
ABSTRACT
Epigenetic alterations underlie various human disorders, including cancer, and this has resulted in the development of drugs targeting epigenetic alterations. Although DNA demethylating agents are one of the major epigenetic drugs, only two compounds—5-azacytidine (5-aza-CR, azacitidine) and 5-aza-2'-deoxycytidine (5-aza-dC, decitabine)—have obtained clinical approval. Here, we aimed to establish a detection system for DNA demethylating agents suitable for a high-throughput screening (HTS) in mammalian cells. We inserted luciferase and EGFP reporter genes under the UCHL1 promoter, which is methylation-silenced in human colon cancers and can be readily demethylated to drive strong expression. Methylated UCHL1 promoter was introduced into HCT116 colon cancer cells, and transfectants with methylated exogenous UCHL1 promoter were obtained. By screening subclones from each of the epigenetically heterogeneous transfectant clones, we finally obtained three optimal subclones that expressed luciferase and EGFP after 5-aza-dC treatment with high signal-to-noise ratios. Nucleosomes with H3K9me2 were present around the exogenous UCHL1 promoter in all three subclones. Using one of the subclones (HML58-3), HTS was conducted using 19,840 small molecules. Two hit compounds were obtained, and these turned out to be 5-aza-dC and 5-aza-CR. The assay system constructed here demonstrates a robust response to DNA demethylating agents, along with high specificity, and will be useful for screening and biological assays in epigenetics.
Introduction
Epigenetic alterations, including aberrant DNA methylation and histone modifications, are deeply involved in cancer development and progression [Citation1–3]. In addition, they are now recognized to be involved in other acquired disorders, such as neurodegenerative diseases and metabolic diseases [Citation4–6]. Therefore, it has become increasingly important to discover novel drugs targeting altered epigenetic statuses for treatment of cancer and other acquired disorders [Citation7–9]. DNA demethylating agents and histone deacetylases (HDAC) inhibitors are already used in practice, and novel classes of epigenetic drugs that target other epigenetic regulators, such as histone methyltransferases, histone demethylases, acetylated histone readers, and isocitrate dehydrogenase 1 and 2 (IDH1/IDH2) are being developed [Citation10,Citation11]. Currently, two DNA demethylating agents—5-azacytidine (5-aza-CR, azacitidine) and 5-aza-2'-deoxycytidine (5-aza-dC, decitabine)—have been approved for clinical use, and a third one, SGI-110, is undergoing clinical trials [Citation12–15]. All three agents induce DNA demethylation by getting incorporated into DNA and depleting DNA methyltransferase [Citation16,Citation17], and DNA demethylating agents with a distinct mode of action is awaited.
The mechanism of DNA methylation-mediated gene silencing in mammalian cells involves multiple steps: maintenance of DNA methylation involves DNMT1, Np95, and other molecules, whereas reading of the methylated status involves MeCP2, MBDs, and other molecules [Citation18–21]. We previously showed that a detection system for DNA demethylating agents could be constructed in mammalian cells using an endogenous promoter as a marker, and diverse molecules could be targeted using an endogenous promoter, not viral promoter [Citation22]. However, the signal-to-noise (S/N) ratio of the detection system was not sufficiently high for its use in high-throughput screening (HTS). In addition, Cui et al. recently reported the use of the SFRP1-GFP recombinant reporter system using the endogenous promoter of the tumor-suppressor gene, SFRP1 [Citation23]. Similarly, Turker's group reported an attractive positive-selection assay system, using the Tet-Off system and the promoter of BRCA1, a well-known tumor-suppressor gene [Citation24]. This system took advantage of i) loss of expression to promote induction of DNA methylation and ii) positive selection to detect rare events of DNA methylation. However, to date, neither of them has been reported to be suitable for use in HTS.
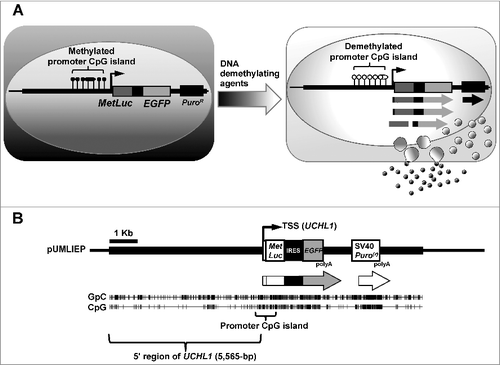
Here, we aimed to construct a highly sensitive detection system for DNA demethylating agents that would be suitable for HTS (A). To obtain a high S/N ratio in the system, we first selected an endogenous human promoter CpG island that was methylation-silenced in colon cancer cells [Citation25] but was readily demethylated by a low dose of 5-aza-dC to drive strong expression. Second, we adopted both luciferase and EGFP reporter genes for the convenience of screening. Third and most importantly, based upon the heterogeneity among a genetically homogeneous transfectant clone, we obtained optimal subclones that expressed the marker genes with a high S/N ratio after 5-aza-dC treatment.
Figure 1. Strategy to construct the detection system for DNA demethylating agents.
Materials and methods
Cell culture and treatment with 5-aza-dC
Human colon cancer cell line HCT116 was obtained from the American Type Culture Collection (Manassas, VA) and cultured in McCoy's 5A (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 1.5 mM L-glutamine (Invitrogen), penicillin/streptomycin (Invitrogen) at 37˚C in a humidified atmosphere with 5% CO2. For treatment with 5-aza-dC, cells were seeded at a density of 3 × 105 cells/10 cm dish or 5 × 104 cells/well in 6-well plate on day 0. On days 1 and 3, the medium was replaced with a medium containing a specific concentration of 5-aza-dC (Sigma-Aldrich), which was freshly dissolved in phosphate-buffered saline (PBS) and filtered through a 0.2-micron filter. On day 4, the cells were observed under a fluorescence microscope, the supernatants were collected to measure luminescence, and the cells were harvested and kept frozen at -80˚C until the extraction of genomic DNA.
Vector construction and transfection
The KpnI genomic fragment including UCHL1 (7,088-bp) and its promoter region (chr4:41,252,812 – chr4:41,259,900 in the GRCh37/hg19 assembly) was excised from a human bacterial artificial chromosome (BAC) clone, CIT987SK-A-635H12, and was cloned into pBluescript II-SK(+) (Stratagene, La Jolla, CA) to construct the pBluescript-UCHL. Separately, the Metridia longa secreted luciferase (MetLuc) cDNA from pMetLuc-Reporter (Clontech, Mountain View, CA) was cloned into the multiple cloning site of pIRES2-EGFP (Clontech). MetLuc-IRES-EGFP sequence was then cloned into pBluescript into which a puromycin resistance cassette (Puro(r)) with SV40 early promoter had been cloned in advance to construct pMetLuc-IRES-EGFP-Puro(r). Finally, the UCHL1 genomic fragment (5,565-bp) including its transcription start site (TSS) was excised from pBluescript-UCHL using the AvrII/Klenow site, and inserted immediately upstream of the translation start codon (ATG) of MetLuc into the EcoRV site of pMetLuc-IRES-EGFP-Puro(r) to complete the pUMLIEP vector (B). The complete sequence data have been submitted to the DDBJ databases under accession number LC175306.
The pUMLIEP vector was linearized by restriction digestion with XhoI (New England Biolabs, Beverly, MA), and then briefly methylated by SssI-methylase (New England Biolabs) for 60 min, which was half the time used in the usual protocol [Citation26], to produce molecules with varying degrees of DNA methylation, including slight methylation of the promoter of puromycin resistance cassette. The briefly methylated vector was transfected into HCT116 cells using Lipofectamine™ LTX (Invitrogen) and Opti-MEM® I reduced serum media (Invitrogen), and stably introduced clones were selected with puromycin (0.6 µg/mL, Sigma-Aldrich).
Fluorescence detection and luminescence measurement
Fluorescence of living cells was analyzed using an Olympus IX71 fluorescence microscopy system (Olympus, Tokyo, Japan) and imaging software Studio Lite (Pixera, Osaka, Japan). Luminescence was measured using 50 µL of the cell culture medium by the Ready-To-Glow™ secreted luciferase reporter system (Clontech, Mountain View, CA) and a multimode plate reader ARVO™MX (PerkinElmer Japan, Kanagawa, Japan).
Methylation-specific PCR (MSP) and bisulfite sequencing
One microgram of genomic DNA was digested with EcoRV (New England Biolabs). Sodium bisulfite modification was performed using 1 µg of EcoRV-digested genomic DNA as previously described [Citation26], and then the modified DNA was suspended in 40 µL of Tris-EDTA (TE) buffer. Conventional MSP was performed using 1 µL of the solution and primers specific to the sequence produced from unmethylated (UM) DNA for 38 cycles, and methylated (M) DNA for 30cycles (Supplementary Table 1). Since the UM reverse primer and the M forward primer were located in the MetLuc sequence, we were able to distinguish between exogenous and endogenous UCHL1 promoters.
For bisulfite sequencing, sodium bisulfite-modified DNA was amplified with primers common to the sequences obtained from methylated and unmethylated DNA (Supplementary Table 1). Similarly to MSP, the reverse primer was located in the MetLuc sequence. The PCR product was cloned into a pCR4-TOPO vector (Invitrogen) and approximately 15 clones were sequenced using an ABI PRISM 310 sequencer (PE Biosystems, Foster City, CA).
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR)
ChIP was performed as described previously [Citation27]. Briefly, 30 µg of chromatin extracted from cross-linked cells was immunoprecipitated using 2 µg of antibody against histone H3 (Abcam, Cambridge, MA, ab1791), acetylated histone H3 (H3Ac; Millipore, Billerica, MA, 06-599), dimethylated histone H3 lysine 9 (H3K9me2; MAB Institute, Inc., Hokkaido, Japan, 308-32361) or RNA polymerase II (Pol II; Abcam, ab5095). Immunoprecipitated chromatin was treated with RNaseA and proteinase K, and DNA was recovered by phenol/chloroform extraction and isopropanol precipitation. The precipitated DNA was dissolved in 50 µL of TE buffer.
One microliter of immunoprecipitated and input DNA was used for quantitative PCR (qPCR). qPCR was performed using an iCycler iQ detection system (Bio-Rad Laboratories, Hercules, CA) with SYBR Green I nucleic acid gel stain (Lonza Rockland, Rockland, ME). We selected the three genes, PCDH7, D4S234E, and LIN28B, as control genes methylation-silenced in HCT116 [Citation25] (http://www.ncbi.nlm.nih.gov/geo/). The primer sites, their sequences, and the PCR conditions are shown in Supplementary Figure 1 and Supplementary Table 1. The numbers of DNA molecules in input and immunoprecipitated samples were obtained by comparing their amplification curves with those of standard samples with known numbers of DNA molecules. Each ChIP-qPCR was carried out 3 times.
HTS
The RIKEN Natural Products Depository (NPDepo) chemical library, which consisted of 19,840 compounds, including synthetic compounds, synthetic peptides, natural compounds and their derivatives [Citation28], was screened. Cells (5 × 103) in 384 well plates were exposed to 5 μg/mL (0.5% from the stock solution) of compounds dissolved in DMSO for 4 days, and this condition was confirmed to be sufficient for induction of luminescence by 5-aza-dC, a positive control. The luminescence generated by adding coelentarazine (Biotium, Fremont, CA) into the culture media was measured using a Synergy H4 hybrid reader (BioTek, Winooski, VT). Compounds that showed an actual value of luciferase activity of ≥ 3,000 (arbitrary units) were considered to be positive. All the positive compounds in the screening were confirmed for their activity in the second analysis. Cell viability was measured by WST-8 assays (Cell Count Reagent SF; Nacalai Tesque, Kyoto, Japan).
Statistical analysis
Pairwise comparison of mean luminescence was conducted using an unpaired Student t-test, in which a prior F-test was performed to determine whether the assumption of equal standard deviations could be used. We considered P < 0.05 to be statistically significant. All the calculations were performed using Microsoft Excel software (Microsoft Corp, Seattle, WA).
Results
Construction of cells highly responsive to DNA demethylating agents
We had previously identified UCHL1 as a promoter in a CpG island that is methylation-silenced in colon cancers and readily demethylated to drive strong expression of its downstream gene after 5-aza-dC treatment [Citation25]. We inserted two kinds of reporter genes, the EGFP gene and the MetLuc gene, downstream to the UCHL1 promoter to construct pUMLIEP (A and B).
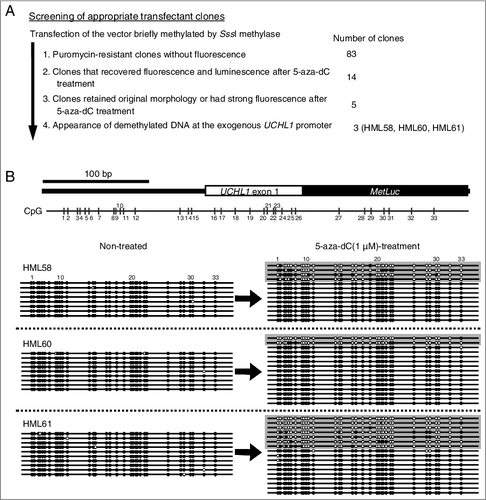
To obtain transfectants with methylated exogenous UCHL1 promoter and unmethylated SV40 early promoter of puromycin resistance cassette, the pUMLIEP vector was briefly methylated by SssI-methylase before transfection into HCT116 cells. After transfection, 83 puromycin-resistant clones without fluorescence, indicative of the presence of methylated exogenous UCHL1 promoter, were obtained (A). Among them, 14 transfectant clones showed induction of fluorescence and luminescence after 5-aza-dC treatment (Supplementary Figure 2). Among these 14 clones, 5 clones retained their original morphology or had strong fluorescence after 5-aza-dC treatment. Among the 5 clones, 3 clones (HML58, HML60 and HML61) showed complete methylation of the exogenous UCHL1 promoter and its demethylation after 5-aza-dC treatment (B).
Figure 2. Screening of appropriate transfectant clones.
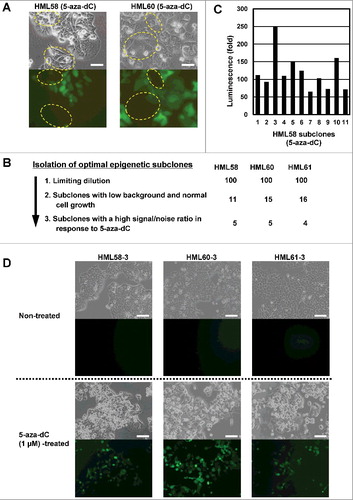
Isolation of three optimal epigenetic subclones
Even among a transfectant clone, the presence of epigenetic heterogeneity was indicated by the variation in fluorescence after 5-aza-dC treatment (A). Therefore, we further screened optimal epigenetic subclones from the transfectant clones. For each of the 3 transfectant clones (HML58, HML60, and HML61), from 100 subclones, we selected 11-16 subclones that had low fluorescence background and showed cell growth similar to that of parental HCT116 cells. Additionally, we excluded clones with low plating efficiency (B). After treatment of 11 subclones from HML58 with 5-aza-dC, some subclones showed strong induction of luminescence with high S/N ratios while others showed weak induction (C). Three of the 14 subclones (HML58-3, HML60-3, and HML61-3) showed homogeneous induction of strong fluorescence after treatment with 5-aza-dC (D).
Figure 3. Isolation of optimal epigenetic subclones from a transfectant clone.
Confirmation of high sensitivity of the three subclones
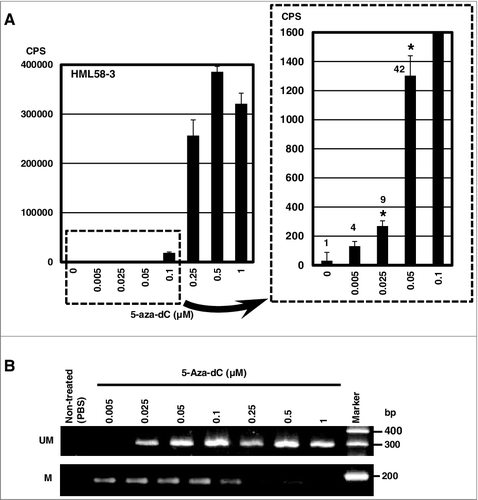
The high sensitivity of the 3 subclones was confirmed using multiple doses of 5-aza-dC. In the HML58-3 subclone, we detected significant increase in the luminescence in the media at 5-aza-dC doses as low as 0.025 µM (A). We were able to detect fluorescence at 5-aza-dC doses of 0.25 µM or higher (Supplementary Figure 3). The detection sensitivity was 10-fold higher using luminescence than fluorescence. We also obtained high sensitivity to 5-aza-dC using luminescence with the other 2 subclones, HML60-3 and HML61-3 (Supplementary Figure 4), but HML58-3 showed the highest sensitivity among 3 subclones. Furthermore, demethylation of the exogenous UCHL1 promoter was clearly observed at 0.025 µM of 5-aza-dC (B). These results demonstrated that HML58-3, HML60-3 and HML61-3 responded to 5-aza-dC with a high S/N ratio.
Figure 4. High sensitivity of the subclone HML58-3 to 5-aza-dC determined by luminescence assay.
Presence of nucleosomes in the marker gene
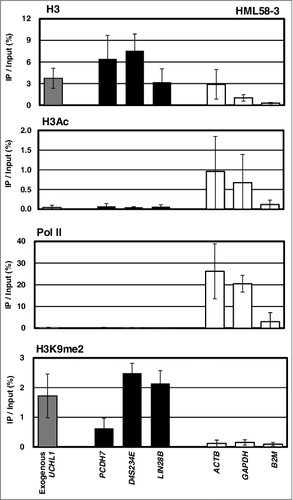
It is not warranted that an exogenously introduced methylation-silenced promoter had nucleosomes as endogenous methylation-silenced genes. Therefore, using ChIP-qPCR, we analyzed whether nucleosomes existed on the exogenous UCHL1 gene, UCHL1-MetLuc, and whether the inactive histone modification H3K9me2 was present. The exogenous UCHL1 gene was specifically amplified using the reverse primer located in the MetLuc sequence. In all the 3 subclones, HML58-3, HML60-3, and HML61-3, histone H3 and H3K9me2 modification were detected in the exogenous UCHL1 gene, as well as in the 3 unexpressed endogenous genes (PCDH7, D4S234E, and LIN28B) (). These results confirmed the presence of a nucleosome with inactive histone modification, H3K9me2, in the exogenous UCHL1 promoter.
Figure 5. Presence of nucleosome with inactive histone modification on the exogenous UCHL1 promoter.
Application of HML58-3 to HTS
Finally, we applied the novel detection system to a HTS of a chemical library with 19,840 compounds. In the first screening, 8 hits were obtained for compounds that showed luminescence of ≥ 3,000 (arbitrary units) (A, Supplementary Table 2 and Supplementary Figure 5). All the compounds identified in the first screening were re-analyzed for their activity in the second HTS analysis. As a result, induction of luciferase activity was observed for the four compounds (Supplementary Table 2).
Figure 6. Application of the subclone HML58-3 to HTS.
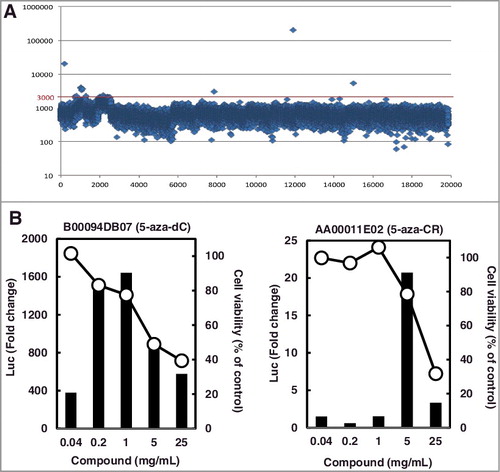
Next, we further confirmed the induction of luciferase activity by these four compounds on a larger scale (5 × 104 cells/well in a 6-well plate and 1-2 × 103 cells/well in a 96-well plate) under multiple conditions, as follows: i) exposure of cells to a compound on days 1 and 3, and measurement of luciferase activity on day 5, similar to the screening of optimal subclones; and ii) exposure on day 0 and measurement on day 2, 3, or 4, similar to the HTS. Although luciferase activity was clearly confirmed for the top two of the four compounds under all conditions, no induction was observed for the remaining two compounds. Therefore, the top two compounds were considered as true hits, and the compounds with the highest and second-highest luciferase activity were identified as 5-aza-dC and 5-aza-CR, respectively (B).
Discussion
In this study, we established a new detection system for DNA demethylating agents that is suitable for HTS. Its high sensitivity was confirmed by the detection of demethylation using a low dose of 5-aza-dC along with a very high S/N ratio of luminescence. These were achieved by the selection of an endogenous promoter with a sensitive and robust response to a demethylating agent, the use of the MetLuc gene as a reporter, and rigorous screening to obtain subclones with high S/N ratios. In fact, an HTS using the novel system identified two previously known DNA demethylating agents, 5-aza-dC and 5-aza-CR, indicating that our system functions as anticipated. In addition, the cell-based screening system is expected to be capable of detecting compounds with a broad range of targets. The presence of nucleosomes with the H3K9me2 modification around the promoter of the marker gene supports this possibility.
A cell-based assay system like ours has considerable potential to detect various compounds with different modes of action, for instance DNMT1 synthesis, degradation, inactivation, stabilization, and recruitment [Citation18–21]. In fact, DNA demethylation can be achieved via multiple modes of action. The majority of known DNA demethylating agents are derivatives of 5-aza-dC, including 5-aza-CR, 5,6-dihydroazacytidine, 5-fluoro-2'-deoxycytidine, zebularine, and SGI-110 [Citation12]. These compounds are converted to the activated triphosphate 5-aza-dCTP, get subsequently incorporated into DNA, form a covalent complex with DNMT1, and then deplete it [Citation16,Citation17]. Direct inhibitors of DNMT1 have also been developed, including small molecules, RG108 [Citation29] and SGI-1027 [Citation30,Citation31], and an antisense oligodeoxynucleotide to DNMT1, MG98 [Citation32]. An antibiotic, Nanaomycin A, was shown to repress DNMT3B transcription and thus induce DNA demethylation [Citation33]. Further, chemicals that alter calcium signaling were shown to affect readers of DNA methylation and induce expression of methylated genes [Citation34].
In this study, we were unable to identify novel compounds with a unique mode of action. In general, the output of HTS is heavily influenced by the number and type of compounds in the chemical library [Citation35], and the outcomes of this study could be explained by the limitations of the library used. Alternatively, the assay system used here may have been optimized for 5-aza-dC, which was used to select optimal subclones. Nevertheless, the assay system was highly efficient and showed high specificity. Thus, the use of additional libraries will increase the chance of identifying novel compounds with different modes of action.
Our assay system can also be applied to the research field of toxicology, especially to the study of chemical carcinogens. There are many environmental non-genotoxic carcinogens [Citation36], and the mutagenic potential of a chemical is not necessarily correlated to its carcinogenic potential [Citation37,Citation38]. Tissue damage and induction of cell proliferation are important factors that explain such discrepancy; however, the presence of “epimutagens”, which induce epigenetic abnormality, has been indicated [Citation39,Citation40]. Nevertheless, our current knowledge on such epimutagens is limited, mainly owing to the lack of an efficient system to identify such chemicals. Our assay system can facilitate detection of novel epimutagens that cause human health hazards.
In conclusion, a novel detection system for DNA demethylating agents that is suitable for HTS was constructed and its high specificity in HTS was demonstrated.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KEPI_A_1267887_supplementary_data.zip
Download Zip (875.5 KB)Acknowledgement
The authors are grateful to Dr. Hiroyuki Osada (RIKEN Center for Sustainable Resource Science) for providing the NPDepo chemical library.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer. 2005;5:223–231. doi:10.1038/nrc1571.
- Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. doi:10.1016/j.molcel.2014.05.015.
- Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284–299. PMID:26972587.
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi:10.1038/nbt.1685.
- Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi:10.1038/nbt.1678.
- Ronn T, Ling C DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics. 2015;7:451–460. doi:10.2217/epi.15.7.
- Dhanak D, Jackson P. Development and classes of epigenetic drugs for cancer. Biochem Biophys Res Commun. 2014;455:58–69. doi:10.1016/j.bbrc.2014.07.006.
- Azad N, Zahnow CA, Rudin CM, et al. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol. 2013;10:256–266. doi:10.1038/nrclinonc.2013.42.
- Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–1204. doi:10.1038/nm.2828.
- Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127:42–52. doi:10.1182/blood-2015-07-604512.
- Shinjo K, Kondo Y Targeting cancer epigenetics: Linking basic biology to clinical medicine. Adv Drug Deliv Rev. 2015;95:56–64. doi:10.1016/j.addr.2015.10.006.
- Yoo CB, Jeong S, Egger G, et al. Delivery of 5-aza-2'-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400–6408. doi:10.1158/0008-5472.CAN-07-0251.
- Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenetics. 2015;7:127. doi:10.1186/s13148-015-0157-2.
- Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia. 2015;29:760–769. doi:10.1038/leu.2014.244.
- Issa JP, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16:1099–1110. doi:10.1016/S1470-2045(15)00038-8.
- Foulks JM, Parnell KM, Nix RN, et al. Epigenetic drug discovery: targeting DNA methyltransferases. J Biomol Screen. 2012;17:2–17. doi:10.1177/1087057111421212.
- Ghoshal K, Datta J, Majumder S, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–4741. doi:10.1128/MCB.25.11.4727-4741.2005.
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi:10.1038/nature14192.
- Nishiyama A, Yamaguchi L, Nakanishi M. Regulation of maintenance DNA methylation via histone ubiquitylation. J Biochem. 2016;159:9–15. doi:10.1093/jb/mvv113.
- Lin RK, Wang YC. Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 2014;4:46. doi:10.1186/2045-3701-4-46.
- Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet. 2014;15:69–81. doi:10.1038/nrg3623.
- Okochi-Takada E, Ichimura S, Kaneda A, et al. Establishment of a detection system for demethylating agents using an endogenous promoter CpG island. Mutat Res. 2004;568:187–194. doi:10.1016/j.mrfmmm.2004.08.011.
- Cui Y, Hausheer F, Beaty R, et al. A recombinant reporter system for monitoring reactivation of an endogenously DNA hypermethylated gene. Cancer Res. 2014;74:3834–3843. doi:10.1158/0008-5472.CAN-13-2287.
- Lu Y, Chu A, Turker MS, et al. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol Cell Biol. 2011;31:3339–3350. doi:10.1128/MCB.01121-10.
- Okochi-Takada E, Nakazawa K, Wakabayashi M, et al. Silencing of the UCHL1 gene in human colorectal and ovarian cancers. Int J Cancer. 2006;119:1338–1344. doi:10.1002/ijc.22025.
- Yamashita S, Takahashi S, McDonell N, et al. Methylation silencing of transforming growth factor-beta receptor type II in rat prostate cancers. Cancer Res. 2008;68:2112–2121. doi:10.1158/0008-5472.CAN-07-5282.
- Takeshima H, Yamashita S, Shimazu T, et al. The presence of RNA polymerase II, active or stalled, predicts epigenetic fate of promoter CpG islands. Genome Res. 2009;19:1974–1982. doi:10.1101/gr.093310.109.
- Osada H. Introduction of new tools for chemical biology research on microbial metabolites. Biosci Biotechnol Biochem. 2010;74:1135–1140. doi:10.1271/bbb.100061.
- Brueckner B, Garcia Boy R, Siedlecki P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65:6305–6311. doi:10.1158/0008-5472.CAN-04-2957.
- Datta J, Ghoshal K, Denny WA, et al. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009;69:4277–4285. doi:10.1158/0008-5472.CAN-08-3669.
- Yoo J, Choi S, Medina-Franco JL. Molecular modeling studies of the novel inhibitors of DNA methyltransferases SGI-1027 and CBC12: implications for the mechanism of inhibition of DNMTs. PLoS One. 2013;8:e62152. doi:10.1371/journal.pone.0062152.
- Klisovic RB, Stock W, Cataland S, et al. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin Cancer Res. 2008;14:2444–2449. doi:10.1158/1078-0432.CCR-07-1320.
- Kuck D, Caulfield T, Lyko F, et al. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther. 2010;9:3015–3023. doi:10.1158/1535-7163.MCT-10-0609.
- Raynal NJ, Lee JT, Wang Y, et al. Targeting calcium signaling induces epigenetic reactivation of tumor suppressor genes in cancer. Cancer Res. 2016;76:1494–505. doi:10.1158/0008-5472.CAN-14-2391.
- Dahlin JL, Walters MA. The essential roles of chemistry in high-throughput screening triage. Future Med Chem. 2014;6:1265–1290. doi:10.4155/fmc.14.60.
- Snyder RD, Green JW. A review of the genotoxicity of marketed pharmaceuticals. Mutat Res. 2001;488:151–169. doi:10.1016/S1383-5742(01)00055-2.
- Nagao M, Ochiai M, Okochi E, et al. LacI transgenic animal study: relationships among DNA-adduct levels, mutant frequencies and cancer incidences. Mutat Res. 2001;477:119–124. doi:10.1016/S0027-5107(01)00113-0.
- Okonogi H, Stuart GR, Okochi E, et al. Effects of gender and species on spectra of mutation induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in the lacI transgene. Mutat Res 1997;395:93–99.
- MacPhee DG. Epigenetics and epimutagens: some new perspectives on cancer, germ line effects and endocrine disrupters. Mutat Res. 1998;400:369–379. doi:10.1016/S0027-5107(98)00046-3.
- Holliday R, Ho T. DNA methylation and epigenetic inheritance. Methods. 2002;27:179–183. doi:10.1016/S1046-2023(02)00072-5.