691615; http://dx.doi.org/10.1016/j.bbamcr.2008.07.007
- Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol 2006; 26:2019-28; PMID:16507982; http://dx.doi.org/10.1128/MCB.26.6.2019-2028.2006
- Alao JP, Lam EW, Ali S, Buluwela L, Bordogna W, Lockey P, Varshochi R, Stavropoulou AV, Coombes RC, Vigushin DM. Histone deacetylase inhibitor trichostatin A represses estrogen receptor alpha-dependent transcription and promotes proteasomal degradation of cyclin D1 in human breast carcinoma cell lines. Clin Cancer Res 2004; 10:8094-104; PMID:15585645; http://dx.doi.org/10.1158/1078-0432.CCR-04-1023
- Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 2003; 22:3411-20; PMID:12840003; http://dx.doi.org/10.1093/emboj/cdg315
- Magner WJ, Weinstock-Guttman B, Rho M, Hojnacki D, Ghazi R, Ramanathan M, Tomasi TB. Dicer and microRNA expression in multiple sclerosis and response to interferon therapy. J Neuroimmunol 2016; 292:68-78; PMID:26943961; http://dx.doi.org/10.1016/j.jneuroim.2016.01.009
- Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015; 20:3898-941; PMID:25738536; http://dx.doi.org/10.3390/molecules20033898
- Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anticancer Drugs 2014; 25:140-9; PMID:24185382; http://dx.doi.org/10.1097/CAD.0000000000000040
- Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 2009; 15:3958-69; PMID:19509172; http://dx.doi.org/10.1158/1078-0432.CCR-08-2785
- Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol 2009; 46:1222-8; PMID:19118902; http://dx.doi.org/10.1016/j.molimm.2008.11.012
- Singh S, Bevan SC, Patil K, Newton DC, Marsden PA. Extensive variation in the 5′-UTR of Dicer mRNAs influences translational efficiency. Biochem Biophys Res Commun 2005; 335:643-50; PMID:16095561; http://dx.doi.org/10.1016/j.bbrc.2005.07.138
- Irvin-Wilson CV, Chaudhuri G. Alternative initiation and splicing in dicer gene expression in human breast cells. Breast Cancer Res 2005; 7:R563-9; PMID:15987463; http://dx.doi.org/10.1186/bcr1043
- Giles KE, Ghirlando R, Felsenfeld G. Maintenance of a constitutive heterochromatin domain in vertebrates by a Dicer-dependent mechanism. Nat Cell Biol 2010; 12:94-9; sup pp 1-6
- Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, Nakayama T, Oshimura M. Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat Cell Biol 2004; 6:784-91; PMID:15247924; http://dx.doi.org/10.1038/ncb1155
- van den Beucken T, Koch E, Chu K, Rupaimoole R, Prickaerts P, Adriaens M, Voncken JW, Harris AL, Buffa FM, Haider S, et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat Commun 2014; 5:5203; PMID:25351418; http://dx.doi.org/10.1038/ncomms6203
- Ting AH, Suzuki H, Cope L, Schuebel KE, Lee BH, Toyota M, Imai K, Shinomura Y, Tokino T, Baylin SB. A requirement for DICER to maintain full promoter CpG island hypermethylation in human cancer cells. Cancer Res 2008; 68:2570-5; PMID:18413723; http://dx.doi.org/10.1158/0008-5472.CAN-07-6405
- Ho JJ, Metcalf JL, Yan MS, Turgeon PJ, Wang JJ, Chalsev M, Petruzziello-Pellegrini TN, Tsui AK, He JZ, Dhamko H, et al. Functional importance of Dicer protein in the adaptive cellular response to hypoxia. J Biol Chem 2012; 287:29003-20; PMID:22745131; http://dx.doi.org/10.1074/jbc.M112.373365
- Gibbings D, Mostowy S, Jay F, Schwab Y, Cossart P, Voinnet O. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat Cell Biol 2012; 14:1314-21; PMID:23143396; http://dx.doi.org/10.1038/ncb2611
- Nakagawa A, Shi Y, Kage-Nakadai E, Mitani S, Xue D. Caspase-dependent conversion of Dicer ribonuclease into a death-promoting deoxyribonuclease. Science 2010; 328:327-34; PMID:20223951; http://dx.doi.org/10.1126/science.1182374
- Matskevich AA, Moelling K. Stimuli-dependent cleavage of Dicer during apoptosis. Biochem J 2008; 412:527-34; PMID:18289125; http://dx.doi.org/10.1042/BJ20071461
- Guan Y, Yao H, Wang J, Sun K, Cao L, Wang Y. NF-kappaB-DICER-miRs Axis Regulates TNF-alpha Expression in Responses to Endotoxin Stress. Int J Biol Sci 2015; 11:1257-68; PMID:26435691; http://dx.doi.org/10.7150/ijbs.12611
- Jafarnejad SM, Ardekani GS, Ghaffari M, Martinka M, Li G. Sox4-mediated Dicer expression is critical for suppression of melanoma cell invasion. Oncogene 2013; 32:2131-9; PMID:22689055; http://dx.doi.org/10.1038/onc.2012.239
- Levy C, Khaled M, Robinson KC, Veguilla RA, Chen PH, Yokoyama S, Makino E, Lu J, Larue L, Beermann F, et al. Lineage-specific transcriptional regulation of DICER by MITF in melanocytes. Cell 2010; 141:994-1005; PMID:20550935; http://dx.doi.org/10.1016/j.cell.2010.05.004
- Hsieh TH, Hsu CY, Tsai CF, Long CY, Wu CH, Wu DC, Lee JN, Chang WC, Tsai EM. HDAC inhibitors target HDAC5, upregulate microRNA-125a-5p, and induce apoptosis in breast cancer cells. Mol Ther 2015; 23:656-66; PMID:25531695; http://dx.doi.org/10.1038/mt.2014.247
- Cho JH, Dimri M, Dimri GP. MicroRNA-31 is a transcriptional target of histone deacetylase inhibitors and a regulator of cellular senescence. J Biol Chem 2015; 290:10555-67; PMID:25737447; http://dx.doi.org/10.1074/jbc.M114.624361
- Wang JH, Shih KS, Wu YW, Wang AW, Yang CR. Histone deacetylase inhibitors increase microRNA-146a expression and enhance negative regulation of interleukin-1beta signaling in osteoarthritis fibroblast-like synoviocytes. Osteoarthritis Cartilage 2013; 21:1987-96; PMID:24107356; http://dx.doi.org/10.1016/j.joca.2013.09.008
- Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 2006; 66:1277-81; PMID:16452179; http://dx.doi.org/10.1158/0008-5472.CAN-05-3632
- Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol 2000; 165:7017-24; PMID:11120829; http://dx.doi.org/10.4049/jimmunol.165.12.7017